
Abstract
An enzyme-linked immunosorbent assay (ELISA) was developed for detection and quantitation of beta2-toxin in neonatal piglet intestinal contents. Polystyrene plates were coated with polyclonal capture antibodies prepared against consensus recombinant beta2-toxin. The ELISA was developed using consensus recombinant beta2-toxin, atypical recombinant beta2-toxin, purified consensus native beta2-toxin, and field samples of neonatal porcine intestinal contents. Captured antigen was detected using a horseradish peroxidase–labeled monoclonal antibody against consensus recombinant beta2-toxin. The limit of detection of the ELISA for consensus beta2-toxin was between 2.0 and 3.5 ng/ml. The ELISA detected atypical recombinant beta2-toxin only weakly. Optical density was protein concentration dependent. The test confirmed differences between consensus and atypical recombinant beta2-toxin, but similar results obtained when testing pure consensus recombinant beta2-toxin and native beta2-toxin. Results obtained from intestinal content samples, particularly from the small intestine, were highly inconsistent and suggested variable protease activity. Addition of protease inhibitors partially prevented degradation of the toxin; however, sample processing at low temperature, at a lower pH (citrate buffer with 5% of bovine serum albumin, pH 6.1), and “cold incubation” of applied antigens abolished protease activity. The recombinant toxin was preserved in spiked intestinal samples by freezing at −70°C, suggesting that necropsy samples can be stored frozen for periodic testing. With appropriate sample preparation, antigen-capture ELISA can detect beta2-toxin in the intestinal content and feces of neonatal piglets.
Introduction
The worldwide distribution of Clostridium perfringens and its diverse arsenal of toxins and enzymes give it the ability to cause disease in several different animal species, including human beings. 14 Clostridium perfringens is currently classified into 5 toxinotypes (A–E), 13 depending on its production of 4 mouse lethal toxins; different types are linked with specific clinical diseases of domestic animal species. Clostridium perfringens toxinotype A is considered by some to be one of the most important enteric pathogens in neonatal piglets.2,12
During purification of beta-toxin (CPB) from C. perfringens toxinotype C, a protein of approximately 28 kDa was isolated. 6 It was originally thought to be a cleavage product of CPB, and because it was toxic for both Chinese hamster ovary cells and for mice, it was named beta2-toxin (CPB2). Later the amino acid sequence was found to be distinct from that of CPB. 6 The toxin is lethal for mice6,17 and for various cell lines3,6 and causes hemorrhagic necrosis when injected into intestinal loops of guinea pigs. 8 Subsequently, it was discovered that there are 2 forms of the CPB2 toxin, the consensus form 6 and a less toxic “atypical” variant. 9 Despite extensive studies of the distribution of CPB2 toxin gene (cpb2) in isolates of C. perfringens from numerous animal sources, no general conclusions have been drawn as to the role of CPB2 in enteric diseases of animals or human beings. 16
In swine, in contrast to many other species, cpb2 is almost invariably the consensus type and is almost invariably expressed.2,19 In addition, multilocus sequence typing has shown that porcine C. perfringens toxinotype A strains belong to a common clonal complex. 11 Diarrheal disease isolates almost always carry cpb2, compared to a smaller proportion of cpb2-positive isolates from healthy pigs.2,5,19 However, there is no convincing evidence to date that CPB2 itself is responsible for C. perfringens toxinotype A diarrhea in neonatal piglets. 15 It is possible that cpb2 is a marker of, rather than being directly involved in, virulence. 10 If C. perfringens toxinotype A causes diarrhea in neonatal piglets, as is widely believed, 16 most aspects of the disease are poorly understood and the diagnosis is equivocal, made only by exclusion of other enteric pathogens in combination with the detection of large numbers of cpb2-positive C. perfringens in the small intestine. As an approach to improving the diagnosis of C. perfringens toxinotype A diarrhea of neonatal piglets, the current study describes the development of an antigen-capture enzyme-linked immunosorbent assay (ACE) for the detection of CPB2 in the neonatal pig intestine.
Material and methods
Purification of recombinant and native CPB2
Purification of His-tagged consensus and atypical recombinant CPB2 (rCPB2) from Escherichia coli DH5α (containing plasmids pJGS197 and pJGS659, respectively) was performed by a standard approach a from bacterial cells grown in lysogeny broth. b Purification of consensus native CPB2 (nCPB2) from a C. perfringens porcine isolate (P18) was performed using anion exchange, followed by size exclusion chromatography.4,6 Fractions showing a single CPB2 band by Coomassie blue c staining were pooled and confirmed as being CPB2 by Western blot (using monoclonal antibody [mAb] Songer 9E4B).
