
Abstract
The study emphasizes the importance of Knoevenagel condensation for forming carbon—carbon bonds to create biologically active α, β-unsaturated compounds. In this research, arylidenemalononitriles (AMN) (
Keywords
Introduction
The most common complications of antibiotic resistance are neglect and maltreatment. In the world, antibiotic resistance is responsible for more than 35,000 deaths yearly. 1 It is essential to search for a new drug due to the availability of medicines and the growing incidence of drugs for inhibitors. In general, malononitriles and cyano-esters are highly reactive chemicals. In condensation reactions, the unstable methylene group is used commonly as a nucleophile. The Knoevenagel condensation frequently uses the reagents malononitrile and cyano-esters, which are methylene-active compounds. This feature makes them a vital chemical in industrial, agricultural, and medicinal applications.2–5 For aldol condensation, base catalysts are commonly employed. Novel solid bases can be utilized to study several base-catalyzed reactions, such as the Knoevenagel condensation between benzaldehyde and malononitrile or benzaldehyde and cyano-esters. 6 The end product of this reaction, arylidenemalononitriles (AMNs) and also ethyl cyano-phenyl-acrylates (ECPAs), have many applications in the pharmaceutical, pharmacology, biotech, and fragrance industries. 7 The biological properties of AMNs and ECPAs derivatives, particularly employed in organic chemistry as both a transitional molecule and a target molecule, have been well explored. The relative position of the substitution on the ring structure has a significant as well as considerable impact on the physiological action of AMNs. The most active position is the 2-substituted position, followed by the 3-substituted position's decline in activity, whereas, the 4-substituent's gradually decrease in activity until it completely disappears.4,8,9 They act as essential synthesis intermediates, mainly in cyclization reactions, for the synthesis of different organic compounds. 10 Pharmaceuticals known as carbonic anhydrase inhibitors (CAIs) suppress the catalytic activity in carbonic anhydrases (CAs). Carbon dioxide is reversibly converted to carbonate and proton by CAs with Zn2+ at the active site.11,12 Recently, research has demonstrated that AMNs derivatives can effectively suppress the acetylcholine esterase (AChE), CA-I and CA-II in erythrocytes in vitro and silico. They were also investigated for the prospective antibacterial effects. 4 According to reports, AMNs and also ECPAs can be utilized as riot-control agents or as fungicides, insecticides, cytotoxic agents against tumors, and anti-fouling agents. 