
Abstract
Kobuvirus infections are common among humans, rodents, carnivores, pigs, and ruminants. We report herein the complete genome sequence of a novel caprine kobuvirus (MN604700) from diarrheic kids in Minnesota. Whole-genome sequencing revealed a kobuvirus genome of 8,139 nt with a single ORF region encoding a polyprotein of 2,480 amino acids. Further analysis revealed nt substitutions along the genome compared with that of the caprine kobuvirus reference strain, with 93% identity. Phylogenetic analysis indicated that the clade of the caprine kobuvirus was most closely related to porcine kobuviruses rather than bovine or ovine kobuviruses. Using primers designed from this genome, caprine kobuvirus was identified in the stools of other goats. Sanger sequencing of PCR products indicated 3D and VP1 gene nucleotides of this latter strain were 95% and 91% identical with those of MN604700, respectively. There were 35 and 101 nt substitutions in 3D and VP1 genes, respectively. Findings of kobuvirus over a 2-y period may indicate an endemic state, which needs further research. In addition, screening for kobuviruses over large geographic areas is needed to identify the evolutionary connections among different strains.
Introduction
Genus Kobuvirus (Picornaviridae) includes 3 species: Aichivirus A, B, and C. In 2016, 3 new species, namely Aichivirus D, E, and F, were added (http://www.picornaviridae.com/). At present, caprine kobuvirus 1, cattle kobuviruses 1 and 2, and European roller kobuvirus 1 have not been assigned to any of the 6 species of kobuviruses mentioned above. 1 Members of a species share a significant degree of amino acid identity in the P1, 2C, 3C, and 3D proteins, and have the same monophyletic group in phylogenetic tree cladistics. 33 The name “kobu” was derived from the Japanese word representing “knobby” appearance, given that the virions appear to be knobby under an electron microscope. 32 Kobuviruses are small (8.2–8.4 kb), spherical, nonenveloped, single-stranded, positive-sense RNA viruses. Genetically, the virus consists of 5′-untranslated regions (UTRs), a single large open reading frame (ORF), and 3′-UTR. A single ORF codes for a polyprotein that is later divided into nonstructural protein L (leader), followed by 3 regions, P1–P3. Region P1 includes capsid structural proteins (VP0, VP3, and VP1); regions P2 and P3 include nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D).6,23
Kobuvirus was first isolated in Japan in 1989 from humans with gastroenteritis following the consumption of raw oysters. 27 Subsequently, the virus was isolated from a wide range of hosts including human, ferrets, mice, foxes, bats, dogs, cats, cattle, sheep, pigs, and goats with and without clinical signs from many countries of the world.3,11,14,16,18,20,29,30 Caprine kobuvirus was detected in a black goat with diarrhea in the Republic of South Korea.13,21 In Italy, kobuvirus was detected in both healthy and diarrheic goats, with a higher percentage in diarrheic goats. 17
Most of the kobuviruses cause subclinical infections in their hosts but, in some cases depending on the immunocompetence of the animals, diarrhea and fever caused by viremia are observed.26,27 Next-generation sequencing (NGS) has played a great role in identification of Aichivirus strains. 19 We describe herein the detection and complete genome sequence of a caprine kobuvirus from Minnesota, which to our knowledge, has not been reported previously from goats in the United States.
Materials and methods
Samples
In March 2017 and October 2019, the Minnesota Veterinary Diagnostic Laboratory (St. Paul, MN) received small and large intestinal samples from goat kids (10–14 d old) with a history of diarrhea followed by acute death. The submitting veterinarian reported that multiple kids were dying after developing diarrhea. Samples were received as segments of unfixed intestine and contents in Ziplock bags plus segments of formalin-fixed intestine. On gross examination, the large and small intestines were dilated and filled with gas and yellow, watery contents. No blood or mucus were observed in the submitted samples. Curdled milk was present in the abomasum; all other tissues appeared normal.
