
Abstract
Prompt detection of virulent strains of Newcastle disease virus (vNDV) using real-time reverse transcription polymerase chain reaction (RT-PCR) is challenging because of the broad genetic variability across 2 clades comprising 18 recognized genotypes. A large proportion of class I low virulence ND viruses recently identified in samples recovered from wild birds and from poultry in live bird markets are not detected by the validated real-time RT-PCR assay that targets the matrix gene (M-gene assay). This study describes the identification and sequencing of a conserved region from the polymerase gene of class I NDV and the design and evaluation of a real-time RT-PCR assay (L-TET assay) that identifies a broad range of NDV, demonstrates a 10-fold increase in sensitivity over a previously reported L-gene assay, and works in conjunction with the existing M-gene assay using the same protocol. The L-TET assay detects ≤1 fg of homologous transcribed RNA from genotypes 5, 7, and 8 of class I, and from class II genotype II in either single- or multiplex format. Differential detection of mixed class I and II viruses down to 100 fg is possible because L-TET uses an alternate fluorophore from the M-gene assay. The multiplexed assay is capable of detecting a broad range of class I and II ND viruses with <1 threshold cycle decrease in sensitivity compared to the single probe. A total of 140 class I (n = 108, genotypes 1–2 and 4–9) and class II (n = 32, genotypes I–VII) were correctly identified by both the single- and multiplex formats.
Introduction
Newcastle disease (ND) is a highly contagious disease of major concern to poultry producers around the world. The causative agent, Newcastle disease virus (NDV; order Mononegavirales, family Paramyxoviridae, subfamily Paramyxovirinae, genus Avulavirus; also known as Avian paramyxovirus 1), is a diverse group of single-stranded, negative-sense, nonsegmented RNA viruses of approximately 15.2 Kb 11 that can infect more than 200 species of birds. 12 Newcastle disease viruses, which exhibit an intracerebral pathogenicity index of ≥0.7 and/or contain multiple basic amino acids at the C-terminus of the F2 protein and phenylalanine at residue 117, are defined as virulent viruses that are notifiable according to the standards of the World Organization for Animal Health (Anonymous: 2004, Newcastle disease. In: OIE manual of diagnostic tests and vaccines for terrestrial animals, Chapter 2.1.15. Available at: http://www.oie.int/Eng/Normes/Mmanual/A_00038.htm. Accessed January 1, 2008). Virulent ND viruses (vNDV) can cause high morbidity and mortality, and the severity of the disease depends on the species infected and immune status. The nature of the clinical disease depends on the predilection of the infecting virus strain for respiratory, digestive, and/or nervous systems. Poultry in the United States are considered free of vNDV; however, the low virulence (loNDV) strains are commonly recovered from domestic poultry 10,13,17 and wild bird populations. 7,15
Because of the highly contagious nature of NDV and its clinical similarity to highly pathogenic avian influenza, accurate monitoring and rapid diagnosis of an outbreak are crucial to any control program. Prompt detection of vNDV and differentiation of these viruses from loNDV are challenging because of their broad genetic variability and because these viruses are serologically indistinguishable. Recent phylogenetic analysis has separated NDV into 2 sister clades, class I and II, which contain several genotypes each. 4,7 The majority of reported virulent viruses are found in class II, while loNDV predominate in class I. Rapid identification of NDV in the United States is currently accomplished by 2 validated real-time reverse transcription polymerase chain reaction (RT-PCR) assays: 14,23 the matrix (M)-gene assay is used as an initial screening technique for detecting any NDV, and the fusion gene (F-gene) assay is employed to distinguish virulent isolates from those of low virulence. The M-gene assay is extensively used in the United States with 4,970 samples tested during 2005–2007 at the U.S. Department of Agriculture (USDA) Animal and Plant Health Inspection Service (APHIS) National Veterinary Services Laboratory (Ames, IA) and ∼30,000 samples tested through the National Animal Health Laboratories Network during 2004–2007, as well as international requests from multiple countries (Dennis Senne, personal communication, December, 2007, National Veterinary Services Laboratory).