13 The key reason for further in silico research against various infections is the efficacy of the produced crystals against five human pathogenic bacteria during in vitro experiments, where it could show a promising result. AMN and ECPA derivatives had distinct and optimized chemical structures, so these structures were chosen for further computational analysis. Data on computational techniques, like DFT, MD, chemical stability, protein-ligand interactions, FTIR, UV-vis, ADMET, QSAR, drug-likeness and many chemical descriptors, are computed. Then, to determine the binding score of the ligand with the active site of proteins, the study of molecular docking is the primary tool and the core idea of these studies. To determine the acceptability of these derivatives, they are compared to an FDA-approved drug: ampicillin (AMP). So far, this study clearly shows the assessment of in vitro and silico analysis, molecular docking in addition to molecular dynamics via NMA analyses, and structure-activity relationships.
Materials and methodology
Materials
Structures of arylidenemalononitriles (1–4) and ethyl 2-cyano-3-phenylacrylates (5–7)
In this research, some analogues of arylidenemalononitriles (AMN) (
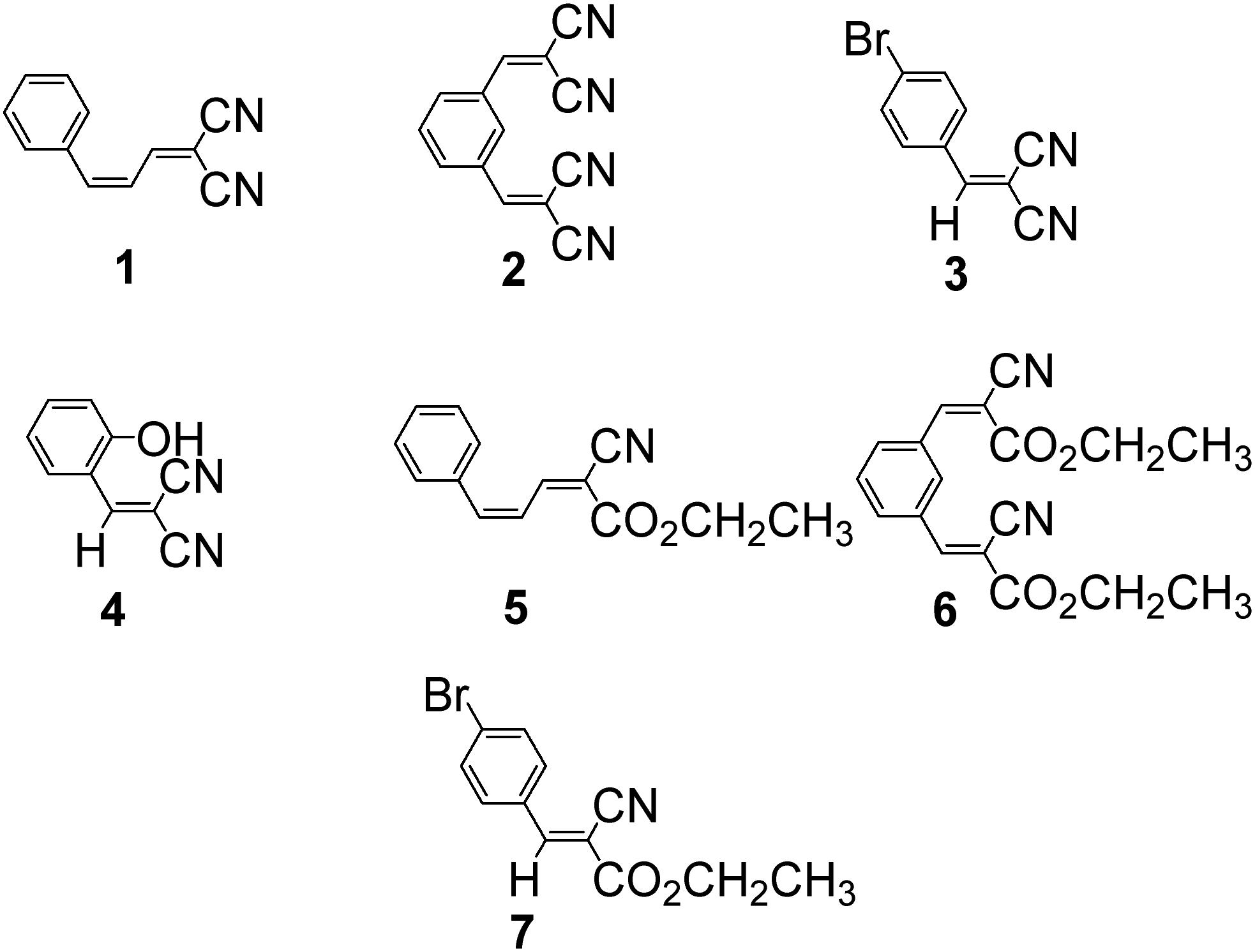
Chemical structures of arylidenemalononitrile (AMN) (
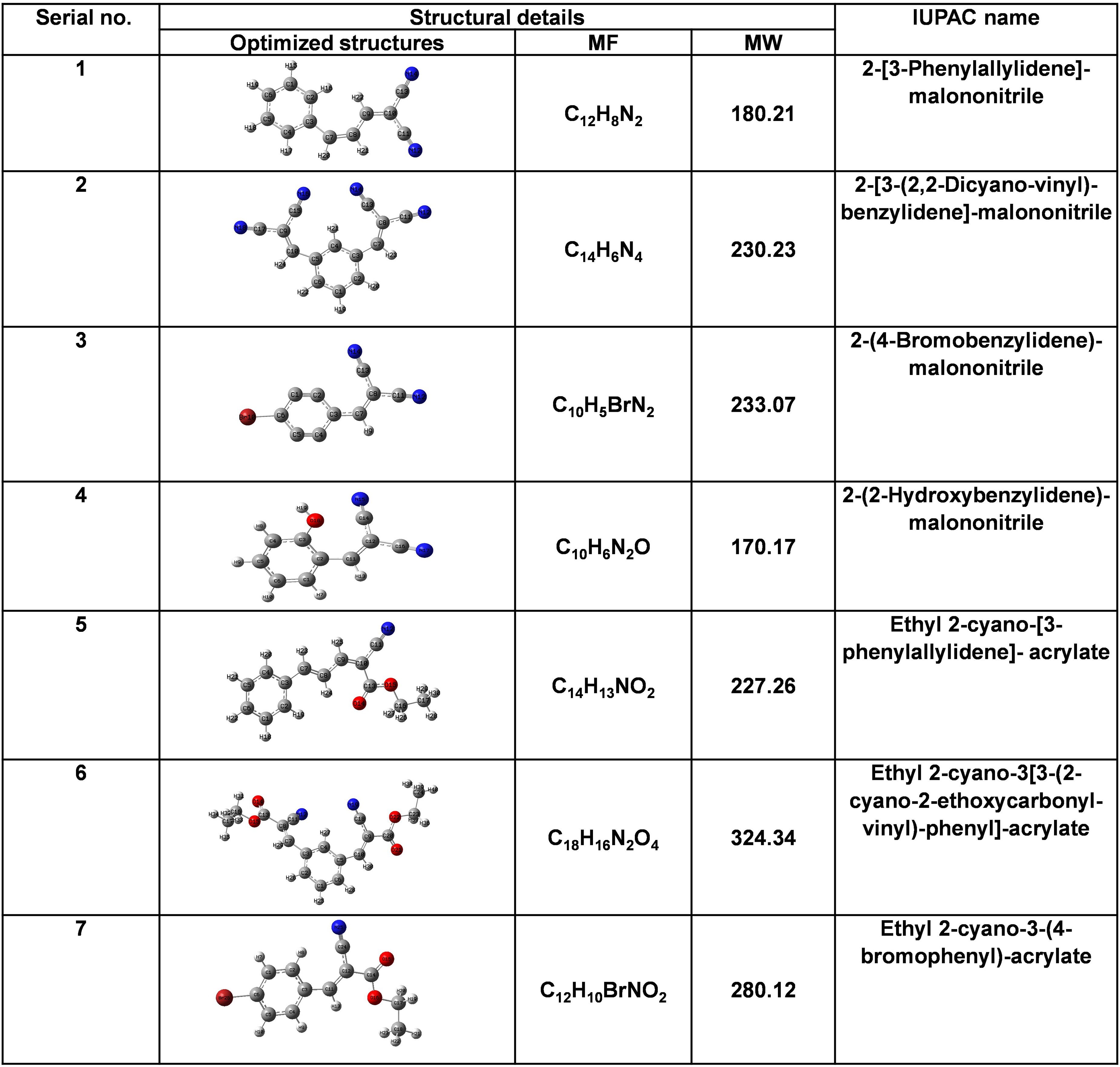
Structural details and IUPAC names of AMN and ECPA derivatives (
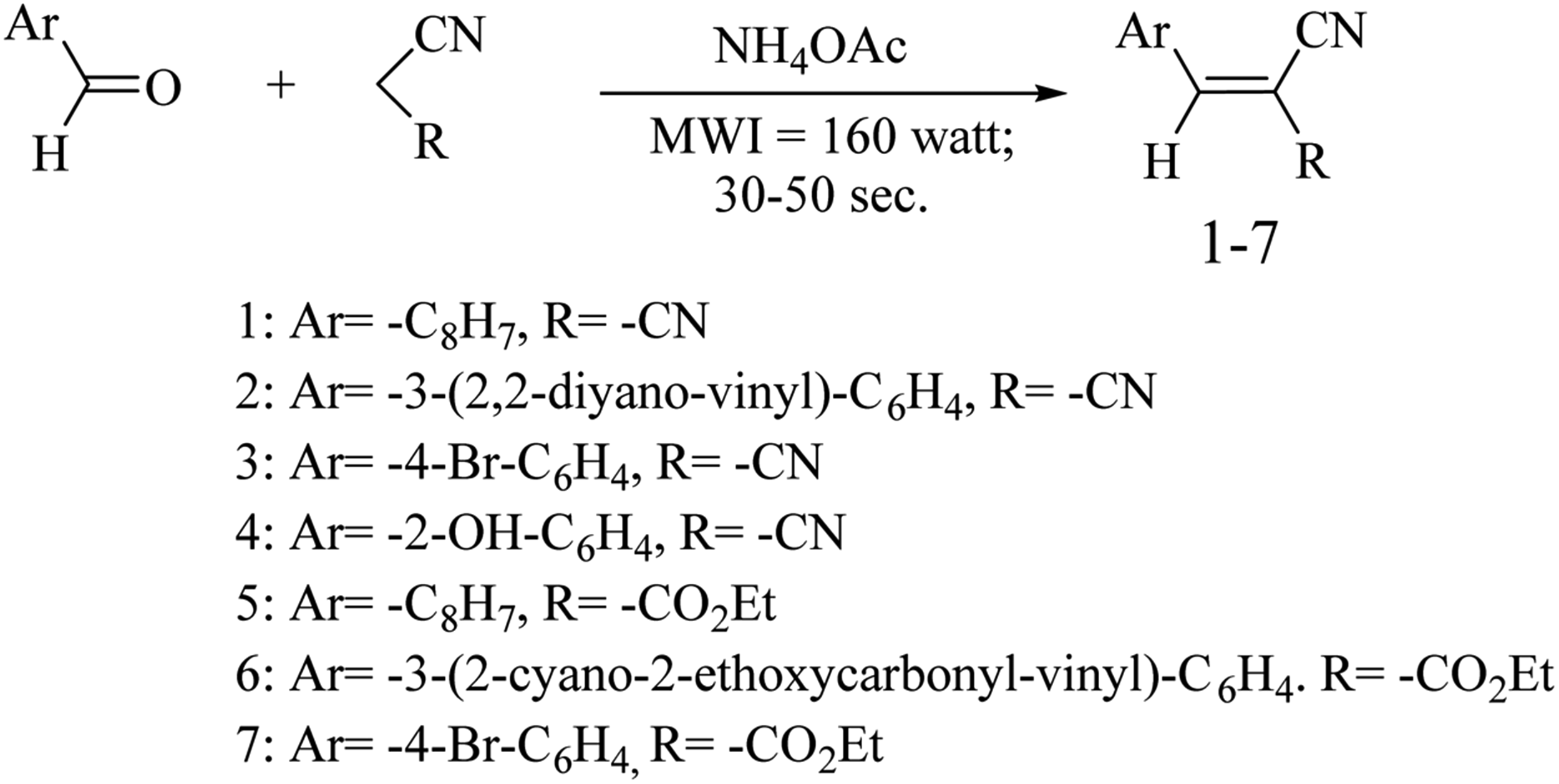
Synthetic method of AMN (
Methodology
In vitro antibacterial evaluation
Five bacteria, including B. cereus, S, typhi, E. coli, S. dysenteriae, and P. aeruginosa, were utilized to investigate the antibacterial efficacy of the tested AMN and ECPA analogs (
Geometry optimization of ligands
People's 6-311G + (d, p) 17 basis set was used in conjunction with density functional theory (DFT) by using Gaussian 09 W (D.01). 18 Lee, Yang, and Parr's (LYP) correlated functional and Backe's (B) three-parameter hybrid equations were also used to ascertain their physicochemical and spectral features. 19 For the electronic absorption calculation, time-dependent (TD) density functional theory (TD-DFT) 20 was applied under the same hybrid functionals and basis set. The inherent reactivity of the highest occupied molecular orbital (HOMO) as well as the lowest unoccupied molecular orbital (LUMO) is a result of their unique characteristics. 21 The HOMO and LUMO values were calculated and converted to electron volts (eV). The following formulas have been used to determine the overall chemical reactivity and molecular orbital features 22 ; energy gap, Δε = εLUMO−εHOMO; ionization potential, I = −εHOMO; electron affinity, A = −εLUMO; electronegativity, χ = (I + A)/2; chemical potential, μ = −(I + A)/2; hardness, and softness, S = 1/2 η.
