
Abstract
Background
Glutamate has been implicated in the pathophysiology of central nervous system diseases, including stroke.
Purpose
In this study, the neuroprotective potential of the newly synthesised thyrotropin-releasing hormone (TRH) analogue [Pyr-
Methods
Cortical neurons isolated from neonatal rats were used to evaluate the effects of the NP-2376. Cortical neurons were pre-treated with NP-2376 (6, 12 and 24 h) prior to glutamate (15 mM) exposure. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and neutral red uptake (NRU) assay, and oxidative stress by chloromethyl-2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) and glutathione assays.
Results
NP-2376 protected against glutamate-induced cortical neuron death and oxidative stress in a dose-dependent manner.
Conclusions
This study demonstrates the neuroprotective potential of TRH analogue NP-2376 against glutamate-induced toxicity, which is attributed to a decrease in oxidative stress.
Keywords
Introduction
Thyrotropin-releasing hormone (TRH) and its receptors bear widespread expression within the brain and spinal cord region as a neurotransmitter or neuromodulator.1, 2 TRH and its analogues modulate the central actions of neurotransmitters or neuromodulators like glutamate. The neuromodulatory role of TRH against diseases like neurotrauma, seizures and stroke/brain ischemia has been well reported.3–5 However, owing to the short half-life, poor stability and permeability across the blood–brain barrier, TRH has not been used much in the clinics due to its limitations.6, 7 There are studies in which TRH analogues have been investigated for neuroprotective potential.
8
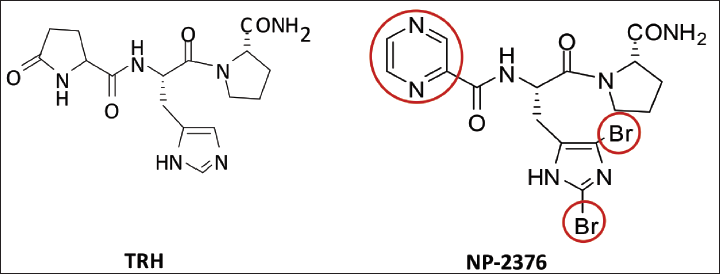
Hence, there is a clinical need to develop novel, safe and potent TRH analogues devoid of the above limitations.9, 10 We have synthesised a newer TRH analogue (NP-2376) in which pyroglutamyl residue is replaced with pyrazine ring system and the central histidine residue is substituted at the C-2 and C-4 positions of the imidazole ring with bromine atom, named as Pyr-
Glutamate is a key endogenous excitatory neurotransmitter in the central nervous system (CNS) and is a putative transmitter in the spinal cord. 12 Normal glutamate level (<2 µmol/L) is associated with excitatory neurotransmission during learning and memory, synaptogenesis, neuronal survival and neuronal plasticity; however, high concentration of glutamate leads to neuronal damage.13, 14 Thus, neuroprotective effects against glutamate-induced neurotoxicity have been postulated to be a therapeutic strategy for treating CNS disorders.4, 13, 15, 16 Therefore, we thought it would be worth exploring new TRH analogues, which are not glutamate antagonists but can protect against glutamate-induced toxicity. In the current study, the neuroprotective effect of NP-2376 was assessed on different models, namely cortical neurons from neonatal rats against glutamate-induced injury in the in vitro system by using neuronal viability and oxidative stress [chloromethyl-2,7-dichlorodihydrofluorescein diacetate (DCFH-DA)] and glutathione assays.
Material and Methods
Cortical Neuronal Culture
Sprague Dawley neonatal rats were used from the Central Animal Facility; National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar (IAEC 11/20). After decapitation, the 0–5 days rat pups; brains (n = 3–4) were isolated from the skull and transferred in the oxygenated phosphate buffer saline. The cortical area was separated from optical lobes, hypothalamus and cerebellum and placed in Dulbecco’s modified Eagle’s media (DMEM; 50%))/neurobasal medium (50%), supplemented with fetal bovine serum (FBS) (10%), 2% sodium bicarbonate and anti-biotic–anti-mycotic solution (100×, 1%) for 10 min to remove the residual blood. 17 Trypsinized tissues were dissociated with Pasteur pipettes for 1–2 min to make single-cell suspension. The contents were filtered through nylon cloth (size 70 µM) to get a clear single-cell suspension. Before experiments, cells were screened using a trypan blue dye exclusion test for cell viability, and the batches that showed cell viability >95% were only used in the present studies.