A large number of class I loND viruses have been recently identified in samples recovered from waterfowl, shorebirds, and from poultry in live bird markets (LBMs); however, the M-gene assay failed to detect the majority (∼73%) of these viruses. 7,8 Comparative nucleotide analysis of the 24-bp M-gene assay probe-site identified significant genomic variability between class I and II isolates at the region of the matrix gene that was targeted by the real-time RT-PCR test, suggesting that this region is responsible for the decreased sensitivity of the M-gene assay to the class I viruses. 8 Because loNDV found in waterfowl and shorebirds represents a large and highly mobile reservoir that has previously been shown to transmit to domestic poultry, 7 there is the possibility that these viruses could become more virulent after introduction and replication in poultry. The increase in virulence from loNDV to vNDV was seen in outbreaks in the Republic of Ireland during 1990 2,3 and Australia during 1998–2000. 5,21
To improve surveillance and to increase understanding of the ecology of NDV in wild bird and domestic poultry populations, an alternative to the M-gene assay is needed to detect the broad range of NDV. The current study describes the identification and sequencing of a conserved region of the NDV genome using bioinformatics tools and the design and evaluation of a real-time RT-PCR assay. The assay targets the polymerase gene (L-gene), identifies a broad range of class I viruses, and works in conjunction with the existing M-gene assay as a multiplex assay that is capable of detecting both class I and II ND viruses.
Materials and methods
Viruses and RNA isolation
Newcastle disease viruses were obtained either from the Southeast Poultry Research Laboratory (SEPRL) repository (Table 1, n = 26) or collected during a multi-institutional cooperative waterfowl virus monitoring project between the University of Georgia Southeastern Cooperative Wildlife Disease Study, the University of Alaska Museum, The Ohio State University Department of Veterinary Preventive Medicine, and USDA SEPRL. 18 Viruses from wild bird samples during 1987–2004 (Table 1, class I n = 109, class II n = 8; fusion gene sequences previously published) 7 were isolated from cloacal swab material by standard virus isolation methods in embryonating chicken eggs. 1,19 U.S. LBM environmental and bird samples were obtained during 2005 (Table 1, class I only, n = 14) from surveillance samples submitted to the USDA APHIS National Veterinary Services Laboratory.
Ribonucleic acid was extracted from allantoic fluids using a commercial phenol and guanidine isothiocyanate solution a according to the manufacturer's instructions. Briefly, 750 μl of the reagent was added to 250 μl of allantoic fluid, vortexed, and incubated at room temperature for 7 min. The RNA was separated into the aqueous phase with the addition of 200 μl of chloroform, precipitated with isopropanol, and pelleted by centrifugation. After 1 wash with 70% ethanol, RNA was dried and resuspended in RNase-free water.
Polymerase chain reaction amplification and sequencing
Polymerase chain reaction amplification of the RNA was performed using a commercial kit. b Amplified products were separated on a 1% agarose gel, the bands excised and eluted using an extraction kit, c and the samples quantified using a standard spectrophotometer. A 456-nt fragment at the 5′ end of the L-gene was sequenced for U.S. waterfowl and LBM isolates (n = 21). This homologous region was also sequenced for a European isolate (Shelduck/France/MC-110/1977; fusion gene GenBank accession AF003726). These partial L-gene sequences have been deposited under accession number EU419620–41 (n = 22). All double-stranded nucleotide sequencing reactions were performed with fluorescent dideoxynucleotide terminators in an automated sequencer. d The 5′ end of the L-gene was sequenced using primers targeting a conserved region 456 nt in length (sense primer 8487F 5′-CTCAGCAGGCAATGGAAGAA-3′; antisense primer 8979R 5′-GCTCAGGAGTGATAAAGACTTG-3′). The RT-PCR protocol for sequencing used 50-μl reactions of a commercial kit e with an RT step of 50°C for 30 min followed by 94°C for 2 min. The cycling conditions were 40 cycles of 15 sec denaturation at 94°C, 30 sec of annealing at 54°C, and extension at 68°C for 60 sec, with a final extension at 68°C for 5 min. Nucleotide sequence editing and analysis were conducted with a sequence analysis software package. f
Design of L-gene real-time reverse transcription polymerase chain reaction
The L-gene was chosen for assay development due to the high degree of conservancy across isolates. 8,9,22 A conserved region near the 5′ end of the L-gene 456 nt in length (positions 8550–9005 based upon the class I NDV L-gene sequence from Duck/US/119535–1/2001; GenBank accession AY626266) from 22 class I ND viruses was aligned using ClustalW 20 followed by manual editing with the BioEdit Sequence Alignment Editor software. 6 For reference, previously published class I NDV GenBank sequences from U.S. LBM (Table 1, n = 3; AY 626266–8) 17 for the homologous region were included.