Preparation of proteins, molecular docking and non-bonding calculations
The 3D crystal structures of the studied proteins i.e., PDB ID: 1AH7, 3FHU, 6DR3, 3FHH and 5X6F, respectively were collected from the online RCSB protein data library (https://www.rcsb.org/). PyMol (1.7.4) software was used to eradicate water molecules, heteroatoms, and co-crystalline ligands to build the protein chains.
23
The Swiss PDB program (4.1.0) was used to minimize the energy and reduce bad atom interaction. Here, four analogs of AMN (
Prediction of ADMET, biological activities and drug-likeness
The ADMET (absorption, distribution, metabolism, excretion, and toxicity) parameters are essential in pharmacological studies. Here, the ADMET profile was predicted using the admetSAR (http://lmmd.ecust.edu.cn/admetsar1/predict/) server. 26 The “prediction of activity spectra for substances (PASS)” and Swiss ADME web applications were used to forecast the biological activities of organic compounds that resemble drugs. 27 All results were predicted with the aid of a “simplified molecular input line entry system (SMILES)” and structural data files in each case.
Molecular dynamics simulation
To validate the outcomes of molecular docking as well as investigate the perdurability of protein-ligand complexes, a simulation of molecular dynamics is frequently used. Here, the C-α atoms of the receptor proteins were simulated employing the NMA (normal mode analysis) dynamics utilizing the “iMODS server (https://imods.iqfr.csic.es/)”. 28
Quantitative structure-activity relationship (QSAR)
In computer-aided drug design, QSAR is assessed by comparing it to eight drug-related descriptors from ChemDes, (http://www.scbdd.com/chemdes/) a free online tool for calculating molecular descriptors. 29 “Quantitative structure-activity relationships (QSAR)”, based upon mathematical and statistical correlations, were utilized to constitute links between the physicochemical properties of chemical substances and their biological functions.30,31 Utilizing “multi-linear regression (MLR)” equations, a model was developed to forecast the QSAR. 29
Here,

Results and discussion
In-vitro antibacterial analysis