Experimental Design
Cortical neuronal cells (10,000) were plated in poly-
Assessment of Cell Viability
MTT Assay
Per cent neuronal viability was quantified by measurement of the formazan product formed because of the mitochondrial reduction of MTT, as reported elsewhere.5, 18, 19 Briefly, following different treatments, MTT (5 mg/mL of stock in Phosphate Buffer Saline) was added, and plates were incubated for 4 h. Following incubation, the plates were centrifuged, and 200 µL of dimethyl sulfoxide was added to the wells, and absorbance was measured at 550 nm in the micro-plate reader (Synergy HT, BioTek, USA).
NRU Assay
The viability of rat cortical neurons was measured by NRU assay as reported earlier.12, 20 It was used to measure the accumulation of the neutral red dye within the viable lysosomes of viable cortical neurons. Briefly, after the respective exposures, neuronal cells were washed with PBS. 0.4% Neutral red dye at the final concentration of 50 µg/mL was added to the complete medium and kept at 37°C for the next 3 h. Thereafter, a neutral red solution was discarded, and cells were washed with a solution containing 1% CaCl2 and 0.5% formaldehyde to remove any extracellular dye. Cells were further incubated in a mixture of acetic acid (1%) and ethanol (50%) for 20 min at 37°C to extract the dye from the lysosomes of viable uninjured cortical neuronal cells. The plates were finally read at 540 nm using a micro-plate reader (Synergy HT, Bio-Tek, USA).
Assessment of Oxidative Stress
DCFH-DA Assay
After respective exposures, cells were treated with DCFH-DA-dye (20 µM; 37°C; 30 min). Thereafter, the reaction mixture was removed, and 200 µL of PBS was added to each well. It was shaken in the dark for 10 min at normal temperature. Finally, fluorescence was estimated using multi-well micro-plate reader (Synergy HT, Bio-Tek, USA) on excitation/emission wavelength at 485/528 nm, as reported elsewhere.13, 20
Glutathione Assay
After respective exposure, cortical neurons were harvested with a rubber policeman and centrifuged for 10 min; supernatant was replaced with cold 0.4 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer containing 0.1 M phosphate and 2 mM ethylenediaminetetraacetic acid (EDTA) (pH 6.0) followed by sonication for 2–3 times for 10 s and centrifugation for 15 min at 4°C at 1,000 × g. The supernatant was replaced with an equivalent volume of metaphosphoric acid (MPA) in each tube (1:1 ratio). The tubes were vortexed well and kept at room temperature for 5 min for deproteination. Again, tubes were centrifuged for 2 min, and the supernatant was collected. The supernatant was added to the assay cocktail by mixing the MES buffer, reconstituted cofactor and enzyme mixture, water and reconstituted 5,5-dithionitrobenzoic acid (DTNB) to the wells. The absorbance was measured at 412 nm using multi-well plate reader (Synergy HT, Bio-Tek, USA).13, 21
Chemicals and Solutions
Pyrazine-
Chemical Structure of Thyrotropin-releasing Hormone (TRH) and TRH Analog NP-2376.
Statistical Analysis
Data are expressed as a percentage of control (mean ± standard error of mean). For the comparison of two groups, Student’s t-test was used, and for the comparison of multiple groups, one-way analysis of variance (ANOVA) and post-hoc Dunnett’s test were used. Statistical significance was considered at p < .05. Jandel Sigma Stat 2.0 software was used for the statistical analysis.
Results
Effect of NP-2376 Against Glutamate-induced Toxicity
The neurotoxic response of glutamate and the protective effect of NP-2376 in cortical neurons observed in MTT and NRU assays have been shown in Figures 2 and 3, respectively.