Bacteriologic and parasitologic examination
Swabs of intestinal contents were inoculated in cooked meat enrichment medium to detect anaerobic microorganisms (mainly Clostridium spp.) followed by incubation at 37°C for 24 h. A loopful was streaked onto a 10% sheep blood agar plate containing neomycin sulfate (200 µg/mL) followed by incubation under anaerobic conditions at 37°C for 24 h. The samples were also streaked onto MacConkey agar and eosin methylene blue agar (Merck) to detect Escherichia coli and other bacteria. 31 A simple flotation technique was used to determine the presence of parasites in intestinal contents and intestinal smears. 9
Histopathology
Pieces of formalin-fixed intestine were processed routinely, stained with hematoxylin and eosin, and examined under a light microscope. 28
Electron microscopy
For negative-contrast transmission electron microscopy (TEM), fecal samples from 4 goats were placed in a 20-mL tube, and double-distilled water was added to a final volume of 15 mL. After 10 min at room temperature, 1 mL of the fecal suspension was filtered through 0.45-µm syringe filters (Thermo Fisher). Fifty µL of filtered suspension was transferred to airfuge tubes (Beckman Coulter) with formvar-coated, 200-mesh copper grids (Electron Microscopy Sciences) and centrifuged at 148,000× g (Beckman Coulter) for 10 min. Grids were washed and stained with 1% phosphotungstic acid (Electron Microscopy Sciences) for 1 min. These samples were visualized (1400 transmission electron microscope; JEOL) and images obtained (Capture Engine v.7.00 camera and software; Advanced Microscopy Techniques). Image analysis was carried out using Image J (NIHR public domain).
Next-generation sequencing
Total nucleic acid was extracted from intestinal contents (QIAamp MinElute virus spin kit; Qiagen) followed by submission to the University of Minnesota Genomic Center (UMGC) for complementary DNA synthesis, library preparation, and 250-bp, paired-end cycle sequencing (MiSeq; Illumina). CLC Genomic Workbench v.11.0.1 (Qiagen) was used to analyze the sequence reads. De novo assembled contigs were further confirmed by BLASTn and BLASTx analysis on NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). The ORF finder tool (www.ncbi.nlm.nih.gov/orffinder/) was used to find ORFs in the obtained sequences. Prediction of the presence of cleavage sites and structural and nonstructural proteins was done based on nucleotide (nt) and amino acid (aa) alignments, with reference strain KF793927.1 of caprine kobuvirus in GenBank.
Phylogenetic analysis
The complete genome sequence of caprine kobuvirus obtained in our study was aligned with published sequences of kobuviruses in GenBank using the Clustal W method in MEGA-X. 12 The maximum-likelihood method for analysis of the complete genome (GTR+G+I), 3D gene (TN93 +G), and VP1 gene (GTR+G) sequences was selected on the basis of the lowest BIC score (Bayesian Information Criterion) with 1,000 bootstraps in MEGA-X software.
Primer design and amplicon sequencing
From the newly identified sequence obtained by NGS, we designed primers that could amplify the 3D and VP1 genes. The 3D forward primer was 5′-TCAACTCTTCCTCAAACACAAC-3′; the reverse primer was 5′-GCCGCCAATCATCTCATAC-3′, yielding an amplicon of 703 bp. The VP1 forward primer was 5′-TCACCAACTATACCCTCCCC-3′ and reverse primer was 5′-ACAGCAAGAGCACCAAGAC-3′; the amplicon size was 1,330 bp. Reverse-transcription PCR (RT-PCR) was performed (OneStep RT-PCR kit; Qiagen) in 25 μL of reaction mixture in an Eppendorf thermocycler. The latter was adjusted at 50°C for 30 min for the RT step, and then at 95°C for 15 min for Taq activation. This was followed by 35 cycles of 94°C for 1 min, 1 min at 50°C and 55°C for 3D and VP1, respectively, and then 72°C for 1 min, followed by final extension cycle of 72°C for 10 min. After confirmation of the amplicon on gel, the product was purified (QIAquick PCR purification kit; Qiagen) according to the manufacturer’s instructions. The purified product was prepared for Sanger sequencing using the same forward and reverse primers as used in RT-PCR. The samples were submitted to UMGC for Sanger sequencing. The obtained sequences were assembled using Sequencher v.5.1 (https://www.genecodes.com/) software followed by NCBI BLAST analysis.