Purification of consensus nCPB2 was confirmed by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis staining with Coomassie blue and by Western blot (using mAb Songer 9E4B). Purified protein was concentrated by use of centrifugal filter units (10,000 Da molecular weight cut-off). d Protein quantitation was performed using bovine serum albumin (BSA) as the standard. e
Preparation of polyclonal and monoclonal antibodies
Polyclonal rabbit antiserum was raised against consensus rCPB2 protein. Mouse mAb Songer 9E4B was prepared using purified consensus rCPB2 as antigen. Immunoglobulins (polyclonal antibodies [pAbs]) were purified using protein A columns. f Antibodies were aliquoted in small volumes and stored at −70°C until used.
Development and optimization of ACE
Optimization steps in the process of development of the ACE included assessment of the optimal concentrations of polyclonal capture antibody (rabbit anti-rCPB2), of rCPB2, of horseradish peroxidase (HRP)-labeled monoclonal detection antibody (mouse anti-rCPB2 mAb-HRP), optimization of incubation conditions, selection of blocking buffers, and selection of coating buffers (details not shown).
For the final ELISA, 96-well plates g were coated with 100 µl/well of polyclonal capture antibody (diluted 1:2,500 in carbonate-bicarbonate buffer, pH 9.6) for 2 hr at 37°C followed by overnight incubation at 4°C. After washing the plate twice with wash buffer (phosphate buffered saline [PBS], 0.05% Tween 20, 0.5% fish skin gelatin h ) and once using PBS (pH 7.4), the coated plate was blocked using blocking buffer (PBS, 0.05% Tween 20, 2% BSA) for 2 hr at 37°C. After washing 3 times with wash buffer, 100 μl/well of purified consensus rCPB2 (2-fold dilution series starting from 109.9 to 3.43 ng/ml in wash buffer) or processed intestinal contents samples from neonatal piglets (described in the following) were added to the plate (in triplicate or in duplicate when the sample volume was insufficient). The addition of antigen was performed on ice. Washing buffer with no antigen was used as a negative control. After incubation at 4°C overnight, followed by 5 washings on ice with cold washing buffer, 100 μl/well of enzyme-labeled monoclonal detecting antibody (diluted 1:5,000 in wash buffer) was applied, and the plate was incubated for 1 hr at room temperature. The plate was then washed 3 times with wash buffer and 100 μl/well of the chromogenic substrate 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS) i was added. After 1 hr further incubation at room temperature, the reaction was stopped using 0.5% SDS (50 μl/well) and the optical density (OD) measured at 405 and 490 nm in an ELISA reader (spectrophotometer). j The mean dual wave OD values (OD at 490 nm subtracted from OD 405) of the replicates of each sample were used to calculate the ratios of the positive and negative samples. Two signal-to-noise ratios were calculated. One compared the OD values between wells with the antigen (intestinal sample or rCPB2) and wells with no antigen added (both wells coated with pAb); the other compared the OD values between the pAb-coated and uncoated wells (antigen was applied in both wells). To determine the optimal concentrations for capture and detection antibodies, the assay was performed with different concentrations of each. Concentrations selected were those that gave the best ratios comparing OD values. Serial 2-fold dilutions of rCPB2 standards, starting from 110 ng/ml, were included in each plate to generate a standard curve.
Source of intestinal samples for development of ACE
Piglets from 10 swine farms with a history of suspected C. perfringens toxinotype A neonatal diarrhea were submitted for detailed diagnostic examination. Piglets from each of the farms were less than 3 days of age; at least 3 piglets (2 diarrheic and 1 healthy) from the same farm were submitted live to the diagnostic laboratory. Piglets were euthanized and immediately subjected to a detailed necropsy examination. Contents of the small and large intestine were collected on ice within 30 min of death and submitted to the laboratory within 1 hr. All subsequent intestinal content processing was done on ice, including centrifugation at 4°C. Briefly, each sample was centrifuged at 4°C at 17,500 × g for 30–60 min, depending on the clarification of the supernatant. If the intestinal contents were of a thick consistency, the contents were diluted in sterile cold PBS and the dilution was recorded. These samples were vortexed vigorously before centrifugation. After clear supernatant was separated, it was filtered (0.45 μm pore diameter, low protein binding), k and was then ready for application onto the ELISA plate.
Effect of intestinal content and feces on CPB2 detection
Clostridium perfringens rCPB2 and, in some cases, concentrated supernatant of isolate P18 were used as a source of consensus CPB2 for spiking piglet fecal and intestinal content samples with undetectable C. perfringens counts (<100 CFU/ml). In addition, small intestinal and colonic content samples, obtained at necropsy of the piglets, were spiked using the same procedure. Briefly, 3 ml of sterile cold PBS was added to 3 g of feces or intestinal content, mixed thoroughly and stored on ice. A volume of 3 ml of rCPB2 (1:250 dilution) was added to the sample, vortexed vigorously for 5 min, and then stored on ice. The spiked sample was divided into 4 aliquots (F-0, F-4, F-8, and F-overnight). F-0 and negative control (non-spiked feces) were processed and tested immediately, whereas F-4, F-8, and F-overnight were processed and tested after incubation at room temperature for 4 hr, 8 hr, and overnight, respectively.