Newcastle disease viruses used in the current study and corresponding accession numbers.
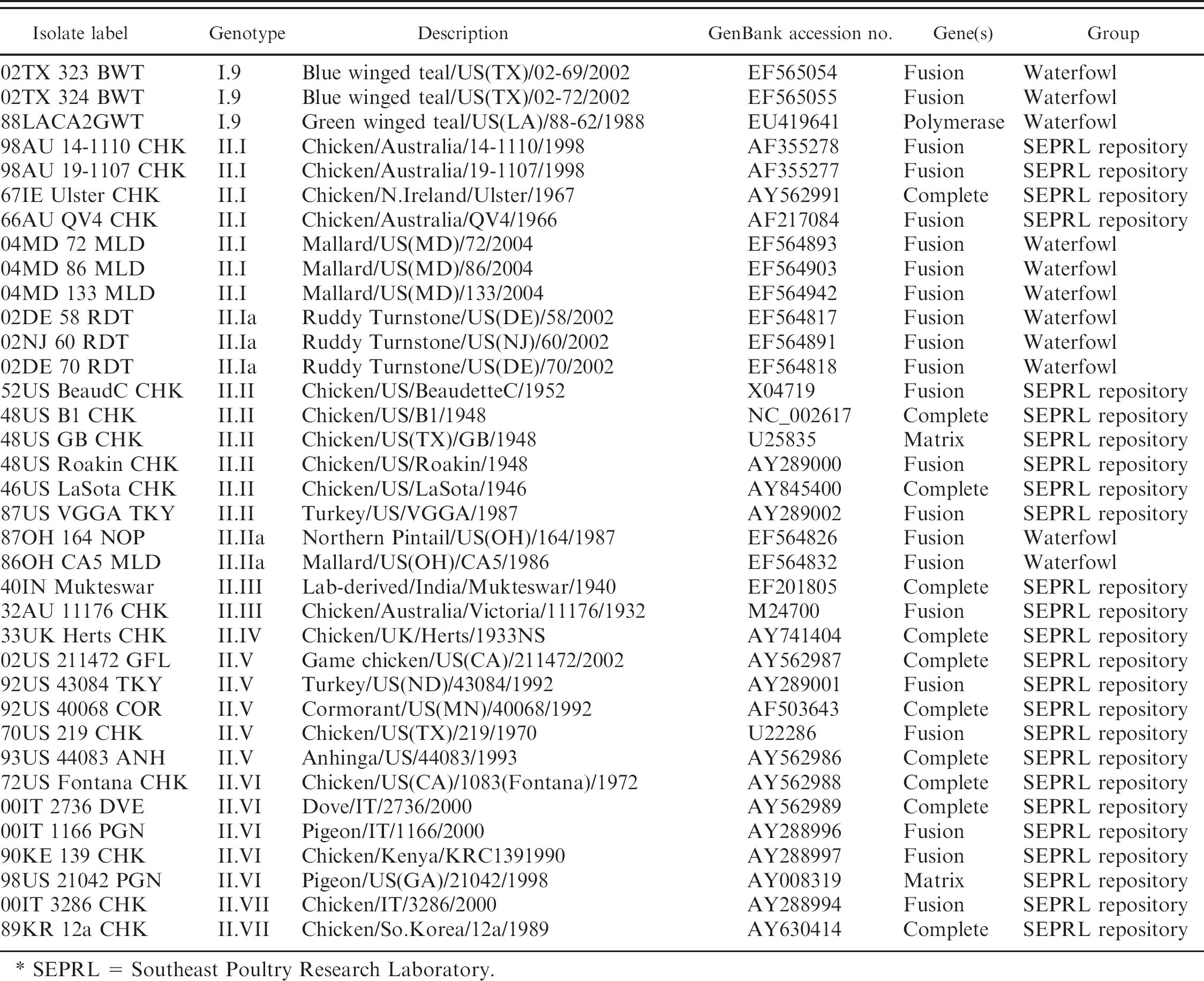
SEPRL = Southeast Poultry Research Laboratory.