Results
Bacteriologic, parasitologic, and histologic investigation
The bacteriologic examination did not result in any significant growth of anaerobic and/or aerobic bacteria. Parasitologic examination yielded no parasitic ova or oocysts. Histopathology revealed intestinal crypt hyperplasia and infiltration of lamina propria of the colon with numerous degenerate and nondegenerate neutrophils (Fig. 1). Postmortem autolysis precluded examination of the surface epithelium for erosions. Evidence of a coccidial infection was not detected in the small or large intestine.
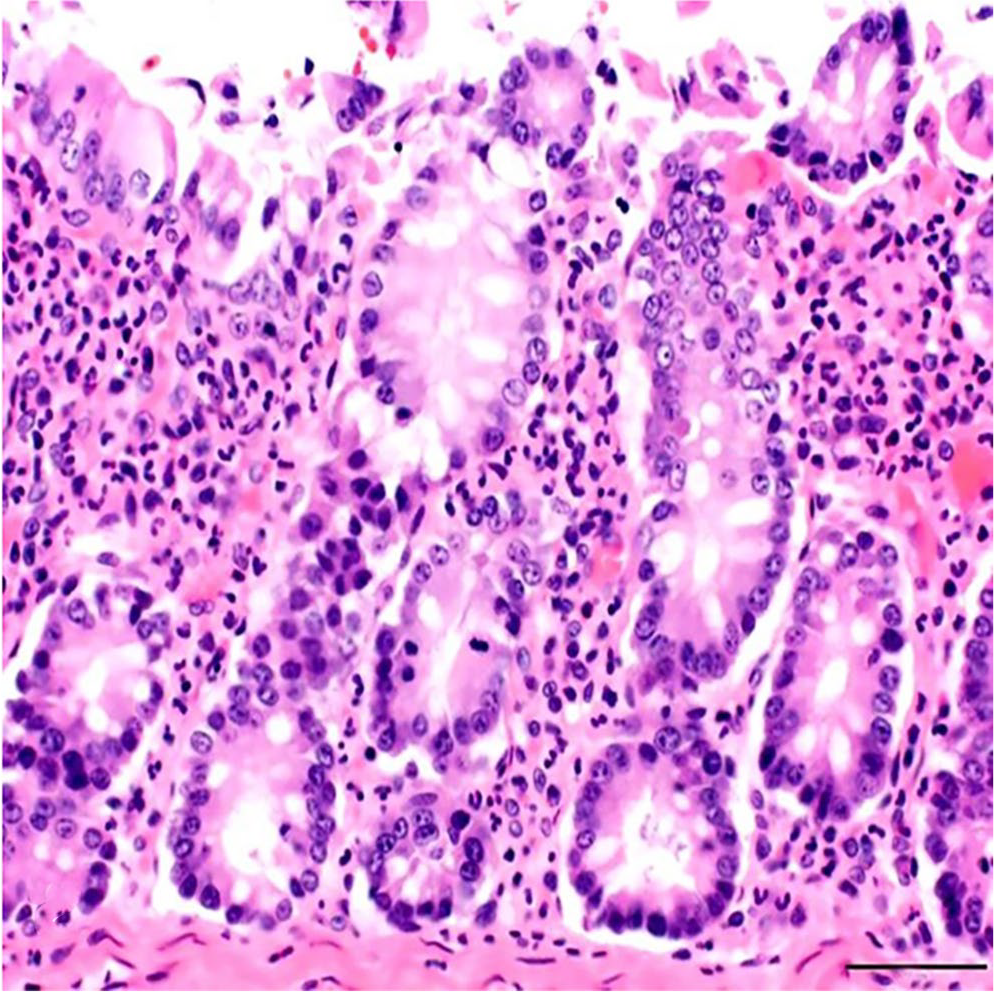
Colon of a goat with kobuviral infection. The lamina propria is markedly infiltrated with neutrophils. H&E. Bar = 50 µm.
Electron microscopy
Large numbers of nonenveloped, spherical particles (30.19–35.55 nm diameter; mean: 33.44 nm, SD: 1.43 nm) were visualized on TEM (Fig. 2).