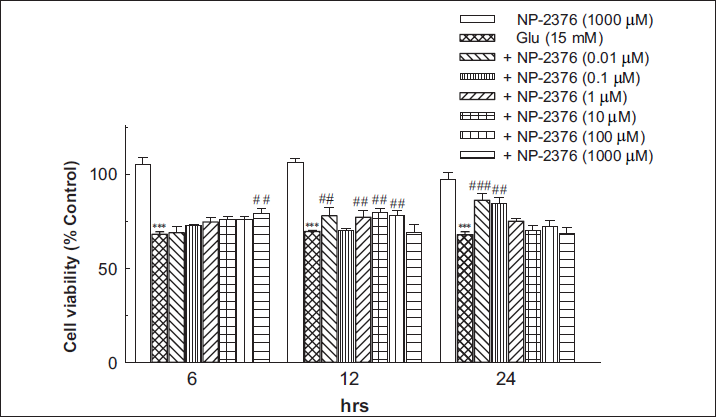
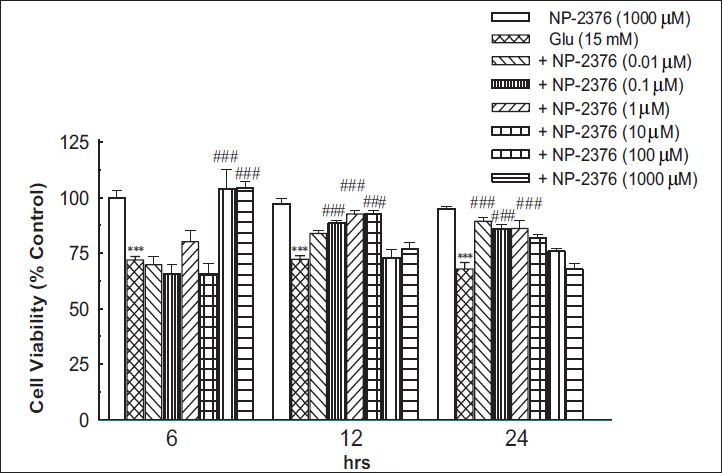
MTT Assay
Glutamate (15 mM) significantly reduced (p < .01) cell viability after 6 h (68.08 ± 1.46%), 12 h (69.64 ± 0.49%) and 24 h (67.91 ± 1.51%) exposure against control. Pre-treatment of neurons with NP-2376 at 1000 µM concentration for 6 h resulted in significantly (p < .01) improved cell viability (79.41 ± 2.14%). In glutamate-exposed neurons, NP-2376, for 12 h, exposure showed significant (p < .01) neuroprotection at 0.01 µM (77.93 ± 4.38%), 1 µM (77.19 ± 3.45%), 10 µM (79.81 ± 2.09%), 100 µM (78.37 ± 2.21%) and 1,000 µM (69.33 ± 3.98%). NP-2376 (0.01 µM and 0.1 µM) exposure for 24 h showed significant (p < .001) neuroprotection (86.16 ± 3.54% and 84.36 ± 3.07% respectively) (Figure 2). NP-2376 (1,000 µM) per se did not affect cell viability after 6 h (105 ± 3.5%), 12 h (106 ± 1.8%) and 24 h (97 ± 3.55%) on cortical neurons.
NRU Assay
Glutamate (15 mM) significantly (p < .001) reduced cell viability after 6 h (71.95 ± 1.49%), 12 h (72.28 ± 1.56%) and 24 h (67.91 ± 2.90%) against control (Figure 3). Pre-treatment of cortical neurons with NP-2376 for 6 h accorded significant (p < .001) neuroprotection against glutamate-induced toxicity at 100 µM (103.6 ± 9.05%) and 1,000 µM (104.35 ± 2.72). NP-2376 for 12 h exposure produced significant (p < .001) neuroprotection at 0.1 µM (88.45 ± 1.25%), 1 µM (92.58 ± 1.45) and 10 µM (92.58 ± 1.59%). 24 h exposure of NP-2376 also produced a significant (p < .001) neuroprotection at 0.1 µM (89.32 ± 1.55%), 1 µM (85.84 ± 2.04%) and 10 µM (86.05 ± 3.67%) respectively (Figure 3). NP-2376 (1,000 µM) per se did not affect cell viability after 6 h (99.81 ± 3.4%), 12 h (97.3 ± 2.3%) and 24 h (94.8 ± 1.3%) on cortical neurons.