Antibacterial activity of AMN and ECPA analogs (

Diameter of zone of inhibition in mm of tested compounds against pathogenic bacteria (100 µg (dw)/ disc). Values are represented as Mean ± SD with all the experiments and indicate the significant differences with (P < 0.05) at the experimental conditions.
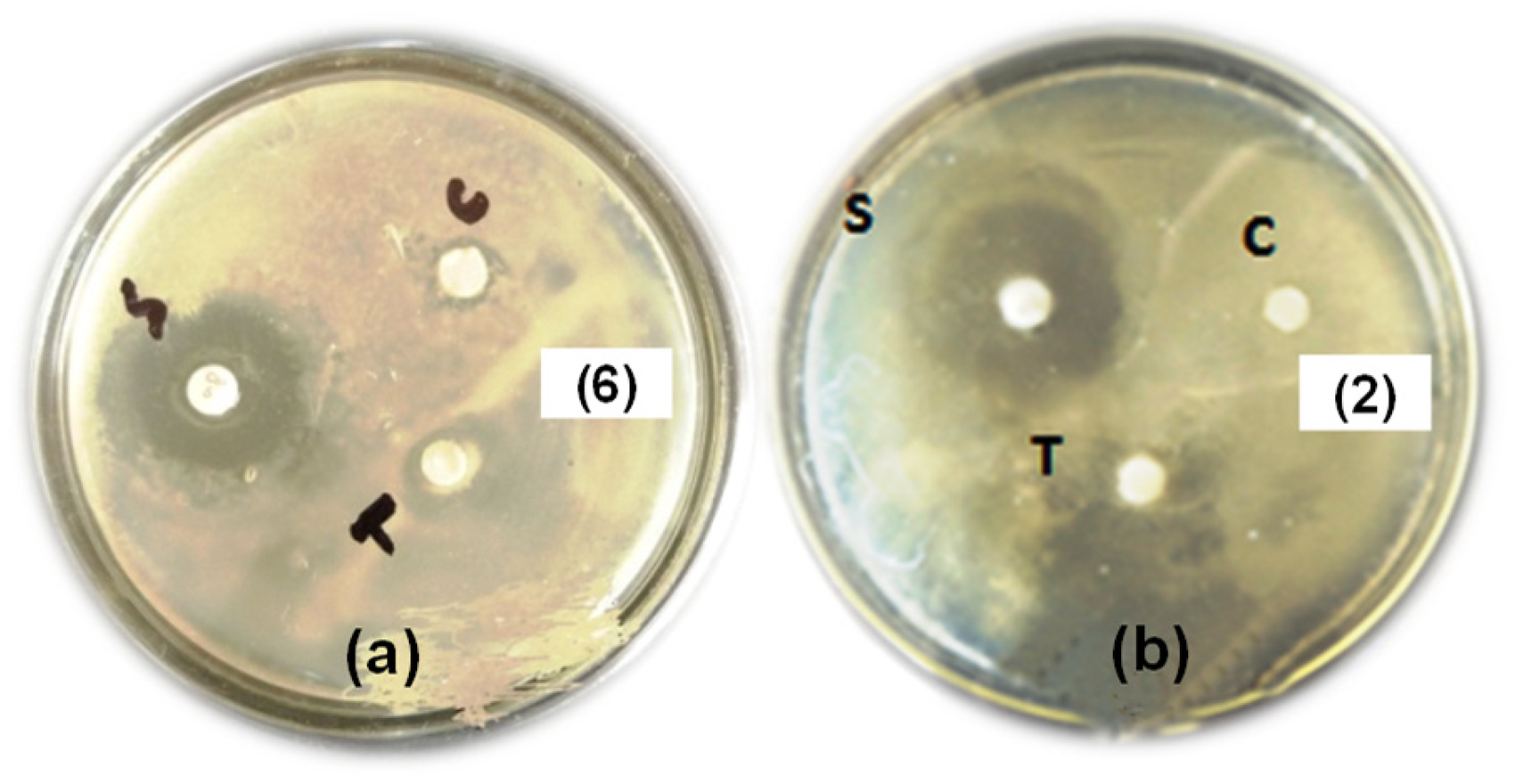
Diameter of inhibitions (mm) (a) compound
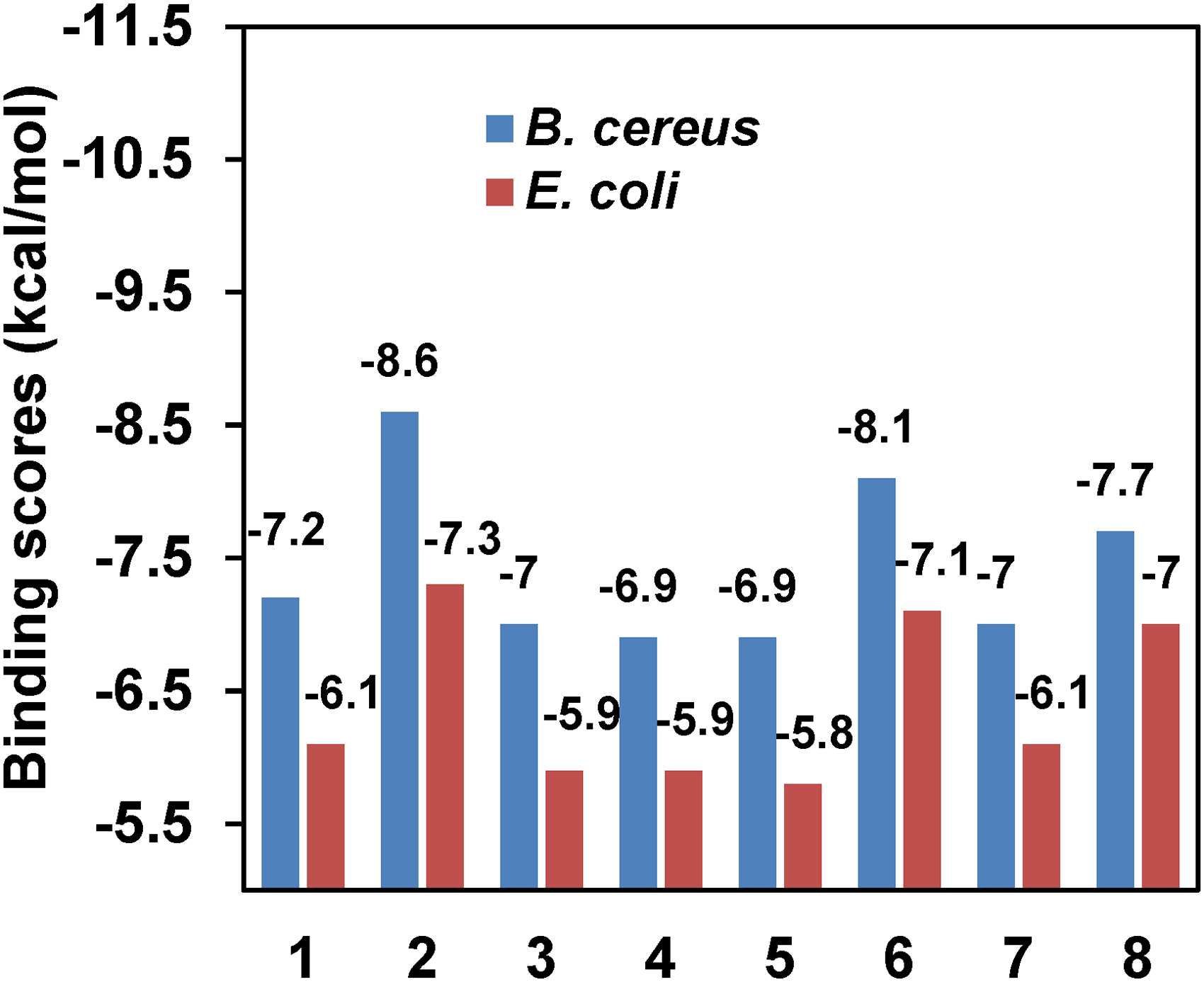
Binding affinities of the tested analogs (
Inhibition against the pathogenic organisms by the test chemicals
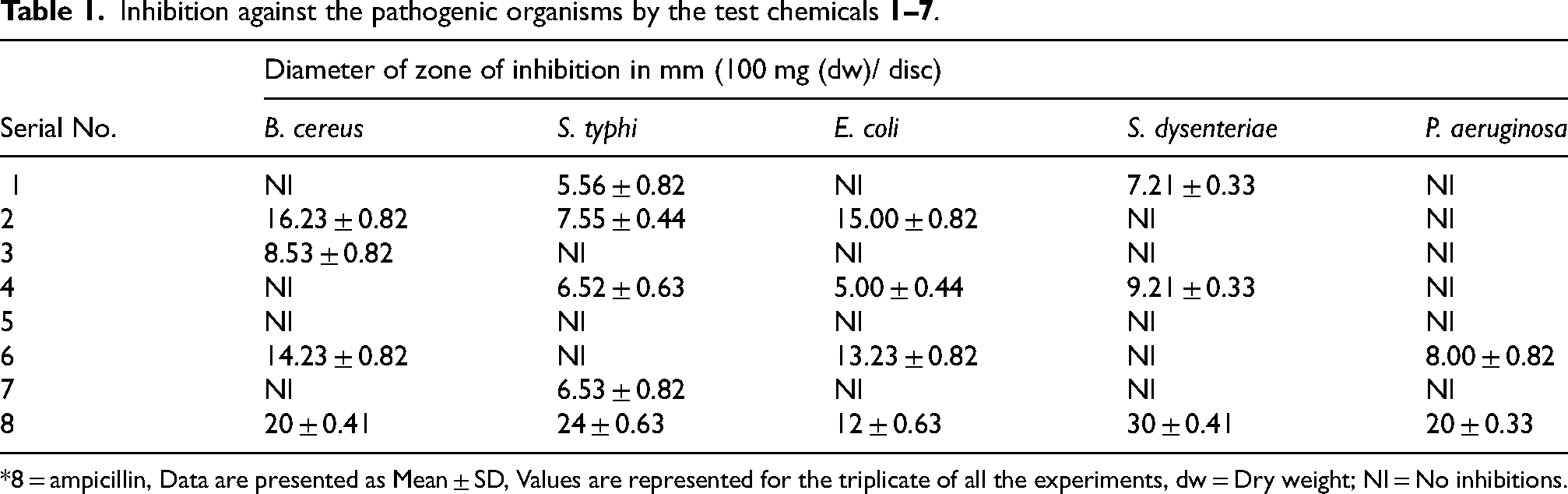
*8 = ampicillin, Data are presented as Mean ± SD, Values are represented for the triplicate of all the experiments, dw = Dry weight; NI = No inhibitions.
Chemistry of the optimized structures and thermodynamic analysis
The optimized structures of AMNs (
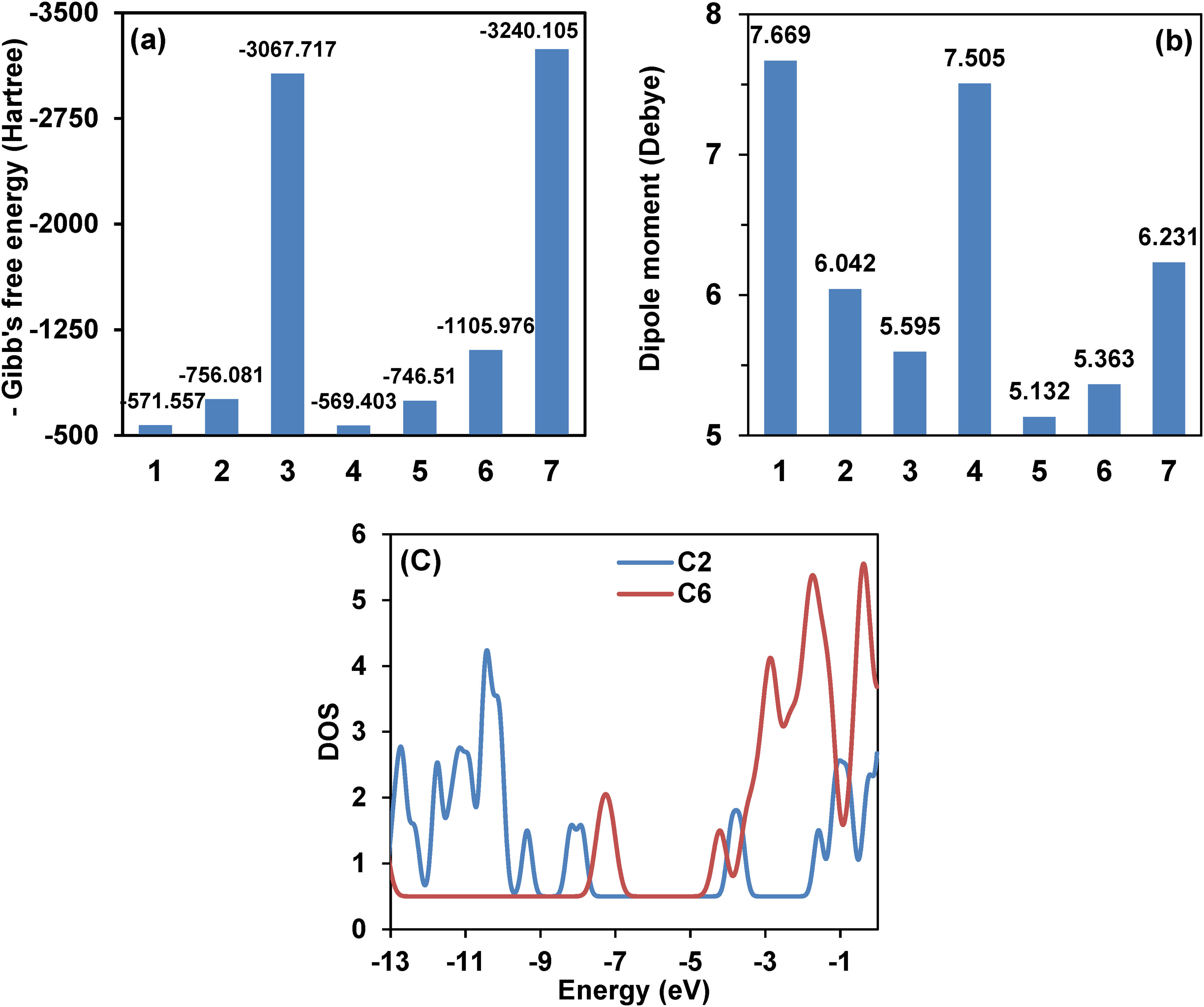
(a) Gibb's free energy (Hartree), (b) dipole moment (Debye) of the AMN (
Frontier molecular orbital analysis
The electronic excitation from HOMO to LUMO explains the electronic transition in a system which is related to the change from the ground state to the first excited state
34
and is also associated with chemical reactivity and stability. The chemical hardness, softness and chemical potentiality are all influenced by the HOMO-LUMO energy gap (Δɛ) of molecules. A compound tends to be more stable when the difference between the energy of HOMO and LUMO increases. For this reason, more energy will be required if one electron needs to be excited so that it goes to the higher excited state LUMO from the ground state HOMO. Table S1, Figure 6(c), Figures S1 and S2 showed the energies that relate to the reactivity of chemicals, thermal stability of a portion of the electrophilic and nucleophilic attractive region, as well as biological segregation or association with protein. Energy is described in terms of chemical reactivities, hardness, softness, participation in the electrophilic or nucleophilic region, and biological dispersion or affiliation with protein.35,36 It is believed that the energy gap (Δɛ), which covers from −3.00 eV to −5.00 eV, is the range that best fits organic molecules.