The L-gene sequence from Northern Pintail/US(AK)/196/1998 (98AK6NOP; EU419625) was used as the template to generate candidate primer-probe sets with beacon design software g using default program settings and applying the melting temperature (Tm) of the M-gene assay (56°C) as a guideline to ensure compatibility in the multiplex format. The final dual hydrolysis probe h and primers were manually selected based upon the L-gene alignment. The L- and M-gene assay primers and probes were tested for their thermodynamic properties, secondary structures (including BLAST search of the primers/probe and amplicon followed by a template structure search, which avoids secondary structure while designing primers), cross homologies, and primer-primer/primer-probe interactions via the beacon designer software. g For the probe, h fluorophore tetrachloro-6-carboxyfluorescein (TET) was chosen for the L-gene assay (L-TET) to allow for multiplex detection with the M-gene assay, which uses fluorophore 6-carboxyfluorescein (FAM).
The real-time RT-PCR protocol for the L-TET assay (Table 2) was carried out as follows for each 25-μl reaction (17 μl of master mix to 8 μl of template) using a commercial RT-PCR kit: b 1 μl of kit-supplied enzyme mix, 5 μl of kit-supplied buffer (5X), 0.5 μl of sense and antisense primers (final concentration: 0.4 μM), 0.5 μl of probe (final concentration: 0.24 μM), 0.8 μl of kit-supplied deoxynucleoside triphosphates (dNTP; final concentration: 320 μM each), 1.25 μl of 25 mM MgCl2 (combined with MgCl2 in kit-supplied buffer; final concentration: 3.75 mM), and 13 U of RNase inhibitor. i
Sensitivity and specificity of L-TET assay
To determine the limits of detection for the L-TET assay, testing in duplicate was performed on serial 10-fold dilutions of the in vitro transcribed RNA (ivtRNA) for a total of 10 dilutions starting at 100 picograms (pg) per reaction. The ivtRNA was generated from the L-gene of class I genotypes 5 98AK6NOP, 7 98AK9NOP (Northern Pintail/US(AK)/207/1998; EU419634), and 8 77FR354SHD (Shelduck/France/MC110/1977; EU419639), and the matrix gene from class II genotype II 48USB1CHK (Chicken/US/B1/1948; accession NC_002617) using a commercial kit j per the manufacturer's protocol. Other avian respiratory viruses from the Paramyxoviridae, Coronaviridae, Orthomyxoviridae, and Herpesviridae families were also tested to ensure specificity of the L-TET assay.