Three approaches were used to examine small intestinal content for protease activity and how this could be overcome. The first approach examined the effect of protease inhibitors. Two inhibitors, amidinophenyl-methanesulfonyl fluoride (APMSF, serine protease inhibitor–specific) j and a protease inhibitor mix f were added to the samples, in the concentrations recommended by the manufacturer, before processing for ACE. After protease inhibitors were added, samples were vortexed, and the processing was continued according to the previously described protocol. The second approach involved processing of every step at 4°C, including incubation of processed samples at 4°C overnight. The third approach examined pH effects. Two sets of 4 different pH (PBS, pH 7.4; citrate buffers pH 4.0; pH 5.0; pH 6.0) buffers were used for sample preparation and sample spiking using rCPB2 (13.75 ng/ml). Bovine serum albumin (5%) and fetal calf serum (FCS, 50%) were added to each of the buffers in set 1 and set 2, respectively.
Effect of freezing feces and intestinal content on CPB2 recovery
Spiked feces and necropsy intestinal samples previously tested immediately upon receiving and after spiking, were frozen at −70°C. After thawing, samples were assayed by ACE, and results compared to those of the freshly prepared samples. Freezing had no effect on CPB2 recovery from spiked samples (data not shown).
Results
Initial evaluation of ACE using recombinant consensus CPB2
Initial experiments to optimize the ACE used rCPB2. The lowest concentration of captured CPB2 detected was between 2.0 and 3.5 ng/ml. The data demonstrated that incubation of pAb diluted 1:2,500 for 2 hr at 37°C followed by overnight at 4°C was optimal for coating the wells and that a 1:5,000 dilution of mAb–HRP incubated for 1 hr at room temperature was optimal for detecting captured antigen. There was a slightly curvilinear OD (3.5 to 0) CPB2 concentration (110 to 0 ng/ml) relationship in plates incubated for 2 hr at 37°C; the minimum concentration detected was 2–3 ng/ml of rCPB2.
Comparison of recombinant versus native CPB2
When completely purified consensus nCPB2 was used as antigen instead of consensus rCPB2, the ACE results were similar (Fig. 1). In contrast, when atypical rCPB2 was used as antigen, the limit of detection was higher than for consensus protein (data not shown). Optical density readings of the signal that resulted from the capture of atypical rCPB2 (when applied at the same concentration) were approximately 90% lower than for consensus rCPB2.
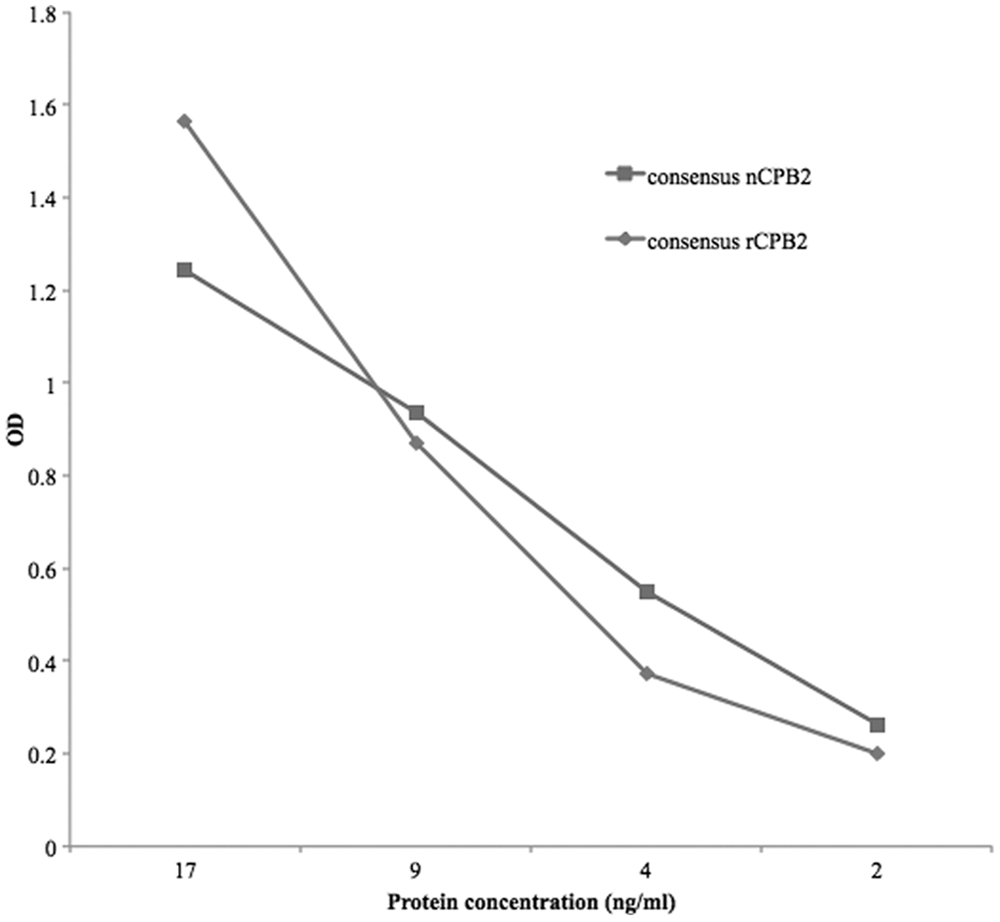
Antigen-capture enzyme-linked immunosorbent assay (ACE) detection of recombinant consensus versus native (consensus) Clostridium perfringens beta2-toxin (CPB2). The ACE detected native consensus CPB2 as well as the recombinant protein using a preliminary protocol (antigen incubation phase at 37°C for 2 hr). OD = optical density.