37
To evaluate the capabilities of atomic electrical transportation, the molecular orbital energy gaps are investigated. Here, Δɛ spans from 3.554 eV to 4.102 eV, indicating a lower Δɛ and denotes that these compounds have greater atomic systems, and thus, significantly lower chemical stability. However, depending on the functional group, results differ significantly.38,39 Chemical potentiality, hardness, and softness can all be used to describe bioactivity. Hardness should often be higher than softness. The rate of absorption depending on the level of softness and hardness should be from 1 kcal/mol to 4 kcal/mol for adequate biological flexibility. In our study, all of the derivatives conform to the softness and hardness rules. Here, softness ranges from 0.244 to 0.272 and hardness ranges from 1.777 to 2.049, which are around the mentioned value. Chemical potentiality ranges from −4.797 eV to −5.884 eV which all are around the reference value of 5.00 eV. Hence, all analogs
Electrostatic potential analysis
The variety and charge distribution in a molecule can be understood and studied with the help of molecular electrostatic potential (MEP). To understand the mechanism, it is important to know where a nucleophile or an electrophile will attack.
40
In a biological system, the specificity and the strength of the binding site are important and the most important bond in such kind of system refers to the hydrogen bond.
41
For a hydrogen bond to be established, it is important to have an electronegative and electropositive site in a molecule. From an MEP map, we can predict the site, and in the maps; the green color represents the zero potential areas, the blue colored area is the more positive, and the red colored one is the electronegative site of the compound. The size and shape of the compound can also be predicted with the help of the MEP. In Figure 7 and S3, the blue-colored area shows the presence of more percentage of carbon chains and the red-colored area indicates the presence of electronegative atoms like nitrogen, oxygen, and bromine. Here, the MEP of the tested compounds ranges from −0.241 Hartree to +0.177 Hartree. These MEP values suggest the possibility of nucleophilic and electrophilic sites of the compound. The amount of positive and negative charge varied based on the number of carbons, hydrogen, and electronegative atoms. Here, the highest negative MEP was seen for analog
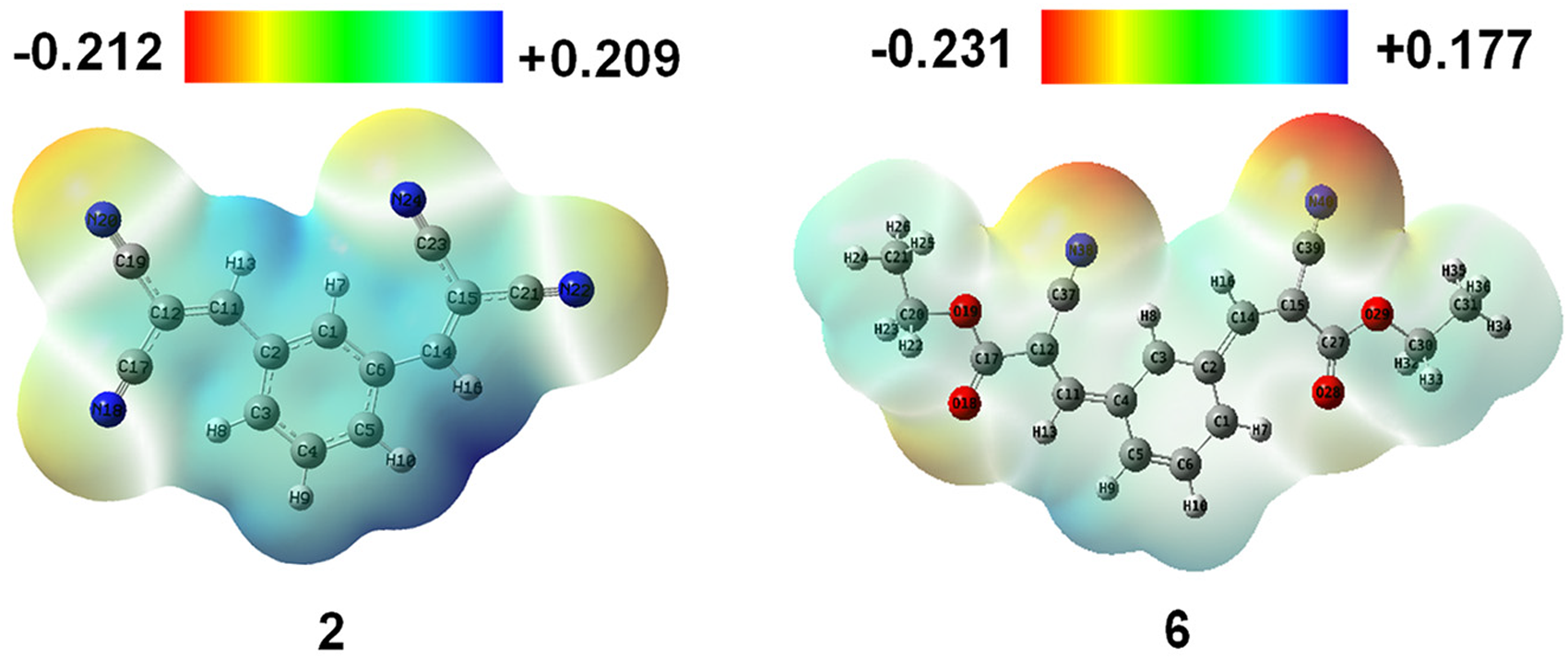
Electrostatic potential map of compounds
FT-IR analysis
Vibrational spectroscopic techniques such as FT-IR, is the most popular techniques that are used in the characterization of the compound, especially to prove the presence of different functional groups in the tested compounds. 42 In our study, vibrational frequencies were calculated from 400 and 4000 cm−1 and compared with our experimental values (Table S2 and Figure 8(a)). The computed values were multiplied by the scaling factor 0.9688 for more accuracy. 17 The most significant functional groups in the study were C = O, C–O, C–H, O–H, C≡N, aromatic C = C, and aliphatic C = C. The peak that is found in the region of 3000–3100 cm−1 is for the presence of C-H bond in the studied compounds and the experimental analysis the region was also found from 2900 to 3350 cm−1 region. Again, the region from 2240 to 2265 cm−1, which matches the result of the experimental vibrational frequency for C≡N of 2200 to 2250 cm−1 confirms the existence of a nitrile functional group in the compounds. The presence of C = C in both the aromatic ring and the aliphatic side chain can be proved by the significant peak in the region of 1500 to 1650 cm−1 which correlates to the experimental value as well. The ester group has two carbon-oxygen bonds, in which the C-O is detected at the region near about 1100 to 1150 cm−1 and C = O is detected at the region of 1650 to 1750 cm−1. Again, the presence of O-H can be confirmed by the significant peak in the region of 3650–3750 cm−1 (Figure 8(a)).
