
Abstract
Background:
Systemic mastocytosis (SM) is a diverse condition characterised by abnormal growth and activity of clonal mast cells. It can have an extensive systemic and cutaneous involvement. Diagnosis is challenging because of variable and non-specific presentation, ranging from pruritus and hypotension to multi-organ manifestations, leading to delayed recognition.
Case details:
A 68-year-old female presented with frequent syncopal episodes, hemodynamic instability, and severe anaemia.
Results:
Initial investigations showed pancytopenia (Hb 2.9 g/dL, TLC 2.19 × 109/L, platelets 7 × 109/L) and splenomegaly (12 cm). A bone marrow biopsy was performed due to persistent pancytopenia, revealing a hypocellular marrow with dense infiltrates of spindle-shaped mast cells (16%). Immunohistochemistry with CD117 and toluidine blue confirmed mast cell infiltration. Total serum tryptase was elevated at 42 ng/mL.
Conclusion:
The patient was diagnosed with aggressive SM with features of mast cell degranulation and bone marrow infiltration. She was managed with low-dose glucocorticoids, packed red cell transfusions, and supportive care. This case highlights the challenges in diagnosing SM due to its non-specific, multi system clinical presentation and the need for high clinical suspicion.
Introduction
Mast cells are tissue-bound immune cells that have a pivotal role in allergic and inflammatory responses. They originate from haematopoietic progenitors and mature at specific sites, where their functions vary. Systemic mastocytosis (SM) represents a spectrum of clonal mast cells.[1] The 2016 classification by the World Health Organization (WHO) categorised SM as a subset of myeloproliferative neoplasms. This framework distinguishes the condition by its clinical presentation, primarily separating it into skin-focused forms and more complex systemic variants.[2] SM is defined by the systemic proliferation of aberrant mast cells across the bone marrow and extra-skeletal organ systems.[3,4]
SM is a rare disorder with an estimated prevalence of around 10 cases per 1 lakh population.[4] Cutaneous mastocytosis classically presents with symptoms ranging from localised urticaria to angioedema, while SM may present with multi-system features include flushing, abdominal pain, tachycardia, diarrhoea, hypotension, musculoskeletal pain and neuropsychiatric symptoms.[1]
Case Details
A 68-year-old lady presented to the emergency department with frequent episodes of loss of consciousness for the past 1 month. Each episode was associated with hypotension, palpitation and fatigue. The course of the episode was delineated by the patient as a sense of fatigue and dizziness, which was followed by an episode of sudden blackout. The patient usually gained consciousness around 5–10 minutes later; however, palpitations, fatigue and nausea persisted after the episode for several minutes.
None of the bystanders reported any abnormal body movement or any involuntary stool and urine passage during the episode. The patient did not report any focal neurological deficits or sensory phenomena during or after the event. The patient denied fever, headache, aural fullness, or any hearing disability. The episodic complaints occurred without diarrhoea or chest discomfort. Additionally, she did not report any skin rash, pruritus, or poor oral intake. Her previous medical history was insignificant.
On examination, she was found to be hypotensive with a blood pressure of 86/52 mmHg, tachycardia (pulse:114/minute), SpO2 of 95%, and random blood sugar levels of 127 mg/dL. With an intravenous fluid bolus, hemodynamic stability was achieved within a span of one hour. Lung and cardiac examination did not reveal any murmur or bilateral bibasilar crepitation. A chest X-ray was normal. Electrocardiogram was suggestive of sinus tachycardia and the transthoracic echocardiography showed normal function of heart. Holter monitoring did not reveal any significant arrhythmia.
Baseline laboratory investigations revealed pancytopenia with a hypo-proliferative marrow with haemoglobin of 2.9gm%, total leukocyte count of 2.19 × 109/L (59% neutrophils, 32.9% lymphocytes, 6.8% monocytes) and platelet count of 7,000 × 109/L with corrected reticulocyte count of 1.1%. Peripheral blood smear revealed lymphocytosis. Notably, her routine nutritional workup was within normal limits. Baseline laboratory evaluations—including metabolic, renal, hepatic, and thyroid panels—were unremarkable. Her urinary and serum metanephrine and 5- Hydroxy indoleacetic acid (5-HIAA) were negative. Ultrasonography revealed a normal liver and mild splenomegaly up to 12 cm in oblique dimension.