Effect of intestinal content and feces on CPB2 recovery
When tested by ACE, the supernatant fluid from strain P18 containing consensus CPB2 showed comparable reaction kinetics to those of rCPB2. Spiking piglet feces or piglet colonic or small intestinal contents with the native CPB2, using the concentrated supernatant of strain P18, showed that the amount of toxin that could be recovered decreased with storage at room temperature over a short time (Fig. 2). The loss of the toxin in colonic or fecal contents was approximately 30–50% after 4 hr at room temperature. If the added CPB2 was detected, as shown for fecal and colonic samples in Figure 2, the dose-response curve was similar to the standard curve but the signal intensity was lower, indicating partial destruction of the toxin. Strikingly, the small intestinal contents from some piglets eliminated the toxin almost immediately, and also caused very high noise in control wells. The OD values in some cases were higher in uncoated than in coated wells (Fig. 3). This was consistent with degradation of capture antibody and/or blocking agent, leading to nonspecific binding to the ELISA plate by the labeled detection mAb.
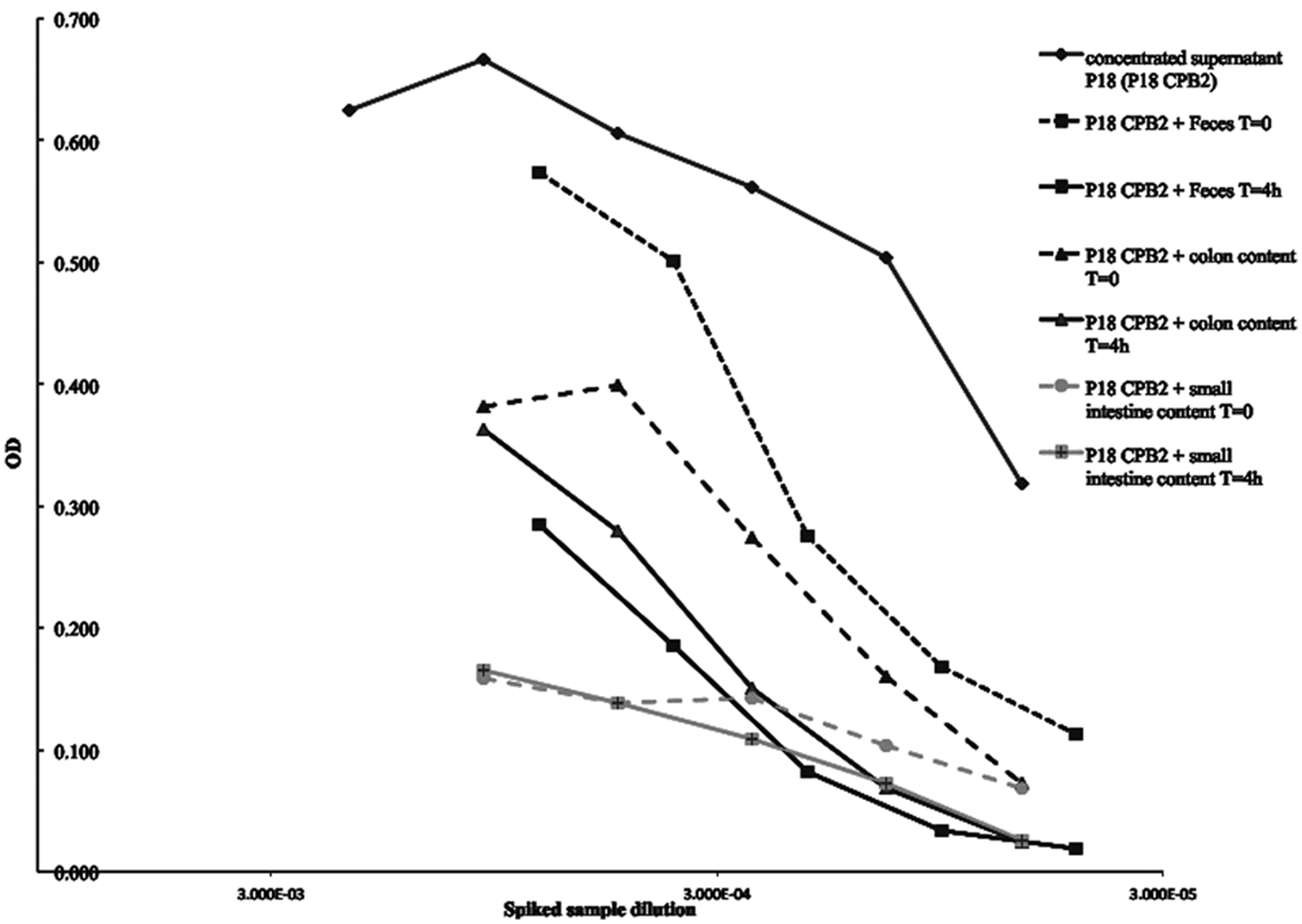
Clostridium perfringens beta2-toxin (CPB2) detection in spiked samples. Porcine feces and colon and small intestinal contents spiked with the P18 isolate concentrated supernatant were tested immediately (T = 0) and after 4 hr (T = 4h) at room temperature using a preliminary protocol (antigen incubation phase at 37°C for 2 hr). Storage for 4 hr in feces or colonic or small intestinal content reduced CPB2 detection. For some samples of small intestinal contents, mixing of toxin with the contents resulted in immediate destruction of CPB2, likely attributable to the presence of proteases in the samples. OD = optical density.