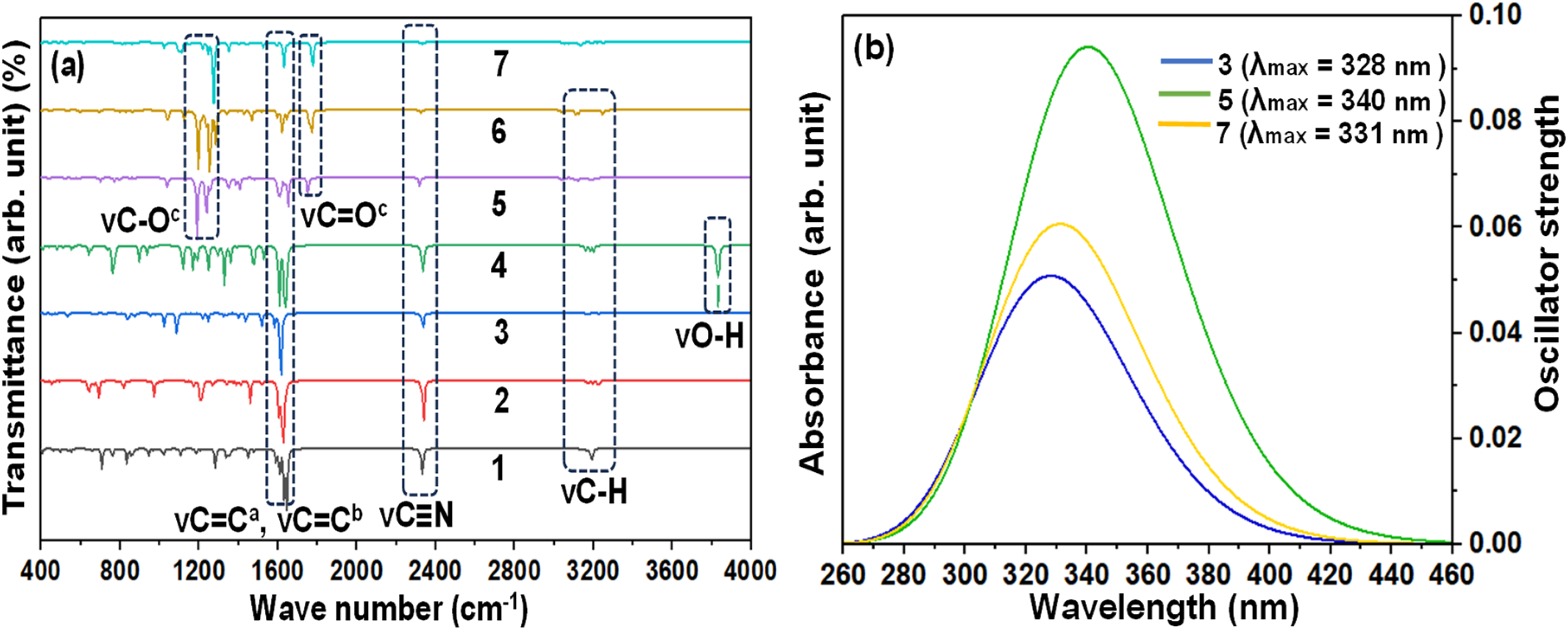
(a) FT-IR and (b) UV-vis spectra of selected derivatives
UV-vis spectral analysis
To know a compound's electronic transition, the UV-visible technique utilizing time-dependent Density Functional Theory (TD-DFT) calculation is the standard benchmark that can ensure a balance between accuracy and computational expense. Two particular electronic transition states of every analog are shown in Table S3, Figure 8(b) and S4. The first transition of an electron from the ground state (S0) to the first excited state (S1) governs the kinetic stability as well as chemical reactivity. All the studied compounds show absorbance at the UV region and no absorption can be seen in the visible region. All the compounds absorb the light between 210 to 460 nm wavelength. The excitation energies are lower and all the transitions are happening comparatively in low regions. Out of all the analogs, 4.298 eV of
Molecular docking and non-bonding interaction analysis
A molecular docking simulation method is commonly used to estimate the binding affinity and the position of ligands in the binding region of a receptor protein.40,44 Here, the docking was carried out using some analogs of AMNs (
Binding affinities of the arylidene malononitrile (AMN) (

*8 = ampicillin
Against the protein 1AH7, some important non-bonding interactions were observed [e.g., two conventional hydrogens (H) with two amino acid (AA) residues (TYR56, TYR79), one pi-cation (PC) with one AA (HIS142) and one pi-pi stacked (PPS) with one AA (PHE66) for analogue
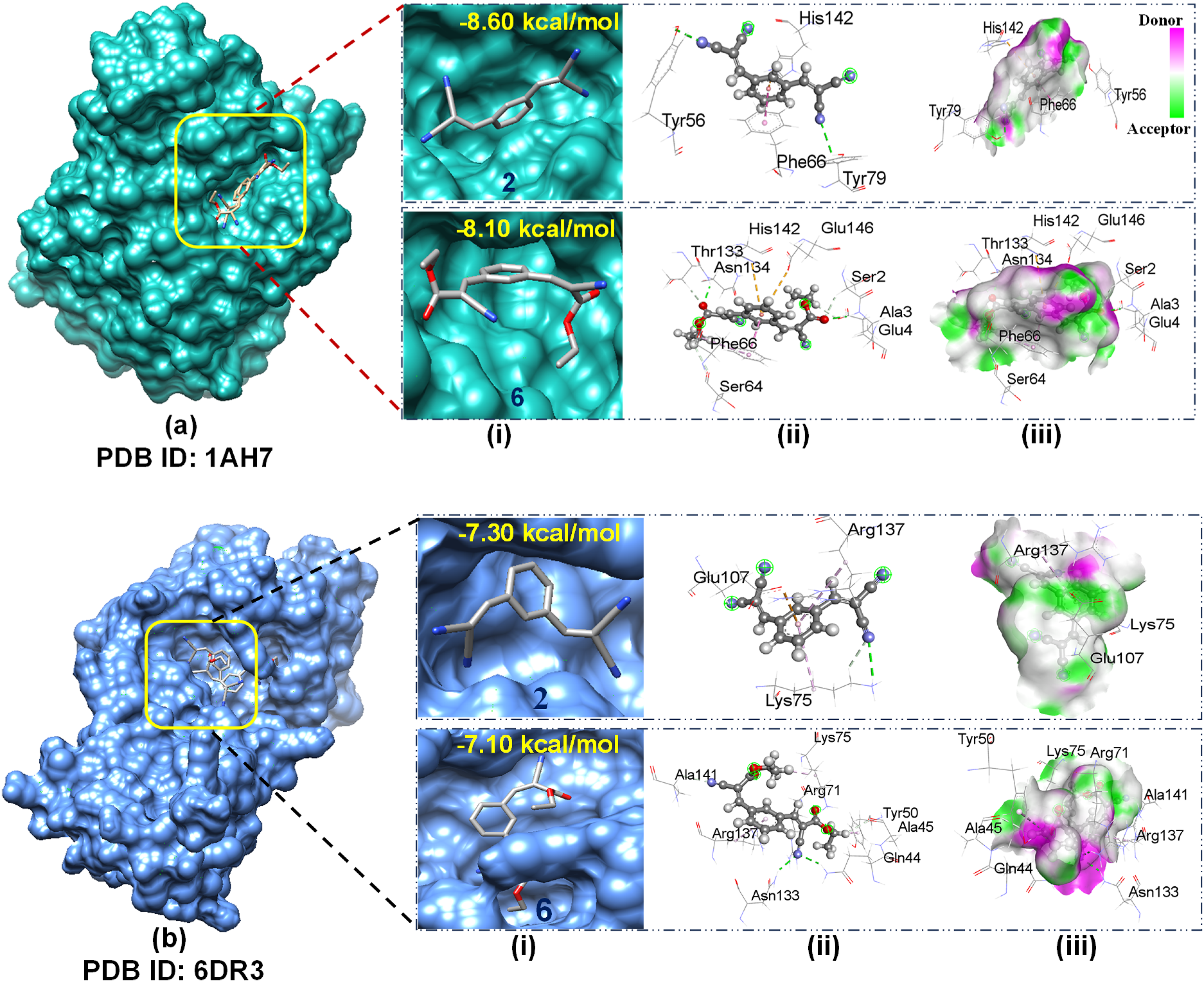
(i) Docked conformer, (ii) non-bonding interaction, and (iii) hydrogen bond surface for compounds
Molecular dynamics simulation analysis
Normal mode analysis (NMA) is commonly used to describe the overall functional mobility of macromolecules. To evaluate the deformability as well as stability of the receptor protein, different parameters including bond distances, positions, dihedral angles, torsions, frictions, and changes in conformation were observed. The ability of a protein chain's residues to deform is measured by its deformability. Here, the most fluctuation was seen for HIS14 for the 1AH7 receptor. The deformation propensity of TRP1, ALA3, GLU9, TRP16, VAL18, ASN19, and ARG20 were moderate to some extent [Figure 10(a)