Ultra-microphotograph of negative-contrasted kobuvirus from goat 1. Nonenveloped, spherical viral particles. Bar = 100 nm.
Molecular investigation
NGS data analysis revealed that the distribution of sequences in the sample was 20% bacteria, 20% eukaryotic, and 49% unknown. However, we do not have the goat (host) genome in our reference dataset yet. Thus, a major portion of the “unknown” sequences are likely goat sequences. Only 1% of all sequences were classified as viral (which is common in metagenomic sequencing). Among the viral sequences, 34% (4,039 reads) of all viral sequences were classified as caprine kobuvirus. In addition, ~3% of viral sequences belonged to the Orthoretrovirinae subfamily.
The almost complete kobuvirus genome sequence was assembled with 8,139 nt including 665 nt in 5′-UTR, 7,440 nt for ORF, and 34 nt in 3′-UTR. The single ORF region encodes a polyprotein of 2,480 aa beginning with a methionine codon and ending with an alanine codon. The complete genome sequence (caprine kobuvirus/MN-USA/2017) was submitted to GenBank (accession MN604700). Detection of nt variations with the closest caprine kobuvirus sequence (KF793927) revealed the insertion of 5 and 3 nt in 5′UTR and 3D genes, respectively. The nt substitutions were detected along the genome compared with the caprine kobuvirus