DCFH-DA Assay
Glutamate (15 mM) significantly (p < .001) increased the ROS generation after 6 h (242.8 ± 40.62%), 12 h (150.99 ± 5.84%) and 24 h (141.95 ± 3.17%) against control. Six hours of NP-2376 pre-treatment to the cortical neurons resulted in significant (p < .001) neuroprotection against ROS generation at 1,000 µM (100.68 ± 5.87%). NP-2376 after 12 h exposure significantly (p < .001) reduced ROS at 0.01 µM (108.51 ± 4.14%), 0.1 µM (103.17 ± 5.99%) and 1 µM (103.51 ± 2.85%). NP-2376 after 24 h exposure significantly (p < .01) reduced ROS at 0.01 µM (118.74 ± 4.71%) and 0.1 µM (123.81 ± 10.86%) respectively (Figure 4). NP-2376 (1,000 µM per se did not produce any significant effects in ROS generation after 6 h (124.2 ± 5.28%), 12 h (119.4 ± 2.97%) and 24 h (119.7 ± 4.11%) in cortical neurons (Figure 4).
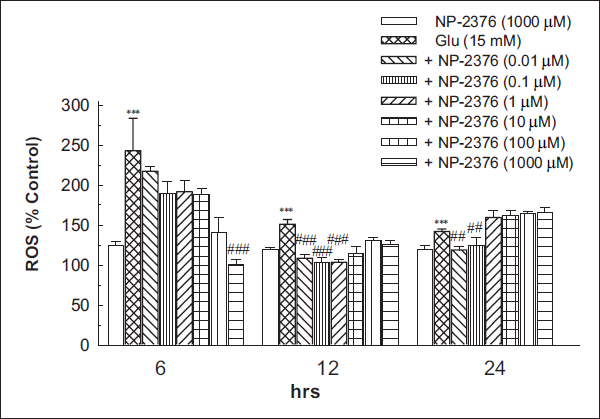
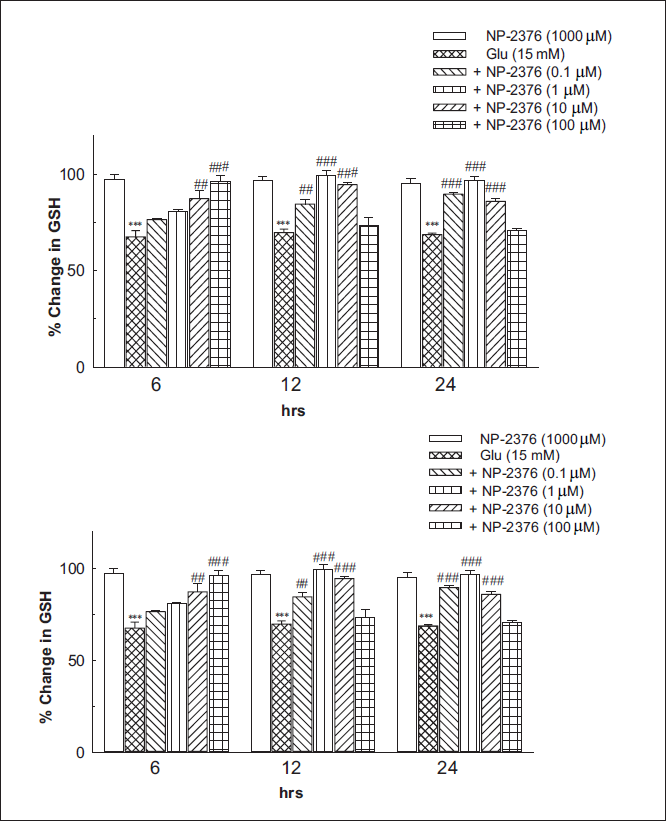
Glutathione Assay
Glutamate (15 mM) exposure for 24 h was found to deplete glutathione levels by 30% after 6 h (p < .001), (67.51 ± 3.26%), (69.73 ± 1.78%) after 12 h and (68.64 ± 0.82%) after 24 h against control. Pre-treatment of neurons with NP-2376 for 6 h resulted in significant (p < .001) reversal of glutathione depletion at 10 µM (87.24 ± 4.49%) and 100 µM (96.38 ± 2.65%). NP-2376 after 12 h exposure significantly (p < .001) reversed glutathione depletion at 0.1 µM (84.51 ± 2.12%), 1 µM (99.4 ± 2.56) and 10 µM (94.5 ± 0.96). NP-2376 after 24 h exposure significantly (p < .001) reversed glutathione depletion at 0.1 µM (89.59 ± 1.13%), 1 µM (96.69 ± 2.28) and 10 µM (85.87 ± 1.40), respectively (Figure 5). NP-2376 (1,000 µM) per se did not produce any significant effects on glutathione levels after exposure for 6 h (97.42 ± 2.32), 12 h (96.76 ± 1.85) and 24 h (95.31 ± 2.43) on cortical neurons (Figure 5).