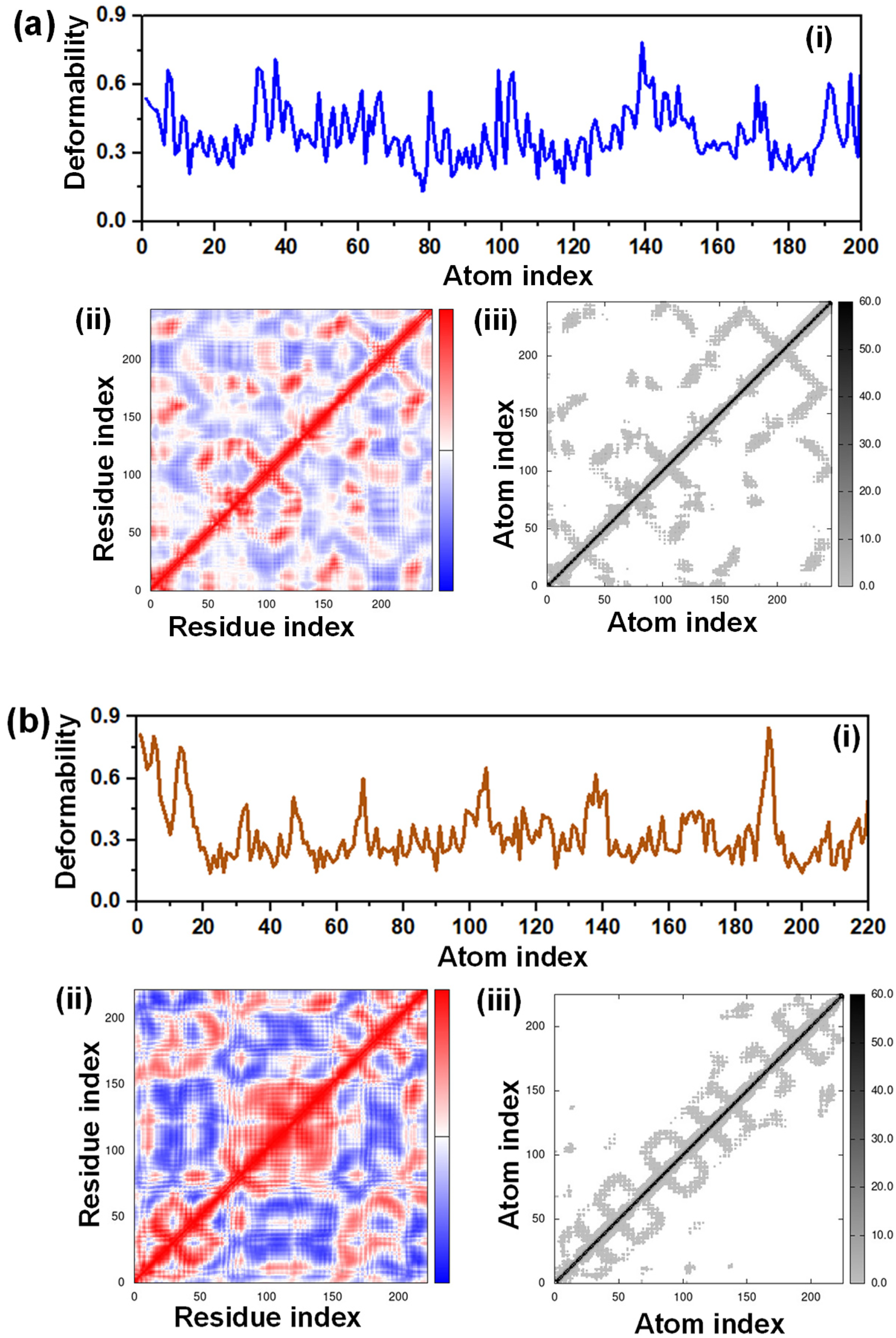
Deformability and interaction index of the receptor proteins (a) 1AH7 and (b) 6DR3.
ADMET analysis
The body's reaction to a drug is explored in pharmacokinetics and referred to as “absorption, distribution, metabolism, excretion, and toxicity (ADMET)”.
48
Now, it is important for the early development and research of medications. Thus, ADMET forecasting is used to analyze all of the potential drug's features (Table 3).49,50 The ADMET features of the tested AMN and ECPA derivatives (
Absorption, distribution, metabolism, and toxicological properties of some AMNs (
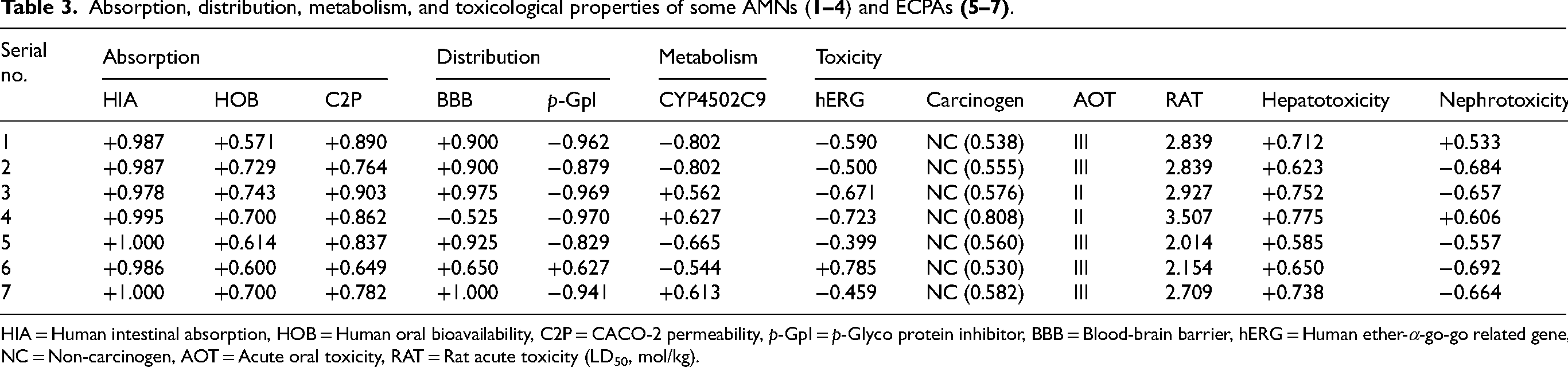
HIA = Human intestinal absorption, HOB = Human oral bioavailability, C2P = CACO-2 permeability, p-GpI = p-Glyco protein inhibitor, BBB = Blood-brain barrier, hERG = Human ether-α-go-go related gene, NC = Non-carcinogen, AOT = Acute oral toxicity, RAT = Rat acute toxicity (LD50, mol/kg).
Biological activities
A computer-aided novel method named “prediction of activity spectra for substances (PASS)” has been used to forecast the results of 1000 biological and toxicological investigations.40,62 Based on the study of “structure-activity relationships (SARs)”, the biological functions of the PASS program showed 90% accuracy. Additionally, biological activity is impacted by the features of the drug, biological entity, and dosage manner.
63
The following rules can be applied to calculate the PASS prediction: (a) “If Pa > 0.7”, the molecule is very likely to exhibit experimental activity, but there has also high pharmacological drug effect; (b) “If 0.5 < Pa < 0.7”, the molecule is likely to exhibit the experimental action, but there has less pharmacological efficacy of the drug; and (c) “If Pa < 0.5”, the molecule is unlikely to exhibit the desired experimental activity.