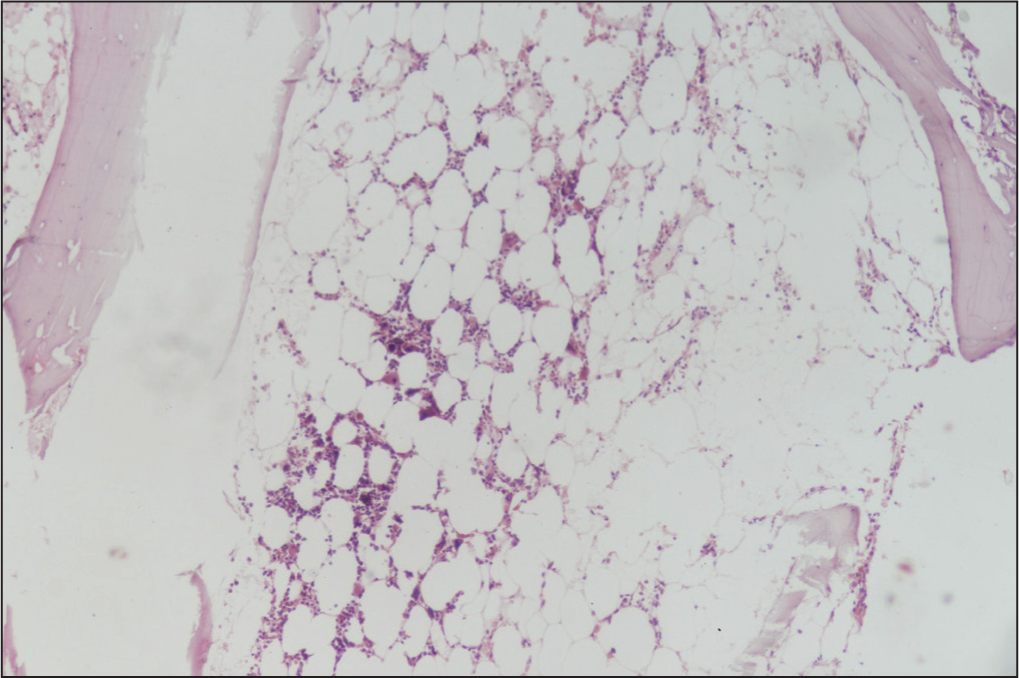
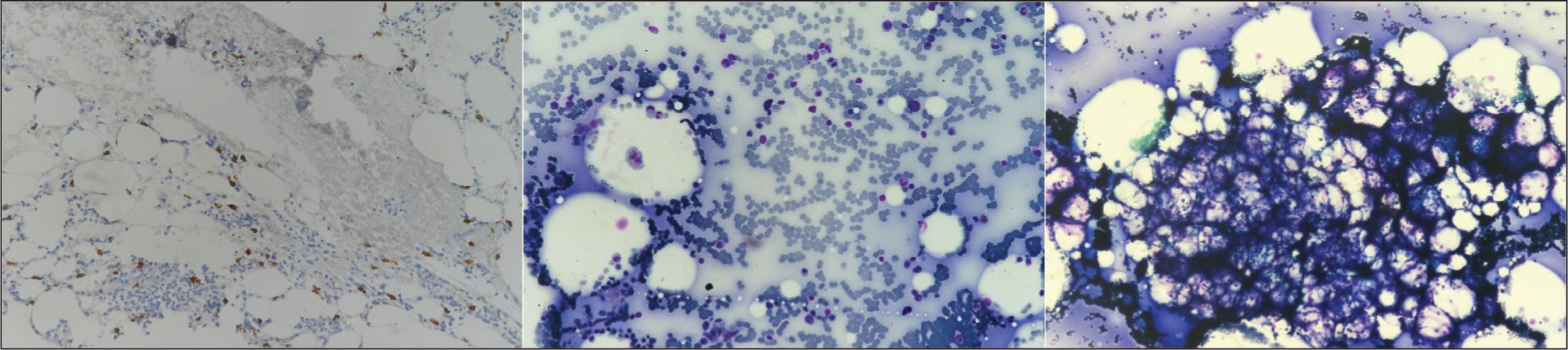
Bone marrow biopsy and aspiration revealed hypocellular marrow with dense spindle-shaped mast cells infiltrate (16%) [Figure 1]. Immunohistochemistry with CD117 marker and toluidine blue staining confirmed mast cell proliferation [Figure 2]. Additionally, total serum tryptase level was elevated at 42 ng/mL. Based on the above clinical findings, she was diagnosed as a case of aggressive SM with features due to mast cell degranulation and bone marrow infiltration leading to evident cytopenia. She was managed with low-dose glucocorticoids and packed cell transfusion, along with other supportive treatment. Histamine antagonists were not prescribed, and she was discharged after counselling and educating her about other related signs and symptoms, including anaphylaxis. She has been on our routine outpatient follow-up and has currently shown significant improvement.