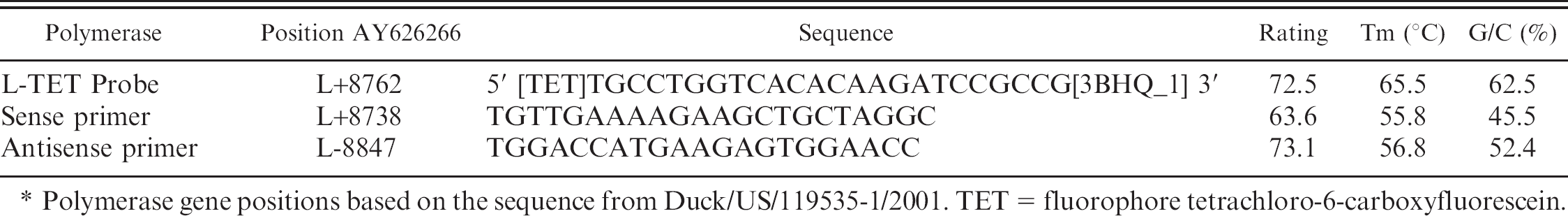
Polymerase gene positions based on the sequence from Duck/US/119535–1/2001. TET = fluorophore tetrachloro-6-carboxyfluorescein.
Multiplex real-time reverse transcription polymerase chain reaction
The multiplex reaction included the M-gene primers/probe (M+4100 5′-AGTGATGTGCTCGGACCTTC-3′, M-4220 5′-CCTGAGGAGAGGCATTTGCTA-3′, probe M+4169 5′-[FAM]TTCTCTAGCAGTGGGACAGCCTGC [BHQ_1]-3′) 14,23 with identical concentrations of primers and probe, dNTP, magnesium chloride, and RNase inhibitor per 25-μl reaction. To determine the limits of detection for NDV class I and II mixed infection, ivtRNA from 98AK6NOP (class I, genotype 5) and 48USB1CHK (class II, genotype II) was used. To prepare the samples, a known quantity of ivtRNA from one virus (0.1 pg ≌ 105 copies) was spiked with serial 10-fold dilutions of the other virus (starting at 101 = 100 pg/25 μl reaction ≌ 108 copies).
L-TET single and multiplex assay evaluation
Wild bird class I NDV isolates representing known U.S. genotypes 1–2 and 4–9 (n = 108), class II isolates from wild bird samples representing genotypes I and II (n = 8), and a panel of repository viruses representing genotypes I–VII (n = 24) were tested by the L-TET single probe assay and the L-TET assay multiplexed with the M-gene assay. All class I viruses (n = 108) were also tested using a previously reported L-gene assay 8 for comparison to the L-TET assay. The average threshold cycle (Ct) values obtained from each assay for the class I panel (n = 108) were compared with the quantitative estimates calculated from the standard curves for each assay of the transcribed RNA by the real-time thermocycler software. k
Real-time RT-PCR reactions were performed using a real-time thermocycler, k and cycling conditions were identical to that of the previously reported M-gene assay protocol: the RT step was 30 min at 50°C followed by 15 min at 95°C; cycling conditions were 40 cycles of 10 sec denaturation at 94°C, 30 sec of annealing at 56°C, and extension at 72°C for 10 sec. 14,23 All assays in the current study perform as expected at the default real-time thermocycler k ramp speed (data not shown); however, a slowed ramp speed comparable with other thermocyclers was applied to both the M-gene and L-TET assays alone and in multiplex format. Real-time RT-PCR reactions producing a Ct value of ≥35 were considered negative for the evaluation of the assay.
All RNA and DNA procedures were carried out in biological safety cabinets, and use of RNase-free reagents and standard anticontamination protocols were followed at all times to minimize contamination. Negative (nuclease-free water) and positive controls (ivtRNA) were included for each run.