reference strain (KF793927/caprine kobuvirus/South Korea/2012), with 93% identity. Investigation of the aa substitutions suggested the insertion of proline codon at 2,142 ORF position. Substitutions were high in L and VP1 regions (10 aa) followed by VP0 (5 aa), 3D (4 aa), 2A (3 aa), 2C and 3A (2 aa); only 1 aa substitution was seen in VP3, 3B, and 3C (Table 1).
Substitution of nucleotides and amino acids along the caprine kobuvirus genome compared with the closest reference strain, KF793927.
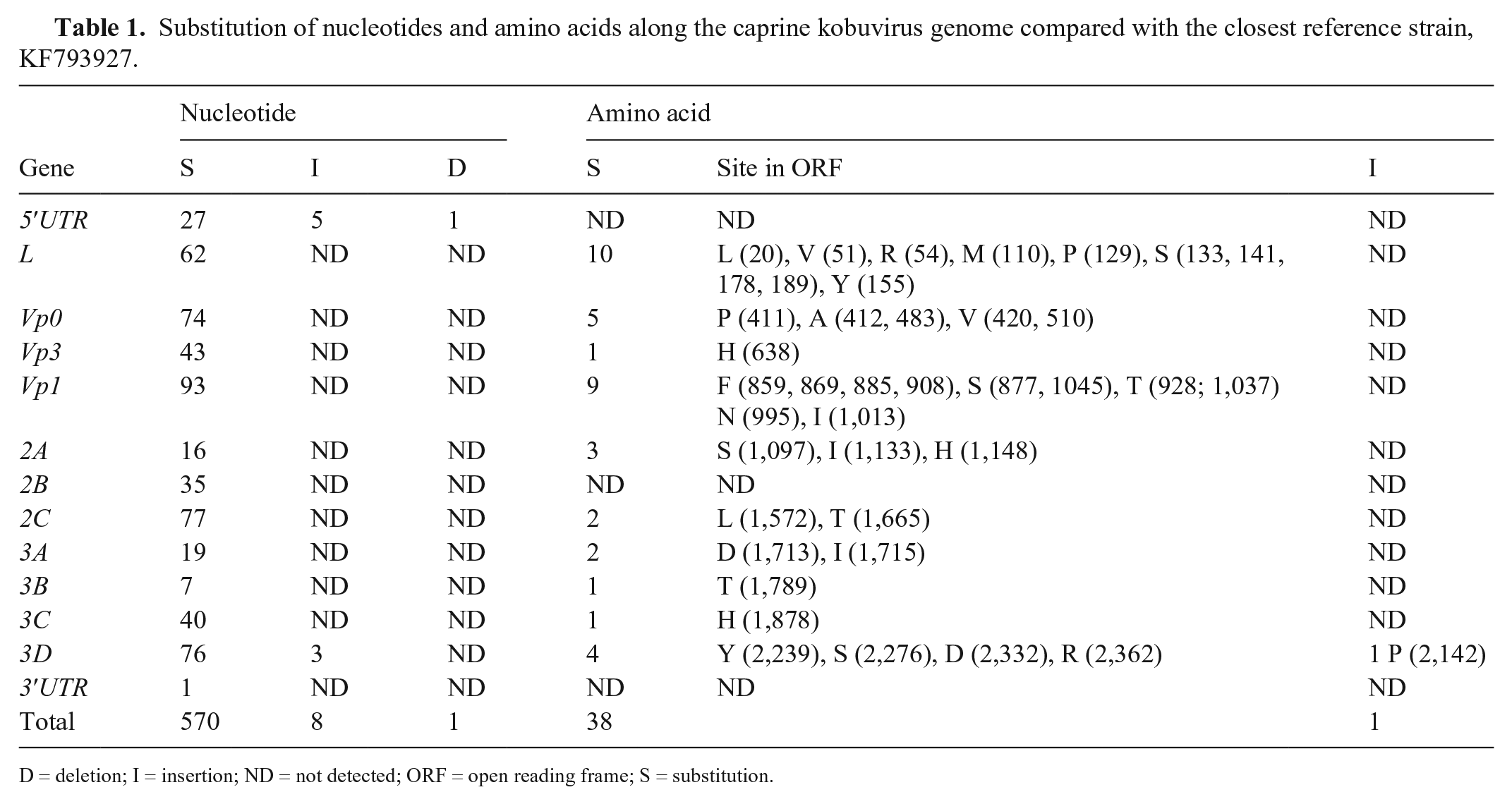
D = deletion; I = insertion; ND = not detected; ORF = open reading frame; S = substitution.
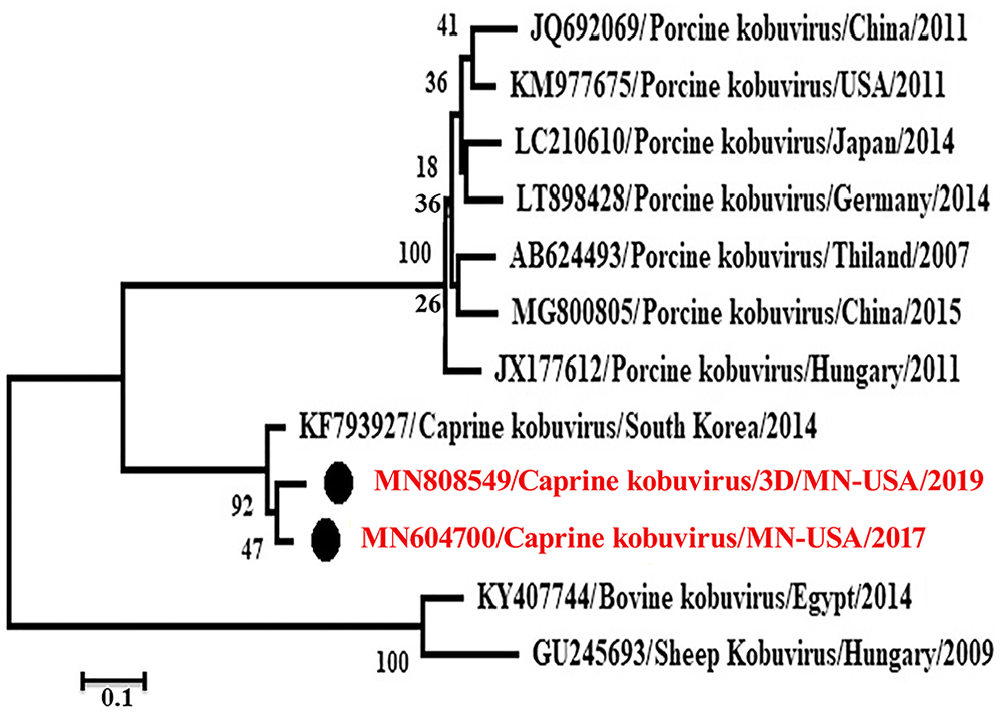
Phylogenetic analysis revealed that these caprine kobuviruses formed a new clade and were closely related to porcine kobuvirus rather than to bovine, and sheep kobuviruses, which are most related to each other (Figs. 3, 4). The P1 region, including the VP1 gene, had low nt and aa identities with the reference caprine kobuvirus sequence. The identity was 89% in VP1 gene nt and 97% among aa. VP1 gene is also the most divergent among kobuviruses of different species (Table 2). The P2 region had the least number of aa substitutions along the genome. The P3 region included genes that are the most conserved and have the least variation among different species.