Bone marrow biopsy and aspiration of the patient showing pancytopenia and hypo-proliferative marrow with dense infiltrates of spindle -shaped mast cells[4]
Immunohistochemistry with CD17 marker and toluidine blue staining confirming the presence of mast cells in the bone marrow[4]
Discussion
SM is the clonal expansion of mast cells triggered by constitutive activation of the KIT receptor tyrosine kinase. This is predominantly attributed to the c-KIT D816V point mutation (80% of cases), which confers ligand-independent signalling.[1,2] Upon activation, the proto-oncogene encodes KIT (CD117), a transmembrane tyrosine kinase receptor. KIT expression is characteristic of early haematopoietic progenitor cells, though it is also maintained in specific terminally differentiated populations, most notably the interstitial cells of Cajal in the gut, respiratory epithelium, skin, etc.[1] These neoplastic cells may also express CD2 and/or CD25 in addition.[2]
Mast cells contain various mediators such as histamine, tryptase and cytokines responsible for diverse systemic manifestations. Triggers include immunologic stimuli, drugs, venoms, physical stress and neuropeptides.[1] The WHO classification distinguishes between cutaneous and systemic disease, with diagnostic criteria based on tissue histology, immunophenotyping and molecular testing [Table 1].
Criteria to diagnose systemic mastocytosis[4]

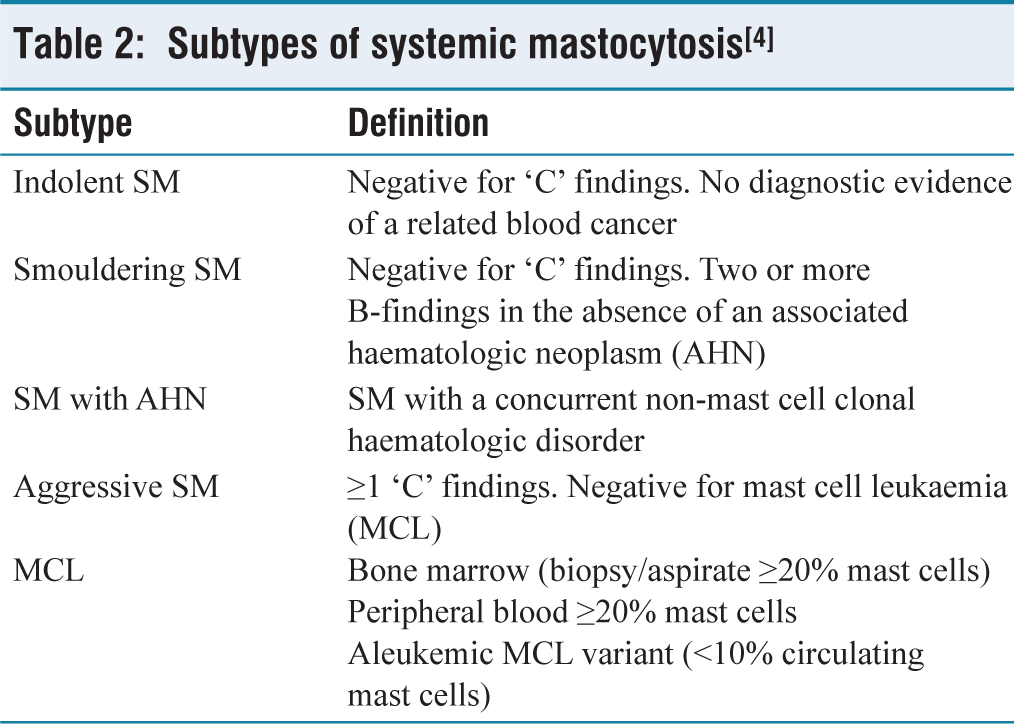
SM is further stratified into distinct subtypes based upon B-findings and C-findings.