Results
The amplicon site for the L-TET primer/probe set (L+8738–8847) located at the 5′ end of the L-gene covered a highly conserved region (Fig. 1) with ≤2 mismatches at the probe site and only 1 isolate with >2 mismatches in either primer region (Duck/US/119535–1/2001 had 3 mismatches in the antisense primer). Real-time RT-PCR testing was performed on serial 10-fold dilutions of ivtRNA (101 = 100 pg/25 μl reaction) to determine the limits of detection for the L-TET assay (Table 3). In single probe format, the L-TET assay detected 102 copies of the target gene or <1 femtogram (fg) of the homologous transcribed RNA from 98AK6NOP (genotype 5) and 98AK9NOP (genotype 7), and 103 copies (1 fg RNA) from 77FR354SHD (genotype 8). The M-gene (FAM) assay detected 103 copies of the target gene (1 fg RNA) from the class II 48US B1 CHK ivtRNA, which is consistent with a previous report. 23 In multiplex format (TET and FAM), the assay detected ≤1 fg of transcribed RNA from 98AK6NOP and 48US B1 CHK. Other respiratory viruses such as Avian paramyxovirus 2 and 4, Avian metapneumovirus (family Paramyxoviridae), Infectious bronchitis virus (family Coronaviridae), Avian influenza virus (family Orthomyxoviridae; also known as Influenza A virus), and Gallid herpesvirus 1 (family Herpesviridae; also know as Infectious laryngotracheitis virus) tested negative on the L-TET assay (data not shown).
The L-TET assay was developed using a reporter dye whose fluorescence did not overlap the FAM channel to enable the detection of mixed NDV infection. To determine the effect of mixed class I and II infections on the limits of detection for the multiplexed assay, dilutions of ivtRNA from 98AK6NOP (class I, genotype 5) and 48USB1CHK (class II, genotype II) were prepared to approximate mixed infections at varying concentrations. The multiplexed assay detected the presence of both viruses down to 100 fg (∼105 copies) of class I RNA spiked into class II dilution, and 10 fg (∼104 copies) of class II RNA spiked into class I dilution (data not shown). At the first dilution (101 ≌ 108 copies), the multiplexed assay detected only the RNA present in the highest concentration regardless of the isolate.

Alignment for class I Newcastle disease virus (NDV; n = 25) near the 5′ end of the polymerase gene (positions 8738–8847 based upon the class I NDV L-gene sequence from Duck/US/119535–1/2001, GenBank accession AY626266) to Northern Pintail/US(AK)/196/1998 (GenBank accession EU419625) identifying sites of primers (arrow with circle) and probe h (arrow with diamond). Virus designations represent an 8–14 character name containing the 2-digit year of collection, location abbreviation, unique virus identification, and species abbreviation (Table 1).
Line equations, limits of detection in femtograms (fg), and copies of the target gene for the L-TET and M-gene real-time reverse transcription polymerase chain reaction assays in single and multiplex format for serial 10-fold dilutions of transcribed RNA (a total of 10 dilutions starting at 100 pg per reaction) from Northern Pintail/US(AK)/196/1998 (98AK6NOP), Northern Pintail/US(AK)/207/1998 (98AK9NOP), Shelduck/France/MC110/1977 (77FR354SHD), and Chicken/US/B1/1948 (48USB1CHK).
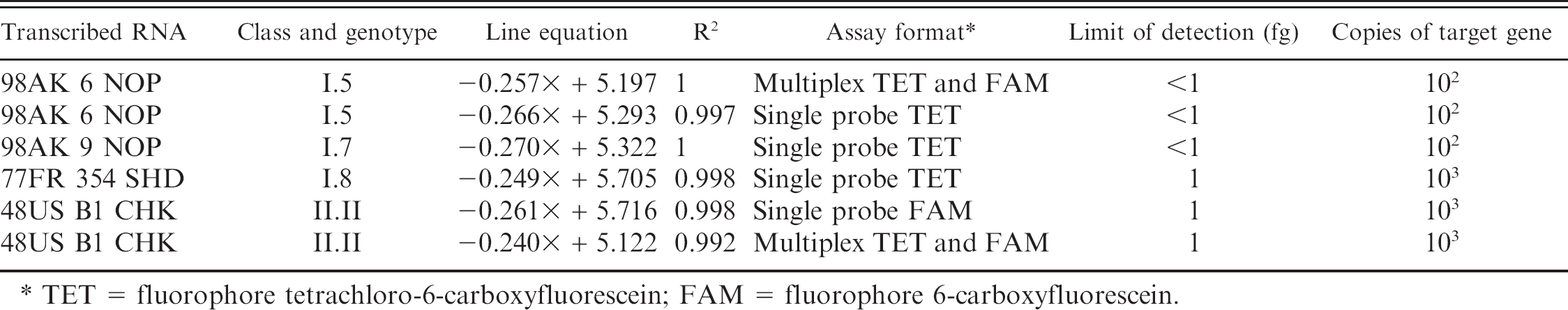
TET = fluorophore tetrachloro-6-carboxyfluorescein; FAM = fluorophore 6-carboxyfluorescein.