Phylogenetic analysis of MN604700 strain (caprine kobuvirus/MN/2017) complete open reading frame with other related viruses through the maximum-likelihood method by using the GTR+G+I model.
Phylogenetic analysis of the 3D gene (MN808549) of caprine kobuvirus/MN/2019 compared with MN604700 strain (caprine kobuvirus/MN/2017) and other related viruses through the maximum-likelihood method by using the TN93+G model.
Identity among nucleotides (nt) and amino acids (aa) along the kobuvirus genome of different species.

The designed primers were used for testing samples from diarrheic goats obtained in 2019. Primers of the 3D and VP1 genes amplify products of the expected sizes, 703 and 1,330, respectively. The amplicons were sequenced and submitted to GenBank (accessions MN808549 and MN808550), for 3D and VP1 genes, respectively. Identity of the 3D and VP1 gene nt of the 2019 strain with MN604700 was 95% and 91%, with 35 and 101 nt substitutions in both genes, respectively. Phylogenetic analysis of the 3D and VP1 genes is compatible with caprine kobuviruses forming a clade that is more closely related to the porcine kobuvirus clade than to bovine and sheep kobuvirus clades (Figs. 4, 5). The 3D gene of the virus newly identified in 2019 (MN808549/caprine kobuvirus/MN/2019) was in the same clade as a virus identified in 2017 (MN604700/caprine kobuvirus/MN/2017; Fig. 4). Interestingly, the VP1 gene (MN808550) was in a different but related clade with MN604700/caprine kobuvirus/MN/2017 virus (Fig. 5).
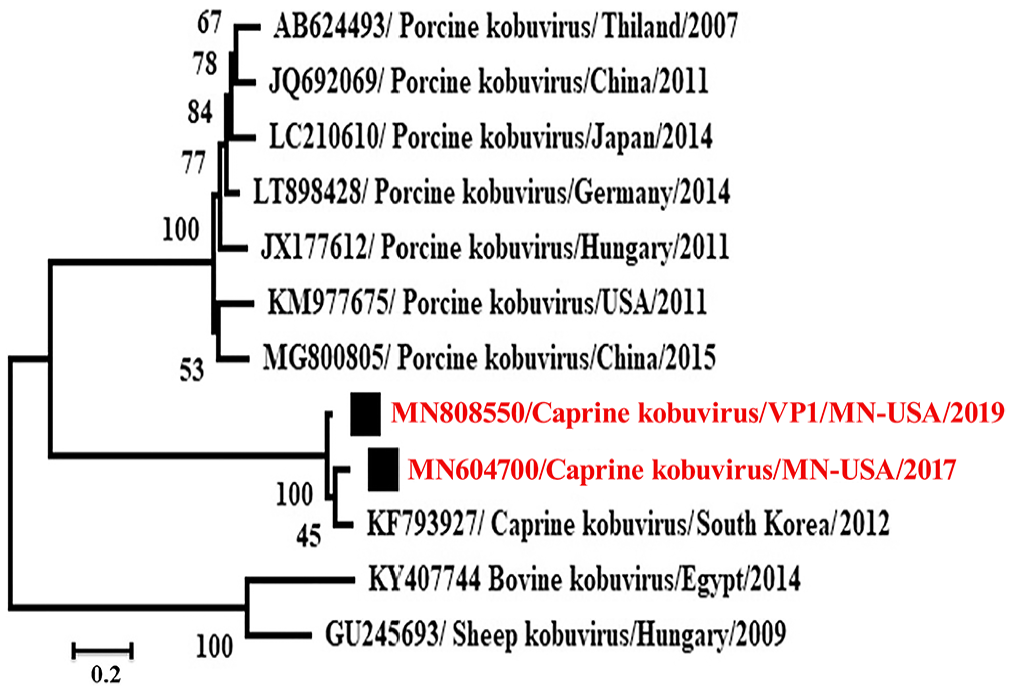
Phylogenetic analysis of the VP1 gene (MN808550) of caprine kobuvirus/MN/2019 compared with MN604700 strain (caprine kobuvirus/MN/2017) and other related viruses through the maximum-likelihood method by using the GTR+G model.