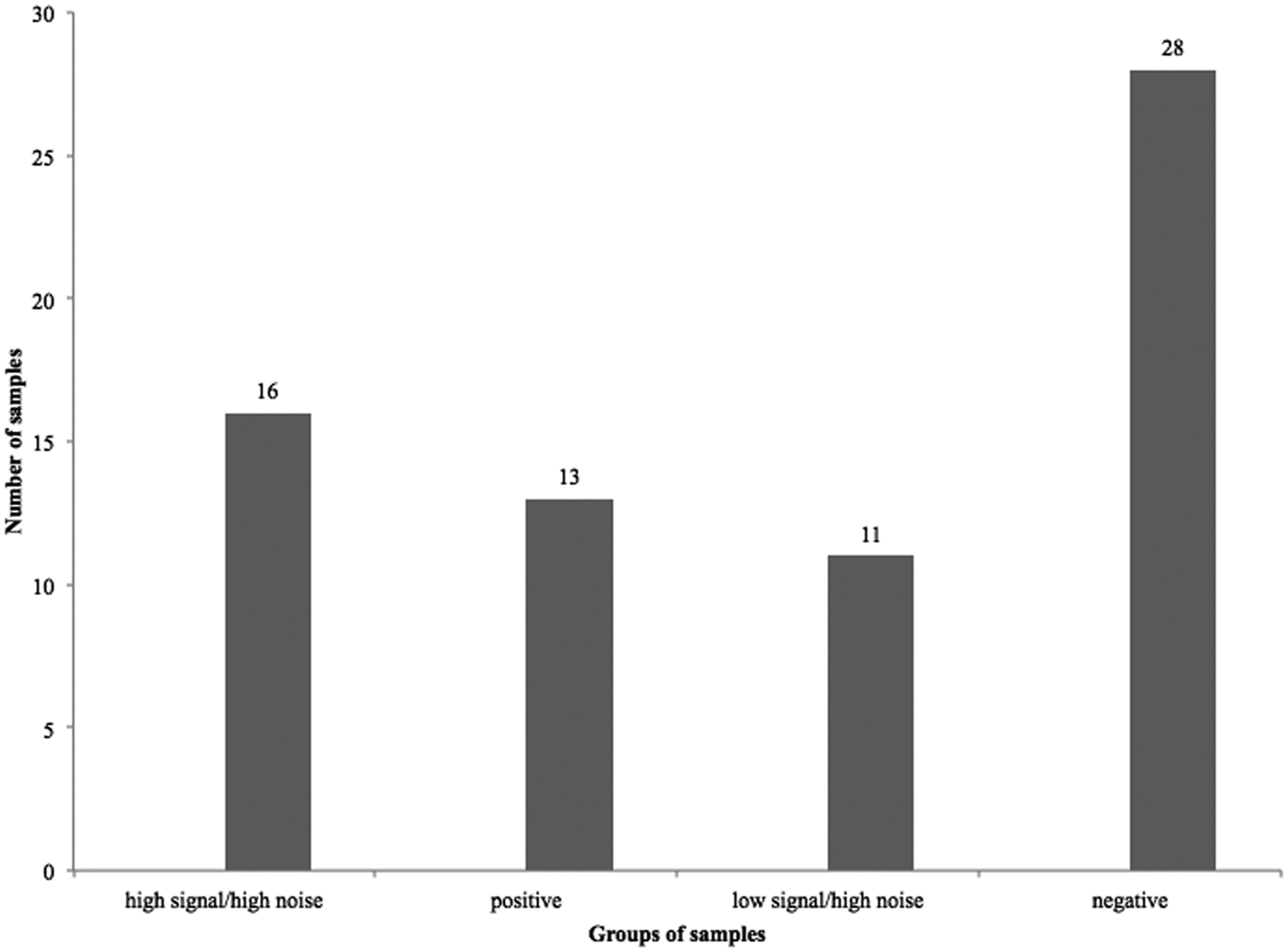
Signal and noise in piglet intestinal samples used in development of the Clostridium perfringens beta2-toxin (CPB2) antigen-capture enzyme-linked immunosorbent assay (ACE). Using a preliminary protocol (antigen incubation at 37°C for 2 hr), the tested samples fell into 4 groups depending on optical density (OD) values of the signal and the noise (3 standard deviations were added to the OD values of the noise in the wells where no antigen was added (the acceptable noise was therefore ≤ OD 0.120): 1. samples with high signal but unacceptable noise in the control wells; 2. positive samples (no noise in control wells); 3. samples with low signal but unacceptable high noise in the control wells; 4. negative samples (no noise in control wells).
Detection of CPB2 in clinical samples
Initial ACE testing of intestinal content samples, from both diarrheic and healthy piglets, showed highly inconsistent results. Some samples apparently contained highly active proteases, which resulted in high OD readings in the control wells lacking capture antibodies (noise). These results suggested that the presence of some other compounds (likely proteases) in the samples caused nonspecific reactions in negative control wells by degrading the blocking agent. Mixing CPB2 with small intestinal contents from some animals resulted in almost immediate toxin loss (Fig. 2). Of 68 intestinal content samples from neonatal piglets that originated from farms with a history of possible C. perfringens toxinotype A diarrhea, 41 samples showed acceptable results (13 positive, 28 negative samples), suggesting