B-findings include:
A bone marrow biopsy demonstrating >30% mast cell infiltration and/or a basal serum tryptase level exceeding 200 ng/mL. Presence of non-mast cell lineage dysplasia or myeloproliferation remains sub-threshold for a formal diagnosis of an associated haematological neoplasm (AHN), often accompanied by normal or only mildly altered peripheral blood counts. Clinical or radiographic evidence of lymphadenopathy and/or hepatosplenomegaly (palpable or detected via imaging) in the absence of significant functional impairment.
C-findings are characterised by:
Bone marrow failure manifesting as one or more cytopenias: an absolute neutrophil count (ANC) < 1.0 × 109/L, haemoglobin (Hb) < 100 g/L or a platelet count < 100 × 109/L. Palpable hepatomegaly with evidence of liver dysfunction, ascites and/or portal hypertension. Skeletal involvement characterised by large osteolytic lesions with/without pathological fractures (excluding fractures caused by osteoporosis). Palpable splenomegaly associated with hypersplenism. Weight loss as a result of malabsorption due to mast cell infiltration in the gastrointestinal system.
Various subtypes of SM have been described further based on B and C-findings [Table 2].
Subtypes of systemic mastocytosis[4]
Identifying SM often requires a high degree of suspicion. The patient can present with a wide range of clinical symptoms, including syncope, light-headedness, abdominal pain, diarrhoea, nausea, vomiting, generalised body aches, osteoporosis, wheezing, shortness of breath, nasal congestion, generalised fatigue, various neuropsychiatric symptoms, etc. Many patients cycle through multiple specialists and facilities without finding the answers or relief they need.[5,6]
Treatment of SM in resource-constrained settings is mainly supportive, focusing on trigger avoidance, use of antihistamines, and oral cromolyn for gastrointestinal manifestations. Supplementary treatments like leukotriene antagonists or H2/PPI blockers may be used as needed. For specific clinical profiles, specialised options including omalizumab, corticosteroids, cytoreductive agents, or tyrosine kinase inhibitors can be evaluated.[7]
Conclusion
With this wide array of symptomatology, often the delayed presentation of the patient to a physician contributes towards the disease being either misdiagnosed or easily missed at the primary care point. A high degree of clinical suspicion is essential for recognising patients with diverse, non-specific symptoms. Careful history examination and a comprehensive laboratory evaluation are required to reach the final diagnosis, which could drastically improve the quality of life of the patient as treatment is so simple and very effective.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Institutional Ethical Committee Approval Number
A proper written consent is present, which was obtained from the patient for the use of the data related to this case.
Informed Consent
Written informed consent was duly signed by the patient. The consent was obtained after explaining to the patient that no identity would be revealed and the case information, including pictures will be used for educational purposes only. The patient gave positive consent for publication, and the authors certify that written patient consent is present and procured for publication.
Credit Author Statement
AD, JK: Case presentation, data collection, investigations and writing of original draft.
MG: Literature review, writing of original draft, including conclusion, references.
MG, AT: Intellectual content, literature search, manuscript final editing and review.
Data Availability Statement
The data supporting the findings of this case report are included within the article. Additional information related to the case may be available from the corresponding author upon reasonable request, subject to patient confidentiality and ethical considerations.
Use of Artificial Intelligence
No artificial intelligence (AI) tools were used in the design, analysis, or interpretation of the data in this study.