The panel of class I (n = 108) loNDV isolates tested by the single probe L-TET assay and the L-TET multiplexed with the M-gene assay (Figs. 2A-C) represented known U.S. genotypes 1–2 and 4–9 isolated during 1987–2005. The majority were from wild bird samples (n = 95) and the remainder were from LBMs (n = 13). All class I isolates (108/108) tested positive by both the L-TET assay and the multiplex format (Figs. 2A-C). The Ct values in the multiplex format had minimal effect on the test sensitivity with an average delay of 0.4 Ct in the TET channel (Ct values: 12–32) when compared with the single probe format (Ct values: 12–29); signal crossover between the fluorescent probes was not observed.
To determine whether the L-TET assay represented an improvement over a previously reported L-gene real-time RT-PCR assay (HK assay), 8 results for testing serial 10-fold dilutions of ivtRNA from 98AK6NOP and the class I panel (n = 108) were compared (data not shown). The L-TET assay demonstrated a ten-fold increase in sensitivity compared to the HK assay (limit of detection 102 vs. 103 copies of the target gene) for the transcribed RNA. This increase in sensitivity was also noted in the class I panel results. For this comparison, the standard curves for each test were used to calculate the pg per reaction of RNA detected based upon the Ct values. The L-TET assay on average detected lower quantities of RNA at earlier Ct values (average Ct of 14.8 representing detection of ∼45 pg of RNA per reaction) compared to the HK assay (average Ct value of 17.4 representing detection of ∼135 pg of RNA per reaction). In addition, the L-TET assay detected isolates in genotype 6 that were missed by the HK assay.
To ensure the multiplex format did not affect the detection of class II ND viruses, isolates were evaluated by both the M-gene assay and the multiplex format (Fig. 3). The class II panel comprised wild bird samples isolated during 1987–2004 representing genotypes I and II (n = 8) and repository viruses representing genotypes I–VII (n = 24). All isolates were correctly identified by the M-gene and multiplex assays (32/32) based upon the FAM channel Ct values with an average 0.25 Ct delay in detection with the multiplex format.
Discussion
While real-time RT-PCR assays may be easily developed by research laboratories, they are often not implemented because of the stringent requirements of the validation process. Additionally, multiplexed assays often result in a considerable decrease in sensitivity that precludes their practical adoption. Use of the L-TET assay, which was specifically tailored to work with the previously validated M-gene assay and demonstrated minimal reduction in sensitivity in the multiplexed format, may be employed without alteration of existing protocols.
A separate real-time RT-PCR assay targeting the L-gene (HK assay) was previously reported to detect LBM isolates from Hong Kong. 8 While the HK assay detected many of the class I genotypes, it failed to detect isolates from all class I genotypes. Another drawback of the HK assay was that, while it could be used in conjunction with the M-gene primer/probe set, the annealing temperature had to be decreased to 52°C from 56°C. This modification of the M-gene assay validated protocol could cause unforeseen changes to the test performance. Additionally, because the probe fluorophore was the same for both the L- and the M-gene assay (FAM), a distinction could not be made between class I and II viruses in a single sample. The L-TET primer/probe set, which starts 46 nt upstream from the HK assay, demonstrates improved sensitivity over the HK assay and was designed to be compatible with the current protocol for the M-gene assay (56°C Tm) allowing for detection of a broad range of class I and II ND viruses in 1 multiplex assay.