that the assay as developed could be applied in diagnosis in approximately 60% of cases. As described in the following, the acceptable OD value for the “noise” was determined to be 0.120. However, 16 samples gave false-positive results (identified by high OD values in the capture antibody–free control wells, with concurrent high OD values in the capture antibody–coated wells) due to the activity of interfering factors present in the intestinal contents (Fig. 3).
Figures 3 and 4 present a summary of the “noise” in the capture antibody–free well of the ACE for colonic and small intestinal samples from diarrheic and healthy piglets, processed before studies involving the addition of protease inhibitors. This was used to define a value above which the noise in the control (capture antibody–free) wells was judged unacceptable. Figure 4 shows that most of the sample values in the control wells ranged between 0.051 and 0.150 OD. An OD of 0.120 was judged to be the “cut-off” value for noise, above which the test results would be considered unreliable. This value was determined by calculation and addition of 3 standard deviations of the OD obtained by the signal in wells where PBS was added instead of the test antigen (intestinal contents).
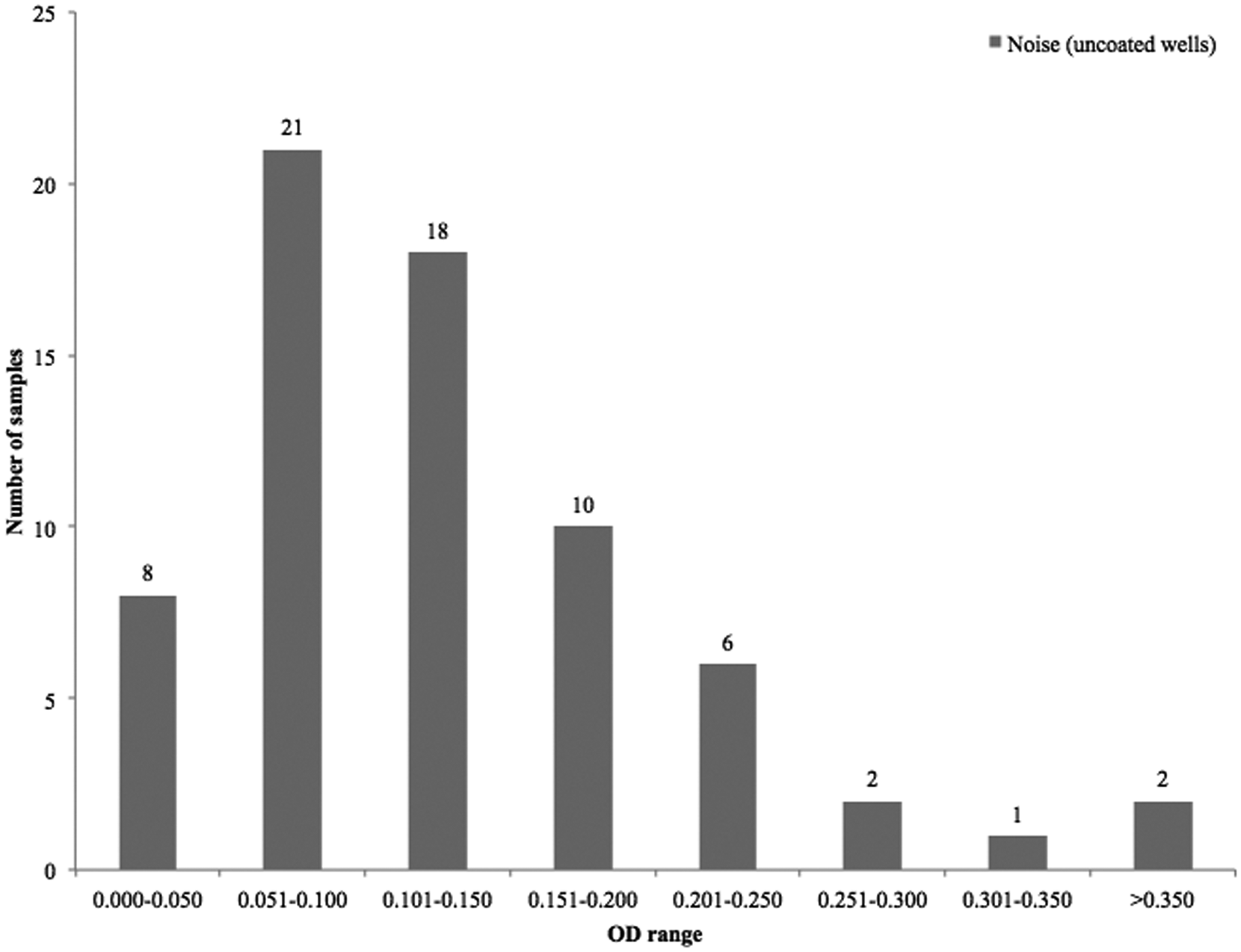
Optical density (OD) range of “noise” in the capture antibody–free control wells. Colonic and small intestinal samples from 68 diarrheic and healthy piglets. Most of the samples clustered between 0.051 and 0.150 OD values.