58
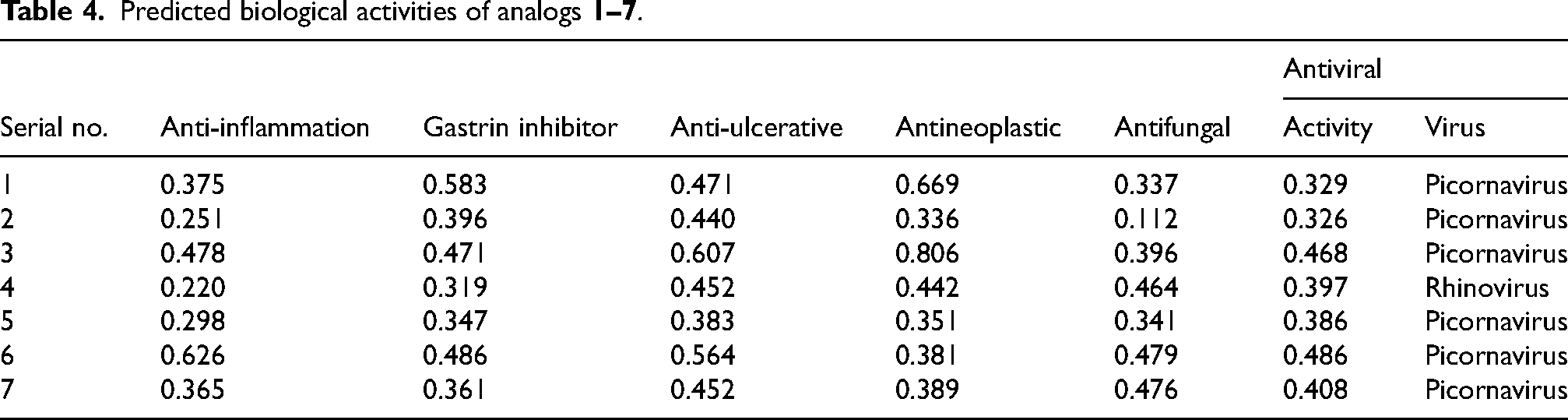
Here, Table 4 illustrates the interpretation of the prediction outcomes. The outcomes of the PASS prediction indicate that AMN analogue
Predicted biological activities of analogs
Drug likeness
The qualitative analysis of a molecule during the early stages of drug design is termed as “drug-likeness”. 64 It could be stated as the similarity between drugs and compounds. 65 This strategy, which is heuristic in nature and depends upon the physicochemical characteristics of the molecules, speeds up the process of developing novel drugs. 65 To evaluate a compound's drug-likeness and establish whether it is consumed as a therapeutic molecule, Lipinski's criterion of five is followed (Table S6). Poor oral bioavailability indicates that a drug violates more than a single rule. 66 According to Lipinski et al., 67 Ghose et al., 68 Veber et al., 53 Egan et al., 69 and Muegge et al., 70 all of the compounds in this study correspond to all drug-likeness standards and have good oral bioavailability scores of 0.571 to 0.729.
pIC50 studies
Utilizing the multi-linear regression (MLR) formula, pIC50 values were examined. A MLR equation is used to analyze the relationship between a dependent variable (pIC50) and a collection of independent variables (descriptors Chiv5, bcutm1, MRVSA9, MRVSA6, PEOEVSA5, GATSv4, J, and Diametert). It has been observed that the pIC50 limit for many drugs is 4.0–10.
71
Here, pIC50 varies from 4.24 to 4.59 (Table S7 and Figure 11). In our study, all drugs have shown the typical range of pIC50 values and analogue
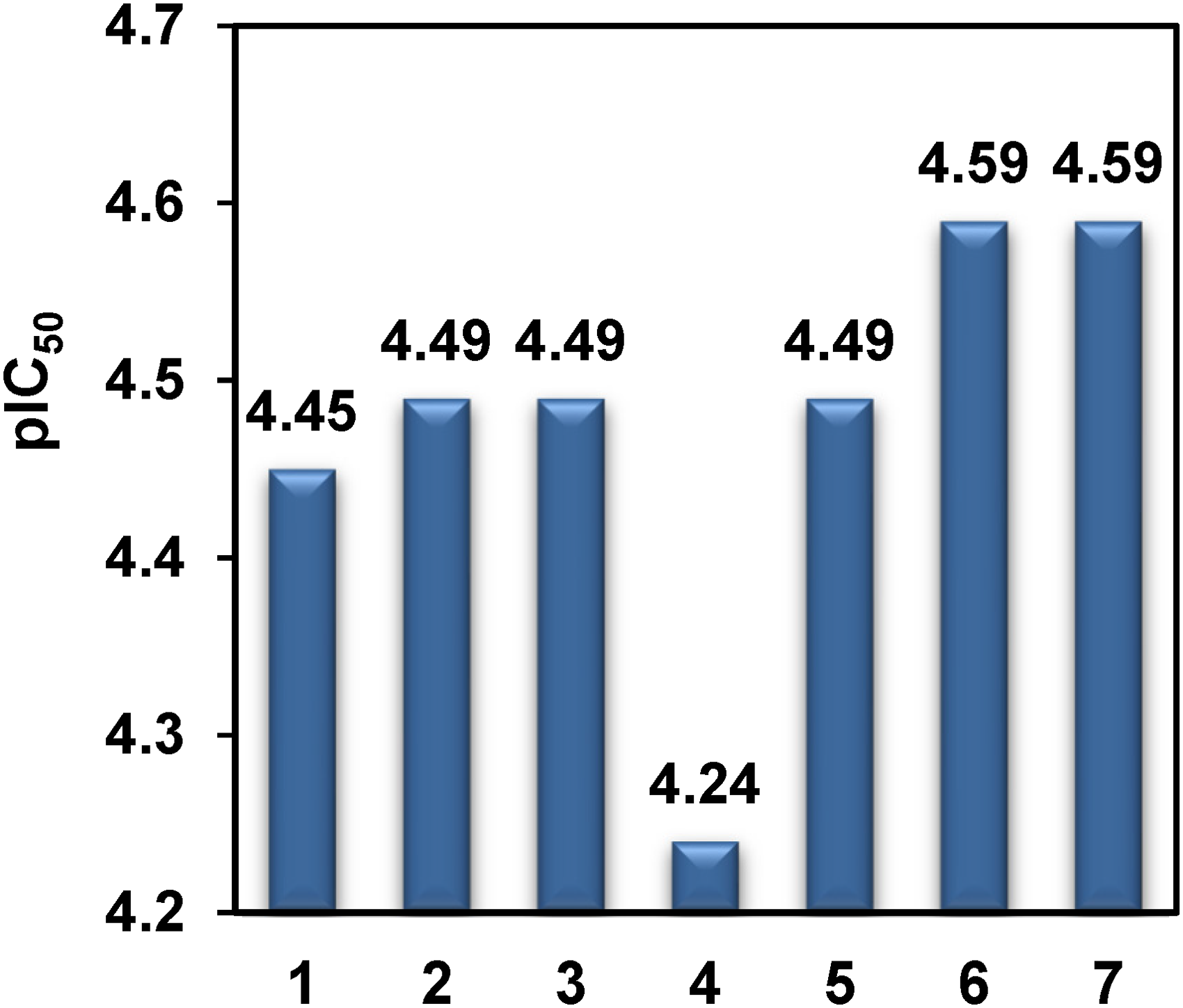
pIC50 of the tested compounds
Conclusion
This work investigated in vitro and in silico antibacterial analyses using some analogs of arylidene malononitrile (AMN) (
Supplemental Material
sj-docx-1-mgc-10.1177_10241221261446763 - Supplemental material for Antibacterial evaluation, quantum chemical computation, and pharmacological studies of some arylidenemalononitrile and ethyl 2-cyano-3-phenylacrylate derivatives: In vitro and in silico approach
Supplemental material, sj-docx-1-mgc-10.1177_10241221261446763 for Antibacterial evaluation, quantum chemical computation, and pharmacological studies of some arylidenemalononitrile and ethyl 2-cyano-3-phenylacrylate derivatives: In vitro and in silico approach by Emranul Kabir, Md. Mosharef Hossain Bhuiyan, M. R. O. Khan Noyon, Monir Uzzaman, Mahbub Alam and S. M. Rahatul Alam in Main Group Chemistry
Footnotes
Acknowledgements
We thank the Computer in Chemistry & Medicine Laboratory, Bangladesh, for their valuable support.
ORCID iDs
Authors contributions
Funding
This research did not receive any external or internal funding.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
Data will be available upon the request to the authors.
Statement of usage of artificial intelligence
This research did not use any support from artificial intelligence.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.