Real-time reverse transcription polymerase chain reaction cycle threshold values from the fluorophore tetrachloro-6-carboxyfluorescein (TET) fluorescence channel for class I isolates representing (A) genotypes 1, 2, 4, and 9 (n = 32), (

Real-time reverse transcription polymerase chain reaction cycle threshold values from the fluorophore 6-carboxyfluorescein fluorescence channel for class II isolates representing genotypes I–VII (n = 32) using the single probe M-gene assay and M-gene with the L-TET assay in multiplex format. Virus designations represent an 8–14 character name containing the 2-digit year of collection, location abbreviation, unique virus identification, and species abbreviation (Table 1).
To allow for differential detection of class I and II viruses, as well as the ability to detect mixed class I and II infections within a single sample, the L-TET primer/probe set uses TET as an alternate fluorescence to FAM (used by the M-gene assay). Although mixed class I and II NDV infections have been observed, the frequency of occurrence is not known nor is the ratio of viruses that are likely to be present under field conditions. In the current study, both class I and II viruses were detected by the multiplexed assay at concentrations down to 0.01–0.1 pg (∼104–105 copies); however, at the 101 dilution (100 pg, ∼108 copies), only the virus present at the highest concentration was detected as its amplification likely out-competed the other isolate and thus consumed available reagents.
Low virulence ND viruses represent a large and highly mobile group of viruses that infect a broad range of wild birds and poultry. 7,16 Newcastle disease outbreaks putatively caused by circulating endemic strains, such as in the Republic of Ireland in 1990 2,3 and in Australia during 1998–2000, 5,21 emphasize the need to monitor wild bird and poultry populations. The class I viruses have been increasingly recognized in wild bird populations and LBMs, and the possibility for these viruses to acquire virulence after introduction and replication in poultry is cause for concern. 2,3
Of the viruses sequenced from wild birds, 74% correspond to class I ND viruses and nearly 75% of these viruses fail detection with current RT-PCR–based diagnostic tests. 7–9 Because of the complex and poorly understood ecology of loNDV in wild birds and the limited range of detection of these viruses by current methods, the L-TET assay could be an invaluable tool to determine the extent of wild birds infected with class I NDV. Additionally, use of the L-TET assay in multiplex with the M-gene assay to monitor NDV among domestic poultry populations in the United States may improve diagnosticians' ability to predict and prevent potential outbreaks caused by circulating strains.
Acknowledgements
The authors would like to acknowledge Dr. Erica Spackman for technical expertise, Dawn Williams-Coplin for technical assistance, and the South Atlantic Area Sequencing Facility for nucleotide sequencing. This work was funded by USDA CRIS project numbers 6612–32000–039–00D, 6612–32000–041–01S, 6612–32000–041–04S, 6612–32000–041–06S, 6612–32000–039–03S, and USPEA (SEPEA #332). Mention of trade names or commercial products in this manuscript is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
a.
TRIzol® LS Reagent, Invitrogen Corp., Carlsbad, CA.
b.
Qiagen® OneStep RT-PCR Kit, Qiagen Inc., Valencia, CA.
c.
QIAquick® Gel Extraction Kit, Qiagen Inc., Valencia, CA.
d.
Applied Biosystems 3730xl DNA Analyzer, Applied Biosystems Inc., Foster City, CA.
e.
SuperScript™ III One-Step RT-PCR System, Invitrogen Corp., Carlsbad, CA.
f.
Lasergene® v5.07, DNASTAR Inc., Madison, WI.
g.
Beacon Designer v7.0, PREMIER Biosoft International, Palo Alto, CA.
h.
TaqMan® probe, Roche Molecular Diagnostics, Pleasanton, CA.
i.
RNasin® Plus RNase Inhibitor, Promega Corp., Madison, WI.
j.
RiboMAXTM Kit, Promega Corp., Madison, WI.
k.
SmartCycler®, Cepheid Inc., Sunnyvale, CA.