The addition of protease inhibitors reduced the noise observed in some wells without capture pAb but the effect appeared to depend on the amount of proteolytic activity present. Only 4 out of 15 samples showed improvement of the test performance, but these improvements in control wells often resulted in lower signal OD by wells with capture antibody, which did not result in overall signal-to-noise ratio improvements. The protease activity in the small intestinal content during ACE testing could also be partially controlled by lowering the temperature to 4°C during the initial sample incubation period. Twenty-one samples previously incubated at 37°C for 2 hr were retested using initial incubation at 4°C overnight. Of these 21 samples, 20 had improved signal-to-noise ratio when coated and “uncoated” (control) wells were compared (data not shown). The results showed that low temperature produced a partial improvement in the overall test performance, although this did not fully remove the effect of protease activity (data not shown).
As another approach to controlling and decreasing the activity of proteases, the effects of pH on test performance and protein saturation of diluents used in intestinal sample processing were assessed using a sample that contained reactive proteases (Fig. 3). Table 1 shows the results of rCPB2 recovery after the small intestinal content spiking using 8 different diluents. Citrate buffer with 5% BSA (pH 6.1) markedly increased recovery of rCPB2 measured by ACE. Overall, rCPB2 recovery was considerably higher when diluents with 5% BSA were used, compared to diluents with 50% FCS (Table 1). These results showed that protease activity could be overcome.
Recovery of recombinant Clostridium perfringens beta2-toxin in the small intestinal content of a neonatal pig with high protease activity by using 8 different diluents.*

PBS = phosphate buffered saline; BSA = bovine serum albumin; FCS = fetal calf serum.