Discussion
This study demonstrated the neuroprotective potential of a TRH analogue NP-2376 against glutamate-induced toxicity and oxidative stress. We observed significant neurotoxicity when glutamate exposure was given to the cortical neurons. Different mechanisms of glutamate-induced cell death were identified; one is the excitotoxicity pathway that relies on the over-activation of glutamate receptors, and another is the oxidative stress pathway, which involves the breakdown of the glutamate/cystine anti-porter with concomitant reduction in glutathione synthesis. The neurotoxic potential of glutamate is well recognised whenever it is released in excess or is not fully recycled. It also results in neuronal death (Figure 6).
22
Glutamate-induced neurotoxicity is known to play its part in stroke, seizures, Parkinson’s disease, as well as several other neurological disorders.23–25 Thus, neuroprotection against glutamate-induced toxicity may be helpful in preventing and/or treating neurological and neurodegenerative disorders.
26
Ischemia triggers the excessive release of presynaptic glutamate from the nerve terminals into the extracellular medium, with subsequent excitation of glutamate receptors. Glutamate activates N-methyl-
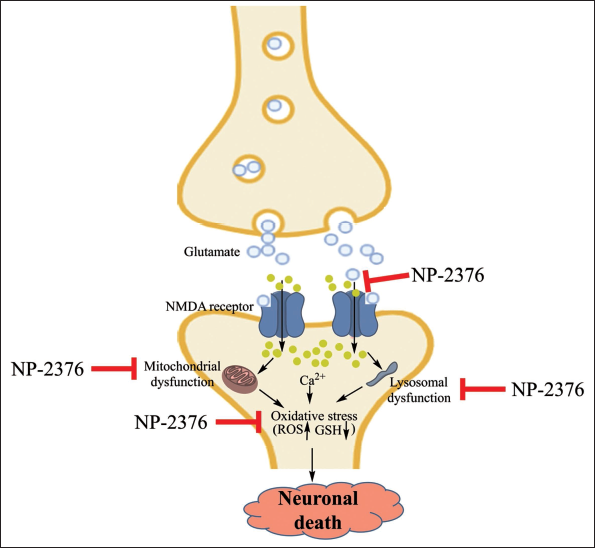
In order to understand the neuroprotective effects of newly synthesised TRH analogue NP-2376, we determined the glutamate-induced mitochondrial and lysosomal damage and oxidative stress-mediated neuronal damage. Glutamate-induced mitochondrial and lysosomal damage was evaluated by NRU assay in cortical neurons. Pre-treatment of NP-2376 showed protection against glutamate-induced neuronal death. The NRU involves the dye’s ability to readily penetrate cell membranes and accumulate intracellularly in lysosomes by predominately non-ionic diffusion. The altered cell surface or the sensitive lysosomal membrane produced by excess glutamate led to lysosomal fragility and other irreversible changes.32, 33 These alterations resulted in decreased NRU and binding, which helped distinguish between viable, damaged or dead cells via spectrophotometric analysis.32, 33 The increased uptake of neutral red after chemical exposure indicates a sensitive, integrated signal of both neuronal integrity and survival. Pre-treatment of cortical neurons with NP-2376 resulted in significant neuroprotection against glutamate toxicity. An interesting effect of NP-2376 appears to be that, at lower doses of the drug, the protection appears later and may be more sustained, reaching a maximum at 24 h; but at high doses, neuroprotection is seen early, is more transient, and is gone by 24 h. A similar effect has been observed with other pharmacological agents, wherein, by increasing the dose and duration, the pharmacological effect is lost. This could be due to the non-specific effects of NP-2376 or the desensitisation/downregulation of the TRH receptor.