Discussion
We detected the presence of a kobu-like virus by using electron microscopy, which is considered an optimal preliminary method for finding an unknown causative agent because it does not use reagents specific for a particular pathogen. However, it cannot be the only approach for virus detection because it requires a high concentration of virus in the sample for optimal sensitivity.4,34
Histopathology showed lamina propria infiltration with neutrophils, which would be consistent with intestinal erosion. Intestinal erosions are difficult to verify histologically unless optimally preserved intestinal samples are used. Molecular confirmation by WGS revealed the presence of kobuvirus in the above lesions. Analysis of the viral genome identified it as a caprine kobuvirus, with the same gene arrangement as the previously identified caprine kobuviruses.13,21 Incompatible with previous studies of VP1 gene sequences of the Aichi viruses, the VP1 domain had the most variation among the viral capsid genes.22,24 VP1 is the most variable structural protein among kobuviruses, and it is the most exposed and immunodominant part of the capsid protein. 25
It is believed that each species’ kobuvirus is different from the others, and that there is a low possibility of cross-species transmission. However, historical transmission between rabbits and bats and between rodents and bats has been reported, which does raise the possibility of cross-species transmission in the future.2,15 Phylogenetic analysis shows that the clade of sheep kobuviruses is adjacent to that of bovine kobuviruses (Fig. 3), although there is no clear evidence of cross-transmission. The recombination rate in the virus is high; a previous study implied that a new porcine kobuvirus arose as a result of recombination between previously identified porcine and bovine kobuviruses. 10 Similarly, 3 strains of sheep kobuviruses in Brazil were near the bovine kobuvirus lineage on the phylogenetic tree. 5 Caprine kobuvirus was also identified in apparently healthy roe deer with 84.2–87.6% nt identity with caprine kobuvirus strain 12Q108. 7 The common finding of kobuvirus among a large class of wild and domestic animals all over the world indicates the widespread nature of these viruses and their potential to cause enteric disease.
Sequences of the 3D gene have a special importance for virus classification within species or genera and for comparing those viruses. 8 Significance of one aa insertion in the 3D gene, plus synonymous and non-synonymous substitutions along the genome of MN604700/caprine kobuvirus/MN/2017 strain, is still unclear (Table 1). The relation of these mutations with virus virulence and pathogenesis needs more investigation. In the absence of virus isolation, electron microscopy and molecular investigation gave clear indication of the presence of kobuvirus in young kids. The close relationship to porcine kobuvirus, which causes diarrhea in young piglets, indicates the potential virulence of caprine kobuvirus-USA strain.
Identification of goat kobuvirus subsequently in 2019 indicates that the virus has been circulating in Minnesota for up to 2 y, sharing in the causation of goat diarrhea. The low identity of 3D and VP1 genes in samples from 2017 and 2019 indicates the possibility of circulation of different caprine kobuviruses in the country. In our study, we have provided the whole-genome sequence of a caprine kobuvirus. Future viral cultivation and experimental infection could be supportive for the presumptive or potential role of caprine kobuvirus in diarrhea incidence. Screening for such viruses over a large geographic area and in different species can help in understanding the temporal and spatial patterns of the virus and its evolutionary connections.
Footnotes
Acknowledgements
We thank Wendy Wiese and Lotus Smasal of the Virology Section of the Minnesota Veterinary Diagnostic Laboratory.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.