Discussion
The 2–3 ng limit of antigen detection of the ACE was almost identical for the purified consensus native CPB2 protein (Fig. 2), but was far lower for the atypical rCPB2 protein, probably because the mAb does not detect the relevant epitope of the atypical protein. Consensus and atypical CPB2 proteins have only 62.3% amino acid identity. 9 The most obvious application of this ACE is in the diagnosis of type A–associated enteric disease in neonatal swine, because C. perfringens isolated from these piglets are commonly consensus cpb2-positive and this gene is commonly expressed, in contrast to isolates from the intestine of other species where cpb2 is often both atypical and apparently not expressed. 9
As others have found, optimizing an ACE by using intestinal content and fecal samples can involve difficulties because intestinal or fecal extracts may affect toxin recovery, either by degradation through proteases7,18 or because of the necessity for dilution.1,20 The effect was less marked in spiked fecal samples (Fig. 2). However, dilution was not an issue because most of the intestinal contents used for the development of the current assay were liquid, and so were applied undiluted or minimally diluted. The major problem identified in development of the ACE was the presence of protease activity in the small intestinal content of some neonatal piglets (Fig. 2). The samples that displayed excessive desorption of the wells, presumably as a result of protease activity, caused a high proportion of nonspecific reactivity. The use of protease inhibitors showed that some intestinal contents had protease activity (likely pancreas-derived trypsin) responsible for “uncoating” antibody-coated wells and/or degrading the blocking agent (BSA) from the polystyrene well. In the absence of coating or blocking, it is likely that mAb-HRP became bound to uncoated wells and gave false-positive signals (“noise”). A “cut-off” of acceptable and unacceptable noise was developed based on the OD of control wells incubated with buffer rather than intestinal samples. Applied to intestinal samples from piglets at necropsy it was shown that, in the absence of protease inhibition, approximately 40% of samples gave unacceptable noise (Figs. 3, 4).
An approach considered for dealing with the protease issue was the use of heat inactivation of proteases, as shown for detection of Clostridium difficile toxinotype A in feces. 7 However, because the CPB2 toxin is very unstable, 6 this would likely have had a destructive effect on the toxin itself, so this approach was not examined.
The low pH buffer combined with high protein concentration and processing at 4°C approach to addressing the protease issue was adopted from a previous study developed for prevention of desorption by intestinal proteases during the detection of C. difficile toxinotype A in feces.7,18 The results of those studies showed that 50% FCS and citrate buffer containing 5% BSA (pH 4.7) significantly reduced desorption of the plates (protease activity) and improved recovery of the toxin A from the samples, compared to the other tested diluents. Fetal calf serum contains various protease inhibitors and high concentration of proteins; in addition to inhibition, it may provide substrate in excess for protease activity. Because the optimal pH for protease activity in the intestinal or fecal content is alkaline or neutral, an acid pH inhibits the activity of intestinal proteases.
One of the difficulties in developing a CPB2 ACE was the absence of a “gold standard.” Clostridium perfringens is considered to be a component of normal microflora, 15 and isolation of the organism does not necessarily mean that it is a cause of the disease. Without a gold standard, a receiver operating characteristic curve could not be used in order to compare the diagnostic performance of the “new” test (CPB2 ACE) with a well-established laboratory or diagnostic test (gold standard). An acceptable OD for noise was therefore determined from the control, PBS-treated, “uncoated” wells.
In conclusion, an ACE was developed to detect C. perfringens consensus CPB2 toxin in feces and intestinal contents of piglets. The ACE was initially confounded by the presence of proteases in the intestinal contents, but this effect was overcome. Control wells lacking capture antibodies allowed detection of proteolytic activity that in some cases could not be overcome by the developed method, as well as identification of the samples to which the ACE cannot be applied. Detection of CPB2 in the intestinal content samples using the newly developed ACE may be valuable in diagnosis of the role of consensus cpb2-associated diarrheal illness in neonatal swine. The advantage of an ACE over an alternative approach, such as real-time polymerase chain reaction quantitation of cpb2 in the intestine, is that the ACE detects expression of antigen, not simply the presence of the DNA or messenger RNA. Further work is required to apply the ACE to assess the role of C. perfringens toxinotype A in enteric disease in neonatal swine.
Footnotes
Acknowledgements
The authors thank Grant Perry for technical support.
a.
Qiagen Inc., Mississauga, Ontario, Canada.
b.
LB agar, BD Difco, Mississauga, Ontario, Canada.
c.
Coomassie brilliant blue R250, Bio-Rad Laboratories Ltd., Mississauga, Ontario, Canada.
d.
Amicon Ultra centrifugal filter units, Millipore Inc., Billerica, MA.
e.
BCA Protein Assay Kit product, Pierce, Fisher Scientific Ltd., Ottawa, Ontario, Canada.
f.
GenScript, Piscataway, NJ.
g.
MaxiSorp, Nunc, Roskilde, Denmark.
h.
Norland Products Inc., Cranbury, NJ.
i.
Roche Diagnostics, Laval, Quebec, Canada.
j.
BioTek Instruments Inc., Winooski, VT.
k.
Fisherbrand syringe filters, Fisher Scientific Ltd., Ottawa, Ontario, Canada.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funded by the Animal Health Strategic Initiative program, Ontario Ministry of Agriculture, Food and Rural Affairs, and by the Natural Sciences and Engineering Research Council of Canada.