We observed that glutamate insult to the cortical neurons led to an increase in oxidative stress, possibly through ROS generation and the depletion of cellular glutathione levels, which may lead to neuronal death. Glutamate-induced production of ROS has also been demonstrated elsewhere, where diethyl maleate caused depletion of neuronal glutathione; this indicates a prominent glutathione defence against ROS production in these cells. Moreover, glutamate has also been shown to increase translocation and activation of protein kinase-C, which is further potentiated by the direct activation of PKC and homologous receptor desensitisation/downregulation. Although the exact procedure of TRH’s role in the neuroprotection of the CNS is not well defined, some evidence indicates the involvement of excess glutamate effects. TRH/analogues directly act on glutamate receptors to exert their effect through selective G-protein coupled receptor (GPCR) and/or through a different receptor-independent mechanism. Glutamate-stimulated increases [Ca2+] i in cortical neurons are shown to inhibit TRH and selectively depress glutamate-mediated neuronal activity. Continuous TRH exposure to its GPCR receptors activates phospholipase C (PLC) through Gαq/11 signalling, causing increased intracellular calcium, protein kinase C activation and homologous receptor desensitisation/downregulation. The mGluR1 and mGluR5 glutamate receptors use the same signalling cascade. TRH does not appear toxic, while metabotropic glutamate R1 receptor-mediated PKC activation, along with an increase in intracellular calcium, is known to potentiate the NMDA receptor, which has been implicated in glutamate excitotoxicity; besides, it has been demonstrated that if two GPCR’s share the same signalling G-protein, downregulation of the homologous receptor may result in downregulation/desensitisation of a heterologous receptor that shares the same signalling G-protein. Furthermore, mGluR1 receptor desensitisation is also known to exert protective effects against neuronal death. Since prolonged TRH exposure results in desensitisation/downregulation in in vitro culture, it may be possible that TRH analogues can have a potential neuroprotective effect, in part, through heterologous downregulation of Group I metabotropic glutamate receptors. In totality, these events could explain the eventual oxidative burst, which caused diminished neuronal glutathione and increased cytotoxicity.20, 34 This was further confirmed by decreased glutamate-induced ROS generation and elevation in the levels of glutathione with pre-treatment of NP-2376. The TRH analogue NP-2376 was found to reduce the dichlorofluorescein (DCF) product formation by raising glutathione levels and inhibiting the generation of ROS, showing its neuroprotective nature (Figures 4–6).
TRH analogues like RX 77368, CG-3509, RGH-2202, JTP-2942 and taltirelin have also been reported for their neuroprotective effects27–31 in rats, supporting that the newer TRH analogues, NP-2376, might be beneficial in the treatment of the CNS toxicity. The binding of glutamate on NMDA receptors leads to an influx of calcium into the neuron, which increases the calcium influx, causing increased excitotoxicity and leading to the production of mitochondrial and lysosomal dysfunctions. These events generate ROS, leading to a reduction in endogenous anti-oxidants such as Glutathione (GSH); these events are attenuated by NP-2376 (Figure 6). In conclusion, the current experimental finding suggests that NP-2376 is neuroprotective in the in vitro models, and its neuroprotective potential may be attributed to a reduction in excite-toxicity and oxidative stress.
Footnotes
Acknowledgement
Mr Bhupesh Vaidya’s assistance in making artwork is kindly acknowledged. The authors are thankful for the financial assistance from the Department of Pharmaceuticals, Ministry of Chemical and Fertilizers, Government of India.
Author’s Contribution
Mallikarjuna R. Sunkara and Jitendra N. Singh have equally contributed in this work.
Statement of Ethics
Not applicable.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Financial assistance is provided by the Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Government of India.
ICMJE Statement
This manuscript conforms to the ICMJE Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals.