
Abstract
Emerging evidence suggests CAR-NK cell therapy shows great promise in cancer treatment. ROBO1 is highly expressed in various cancer types, including glioblastoma, hepatocellular carcinoma, lung cancer, breast cancer, and uterine cancer. Our and other laboratories’ studies have shown that ROBO1 CAR-NK cells exhibit promising tumor therapeutic effects. However, the results still have some limitations. Cbl-b, an E3 ubiquitin ligase, has been reported to negatively regulate NK cell activation, homeostasis, and antitumor immunity. 1 Therefore, we attempted to further enhance the antitumor activity of ROBO1 CAR-NK92 cells by knocking out Cbl-b using CRISPR/Cas9 gene-editing technology. In this study, we conjugated Cbl-b sgRNA with Cas9 protein to form ribonucleoprotein complexes, which were then delivered into ROBO1 CAR-NK92 and NK-92 cells (control cells) via electroporation. Through fluorescence-activated cell sorting, limiting dilution, and sequencing, we obtained monoclonal Cbl-b-knock-out (KO) cell lines. Both in vitro cytotoxicity assays and in vivo tumor xenograft experiments were conducted to examine whether Cbl-b knockout enhances the target cell killing and tumor suppression capacities of ROBO1 CAR-NK92 cells. In this study, monoclonal cell lines of ROBO1 CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO were successfully established. In vitro, at an effector-to-target (E:T) ratio of 0.1:1, ROBO1 CAR-NK92-Cbl-b-KO (50.55%) cells exhibited significantly higher cytolytic activity against ROBO1-positive T47D target cells after 3 h of coculture than ROBO1 CAR-NK92 (34.10%), NK92-Cbl-b-KO (22.22%), and parental NK-92 cells (3.28%). In vivo, tumor volume and weight measurements demonstrated that mice treated with ROBO1 CAR-NK92-Cbl-b-KO cells developed significantly smaller tumors than all control groups, achieving a tumor growth inhibition (TGI) rate of 32.45%, indicating enhanced antitumor efficacy conferred by Cbl-b knockout. In vitro and in vivo data confirmed that Cbl-b knockout potentiates the antitumor efficacy of ROBO1 CAR-NK92 cells. The overall cytotoxic capability ranked as follows: ROBO1 CAR-NK92-Cbl-b-KO > ROBO1 CAR-NK92 > NK92-Cbl-b-KO > NK-92.
INTRODUCTION
Cancer is a leading cause of death worldwide, and its incidence is projected to rise with population growth and aging. 2 Concurrently, adoptive cell therapy utilizing NK cells and chimeric antigen receptor (CAR)-engineered lymphocytes has achieved clinical success. 3 Compared with CAR-T cells, CAR-NK cells offer distinct advantages, including a superior safety profile—characterized by reduced incidences of cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome, and the feasibility of allogeneic infusion without graft-versus-host disease.4,5 Moreover, CAR-NK cells exhibit inherent potential for universal donor applications and diverse tumor-recognition mechanisms. 5 Continued advances in NK cell engineering are expected to further potentiate their therapeutic efficacy.
ROBO1, also known as DUTT1, FLJ21882, and SAX3, is the receptor for SLIT-family proteins, which function in axon guidance and neuronal precursor cell migration. In addition, the abnormal expression of ROBO1 in various tumors suggests that it may be involved in tumorigenesis and progression. Based on prior analyses of ROBO1 expression across different tissues, 6 ROBO1 is proposed to serve as a potential target for tumor therapy. Recent studies have validated ROBO1 as a therapeutically actionable target, with ROBO1-directed CAR-T cells demonstrating potent antitumor activity in preclinical models of glioblastoma and brain metastases. 7 In the present study, we found that, compared with NK-92 cells, ROBO1 CAR-NK92 demonstrated significantly higher cytotoxicity, eliminating over 50% of ROBO1-positive target cells. A recent study has also reported that “ROBO1 BiCAR-NK combined with 125I seed implantation” represents a promising therapeutic strategy for pancreatic cancer. 8
Cbl-b is an E3 ubiquitin ligase composed of three key domains: a conserved N-terminal tyrosine kinase-binding domain, a short linker region, and a RING finger domain. 9 It has been reported that, upon phosphorylation by Gas6/TAM signaling, Cbl-b mediates the ubiquitination and degradation of LAT1, which is required for signal transduction downstream of NK cell activating receptors (e.g., NK1.1 and NKG2D), thus inhibiting NK cell activation. Additionally, Cbl-b knockdown in primary human NK cells enhances cytotoxicity and IFN-γ production, particularly under low (near-physiological) IL-15 conditions. 1 Cbl-b knockout or targeted inactivation of its E3 ligase activity licenses NK cells to spontaneously reject metastatic tumors. 10 All of these studies suggest that Cbl-b can serve as a potential target for NK cell engineering to enhance antitumor capabilities. While ROBO1 CAR engineering and Cbl-b deletion have been investigated independently as strategies to enhance NK cell function, their combination represents a novel approach that integrates CAR-mediated tumor targeting with immune checkpoint disruption to maximize antitumor potency.
In this study, we established ROBO1 CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO monoclonal cell lines. Real-time cell analysis (RTCA) assays for in vitro cytolytic activity demonstrated the following efficacy hierarchy: ROBO1 CAR-NK92-Cbl-b-KO > ROBO1 CAR-NK92 > NK92-Cbl-b-KO > NK-92. In vivo animal studies further revealed that ROBO1 CAR-NK92-Cbl-b-KO cells exhibited superior tumor suppression compared with control groups. Collectively, our findings provide novel insights for developing CAR-NK therapeutics and advancing chimeric antigen receptor-based immunotherapies.
MATERIALS AND METHODS
Reagents
Antibodies for flow cytometry, including CD56-APC (318310) and CD107a-FITC (328606), were obtained from BioLegend (USA). Antibodies used for Western blotting were as follows: β-actin (T0022, Affinity Biosciences) and Cbl-b (D3C12, Cell Signaling Technology). Enzyme-Linked Immunosorbent Assay (ELISA) kits for IFN-γ (DIF50C) and TNF-α (DY210) were acquired from Bio-Techne (USA). The LEGENDplex™ Human CD8/NK Panel (13-plex) (741187) was sourced from BioLegend.
Cell culture and cell lines
The NK-92, T47D, T47D-ROBO1-KO, and H1299 cell lines were maintained in our laboratory. The tumor cell lines T47D and T47D-ROBO1-KO were cultured in complete RPMI1640 medium (BasalMedia, China), while H1299 cells were grown in complete Dulbecco's Modified Eagle Medium (DMEM, BasalMedia). ROBO1 expression in T47D and H1299 cells was confirmed via the Cancer Cell Line Encyclopedia (https://sites.broadinstitute.org/ccle/) database. T47D-ROBO1-KO cells were subsequently generated by CRISPR/Cas9-mediated ROBO1 knockout in T47D cells. NK-92 cells and their derivative lines were maintained in Alpha-MEM (BasalMedia) supplemented with 12.5% horse serum (SF301.02, Lanzhou Minhai Bioengineering Co., Ltd., China), 12.5% fetal bovine serum (FBS, BC-SE-FBS01, Biochannel, China), 0.1 mM β-mercaptoethanol (M8210, Solarbio, China), 0.02 mM folic acid (F7876, Sigma-Aldrich, USA), 0.2 mM myo-inositol (I7508, Sigma-Aldrich), and 200 IU/mL recombinant human IL-2 (Jiangsu Jinsili Pharmaceutical Co., Ltd., China). ROBO1-CAR-NK92 cells were generated from NK-92 cells via infection with a four-plasmid lentiviral system comprising pSF-ROBO1-CAR, pSF-VSV-G, pSF-GagPol, and pSF-Rev. All cell lines were incubated at 37°C under 5% CO2.
Cbl-b gene modification of NK-92 cells
The sgRNA sequences for Cbl-b knockout used were as follows: sgRNA1: TAATCTGGTGGACCTCATGA; sgRNA2: ATTTTAGAGCCATTGCTTCC. The sgRNAs and Cas9-GFP protein (Z03467) were obtained from GenScript Biotech Co., Ltd. (China), and then they were precomplexed at a molar ratio of 3:1 to form ribonucleoprotein(RNP) complexes. These RNP complexes were coincubated with premixed buffer A and buffer B (at a ratio of buffer A:buffer B = 1:1) (1201, Celetrix LLC, USA). The mixture was then delivered into NK-92 or ROBO1-CAR-NK92 cells via electroporation using a Celetrix electroporator (11–0103, Celetrix LLC) with parameters of 540 V and 20 ms. Single-cell clones were isolated sequentially by flow cytometry sorting for GFP+ cells, followed by limiting dilution cloning. The NK92-Cbl-b-KO and ROBO1 CAR-NK92-Cbl-b-KO cells were further amplified and validated through sequencing and Western blot analysis. The sequences of the primers used for PCR validation are as follows: F: 5′-TGATAGCCTAGGACTGTTTGAGAGAA-3′; R: 5′-CCAGGCTCACAGCATCTGATAAC-3′. In brief, total DNA was isolated using a DNA extraction kit (Vazyme Biotech Co., Ltd., China). PCR amplification was performed with 2× TransStart® GoldPfu PCR SuperMix (-dye) (TransGen Biotech, China). The PCR products were purified using the FastPure Gel DNA Extraction Mini Kit (Vazyme Biotech Co., Ltd.). Subsequently, the purified PCR products were transformed with the pEASY®-Blunt Cloning Kit (TransGen Biotech) and submitted to sequence analysis conducted by GENEWIZ (China).
Western blot
Cells were lysed using Radioimmunoprecipitation Assay (RIPA) buffer (Beyotime Biotechnology, China) supplemented with 1% PMSF. The lysates were centrifuged at 13,800 g for 20 min at 4°C, and the supernatant was collected. Total proteins were separated by SDS-PAGE and transferred onto Polyvinylidene Fluoride (PVDF) membranes. The membranes were incubated overnight at 4°C with primary antibodies against β-actin and Cbl-b (Cell Signaling Technology, USA). Horseradish peroxidase (HRP)-conjugated AffiniPure goat anti-rabbit IgG (Proteintech, China) was used as the secondary antibody. After incubation with enhanced chemiluminescence (ECL) substrate, protein signals were detected using a chemiluminescence imaging system.
RTCA
T47D, T47D-ROBO1-KO, and H1299 cells were used as target cells (T). Effector cells (E) included NK-92, NK92-Cbl-b-KO, ROBO1 CAR-NK92, and ROBO1 CAR-NK92-Cbl-b-KO cells. Background impedance of the E-Plate 16 (Agilent Technologies, USA) was measured with 50 μL medium per well. Target cells were added in 100 μL medium to achieve final densities of 4 × 105 or 3 × 105 cells/mL. After 24 h incubation in the xCELLigence RTCA system (Agilent Technologies) at 37°C, 5% CO2, effector cells (80 μL) were added at E:T ratios of 1:1, 0.5:1, 0.25:1, 0.1:1, and 0.05:1. Target cell-only and effector cell-only controls were included. Impedance was monitored for an additional 4 or 16 h. Cytotoxicity was calculated as the percentage of target cell lysis relative to control wells using RTCA Software Pro (Agilent Technologies).
Effector function analysis
Effector and H1299 target cells were cocultured in 24-well plates for 6 h. Following coculture, the levels of TNF-α and IFN-γ in the cell culture supernatants were assessed using an ELISA kit (Bio-Techne, USA) according to the manufacturer’s instructions.
Effector and H1299 target cells were cocultured in 24-well plates at a 1:1 E:T ratio. CD107a-FITC antibody was added to the corresponding wells at the start of coculture. After 1 hour of incubation, brefeldin A (BFA, BioLegend, USA) was introduced to inhibit protein transport, and samples were collected 3 h later. Cells were stained with an anti-CD56-APC antibody and analyzed by flow cytometry.
Establishment of a subcutaneous tumor model and in vivo antitumor efficacy evaluation of engineered NK cells in NCG mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of Soochow University and conducted in accordance with institutional guidelines. Female NCG (NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt) mice (6–8 weeks old, 18–20 g body weight) under specific pathogen-free conditions were purchased from GemPharmatech Co., Ltd. (China).
Four days prior to treatment initiation, H1299-Luc cells (3 × 106) were inoculated subcutaneously into the right axilla of NCG mice to establish tumor xenografts. When tumor volumes reached ∼60 mm³, mice were randomized into five groups (n = 3/group): a. Phosphate-Buffered Saline (PBS) (200 μL/mouse); b. NK-92 (5 × 106 cells in 200 μL/mouse); c. NK92-Cbl-b-KO (5 × 106 cells); d. ROBO1 CAR-NK92 (5 × 106 cells); e. ROBO1 CAR-NK92-Cbl-b-KO (5 × 106 cells). On Day 0, the first tail vein injection of cells or PBS was administered, followed by twice-weekly dosing for 4 weeks. NK-92 and their derivative cells were not subjected to irradiation prior to in vivo experiments, and no exogenous IL-2 was administered to the mice. Tumor volume (calculated as a×b2/2, where a = major axis, b = minor axis) and body weight were measured every 5 days. At designated endpoints, mice were humanely euthanized for analysis. Carcasses were disposed of according to institutional biosafety protocols.
Serum effector molecules (IFN-γ, perforin, granulysin, and granzyme A) were quantified using the LEGENDplex™ Human CD8/NK Panel (13-plex, BioLegend) per manufacturer’s instructions. Tumor growth inhibition (TGI) was calculated as
Statistical analysis
Statistical analyses were performed using Prism 8 (GraphPad Software, USA). Data were presented as mean ± SD. For RTCA, ELISA, and in vivo cytokine assays, comparisons among multiple groups were analyzed by one-way Analysis of Variance (ANOVA). CD107a degranulation and in vivo tumor growth data were analyzed by two-way ANOVA, accounting for both group and time as variables. Replicate information is as follows: RTCA (n = 2 biological, 2 technical); ELISA (n = 3 biological, 3 technical); in vivo cytokines (n = 2 experiments, n = 3 mice/group); CD107a (n = 2 biological, 3 technical); tumor growth (n = 2 experiments, n = 3 mice/group). Unpaired two-tailed Student’s t-tests were used for two-group comparisons. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. A p value < 0.05 was considered statistically significant.
RESULTS
Design and construction of Cbl-b KO ROBO1 CAR-NK92 cell line
The genomic sequence of human Cbl-b (Gene ID: 868) was retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/). The knockout target site was designed within the fifth exon of the Cbl-b gene. Potential sgRNA candidates were predicted using two specialized online tools: CCTop—CRISPR/Cas9 Target Online Predictor (https://cctop.cos.uni-heidelberg.de:8043/) and CRISPOR (http://crispor.tefor.net/). These platforms enabled comprehensive analysis of guide RNA efficiency and specificity. Optimal sgRNAs were selected through systematic evaluation of predicted off-target effects and strategic positioning within the exon to maximize knockout efficacy. Final candidate sequences were prioritized based on their calculated off-target probability scores and alignment with optimal CRISPR/Cas9 targeting requirements (Fig. 1A).

Generation and validation of Cbl-b knockout in NK-92 and ROBO1 CAR-NK92 cell lines.
Cbl-b knockout polyclonal cells were obtained by flow cytometry sorting for GFP+ cells and verified by sequencing. These polyclonal cells were then subjected to limiting dilution for monoclonal expansion. Through sequencing validation, this process yielded six potential Cbl-b-KO clones of ROBO1 CAR-NK92 cells and ten potential Cbl-b-KO clones of NK-92 cells. Subsequent Western blot analysis demonstrated that clones 2F6, 5D11, and 5G6 were true Cbl-b-KO ROBO1 CAR-NK92 cells, whereas clones 1E8, 6E10, 8E3, and 9E5 being true Cbl-b-KO NK92 cells (Fig. 1B, C). Finally, gene editing efficiency was assessed by sequencing purified PCR products of these monoclonal clones. Karyotype analysis confirmed NK-92 cells as triploid, and only the clones 5D11, 6E10, and 8E3 were identified to exhibit triple allelic mutations (Fig. 1D–1F). Therefore, these three clones were selected for further studies.
In vitro function evaluation of ROBO1 CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO cells
To first confirm whether the cytotoxic activity of ROBO1 CAR-NK92 cells is specifically directed against ROBO1-expressing cells, T47D—a human breast cancer cell line highly expressing ROBO1—and its isogenic ROBO1 knockout counterpart (T47D-ROBO1-KO) were used for target cells. Given the inherently strong cytotoxic activity of ROBO1 CAR-NK92 cells against ROBO1-expressing targets, mediated by the CAR structure, we used a relatively low effector-to-target (E:T) ratio of 1:1 to evaluate their cytotoxic activity. RTCA revealed that the killing efficiency of ROBO1 CAR-NK92 cells against T47D cells was significantly higher than that against T47D-ROBO1-KO cells, demonstrating target specificity (Fig. 2A).
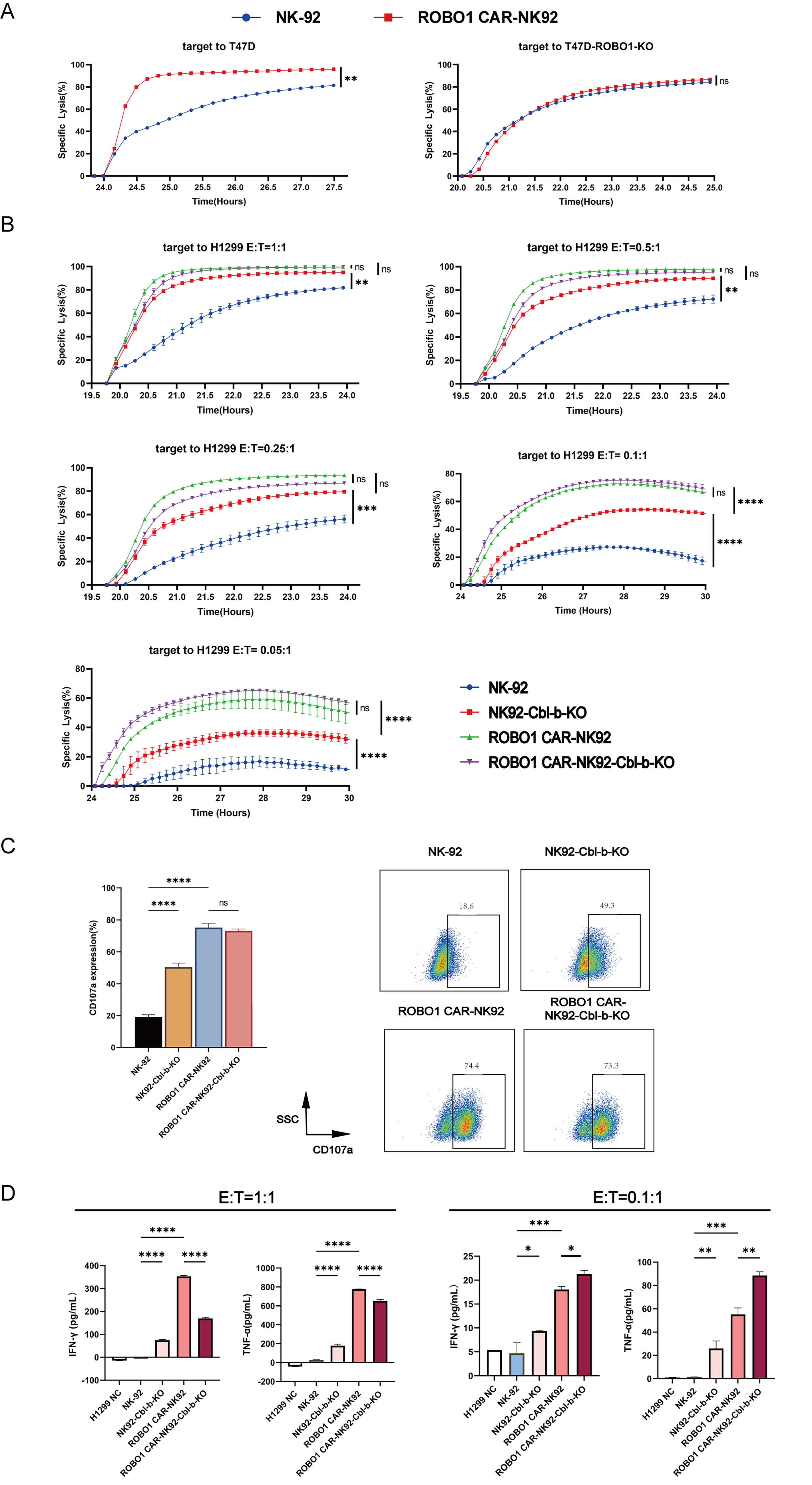
Cytotoxic activity and cytokine secretion profiles of ROBO1 CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO cells across effector-to-target ratios.
Then, the tri-allelically mutated monoclonal clones—ROBO1 CAR-NK92-Cbl-b-KO clone 5D11, as well as NK92-Cbl-b-KO clones 8E3 and 6E10—were evaluated for cytotoxic activity against another ROBO1-expressing cell, H1299. As previously mentioned, Cbl-b deficiency or inactivation enhances the antitumor activity of NK cells; therefore, we used lower E:T ratios (1:1, 0.5:1, 0.25:1) to evaluate cytotoxicity. NK92-Cbl-b-KO clones 8E3 and 6E10 exhibited higher cytotoxicity than parental NK-92 cells (Fig. 2B), which confirmed that Cbl-b knockout enhances NK-92-mediated cytotoxicity in a dose-dependent manner. By contrast, ROBO1 CAR-NK92-Cbl-b-KO clone 5D11 did not exhibit significantly enhanced cytotoxicity compared with parental ROBO1 CAR-NK92 cells, suggesting that the cytotoxic activity conferred by the CAR structure is so strong that it masks the functional differences resulting from Cbl-b knockout. Thus, we assessed cytotoxicity at even lower E:T ratios. As expected, the cytotoxic activity of ROBO1 CAR-NK92-Cbl-b-KO cells gradually surpassed that of parental ROBO1 CAR-NK92 cells as the E:T ratio decreased, establishing a cytotoxic hierarchy of ROBO1 CAR-NK92-Cbl-b-KO > ROBO1 CAR-NK92 > NK92-Cbl-b-KO > NK-92(Fig. 2B).
The upregulation of CD107a surface expression, a marker of NK cell degranulation and perforin secretion, correlates significantly with enhanced cytotoxic activity. Thus, CD107a-positive NK cells are considered as functionally active cytotoxic ones. Flow cytometric analysis of CD107a expression on NK cells after 2-h coculture at a 1:1 E:T ratio revealed elevated CD107a levels on NK92-Cbl-b-KO cells compared with parental NK-92 cells, consistent with the enhanced cytotoxicity of the former (Fig. 2C). By contrast, ROBO1 CAR-NK92-Cbl-b-KO cells exhibited CD107a expression comparable to parental ROBO1 CAR-NK92 cells, likely due to the inherently high baseline cytotoxic activity of CAR-engineered NK cells.
One of the killing mechanisms of NK effector cells involves the secretion of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α). ELISA quantification of these cytokines in supernatants from NK cells cocultured with H1299 cells (E:T ratio of 1:1) revealed no significant difference in IFN-γ or TNF-α secretion between ROBO1 CAR-NK92-Cbl-b-KO and parental ROBO1 CAR-NK92 cells (Fig. 2D). Notably, at a limiting E:T ratio (0.1:1), ROBO1 CAR-NK92-Cbl-b-KO exhibited markedly increased cytokine production compared with its parental counterpart (Fig. 2D), indicating that Cbl-b ablation specifically enhances cytokine release when effector cell availability is suboptimal.
In vivo antitumor efficacy evaluation of ROBO1 CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO cells
To investigate the antitumor efficacy of ROBO1-CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO cells in vivo, an H1299 xenograft model was established in NCG immunodeficient mice, with a schematic diagram of the experimental workflow provided in Figure 3A. Throughout the treatment period, no significant alterations in body weight were observed across all groups (Fig. 3B), indicating minimal systemic toxicity associated with cell administration. Quantitative analysis of tumor growth dynamics revealed that ROBO1 CAR-NK92-Cbl-b-KO cells exhibited superior therapeutic efficacy compared with both ROBO1 CAR-NK92 and NK92-Cbl-b-KO cells, as demonstrated by significantly reduced tumor volume trajectories over time (Fig. 3C). Ex vivo tumor weight measurements following euthanasia (Fig. 3D) corroborated these findings, establishing a consistent therapeutic hierarchy: ROBO1 CAR-NK92-Cbl-b-KO > ROBO1 CAR-NK92 > NK92-Cbl-b-KO > PBS control.
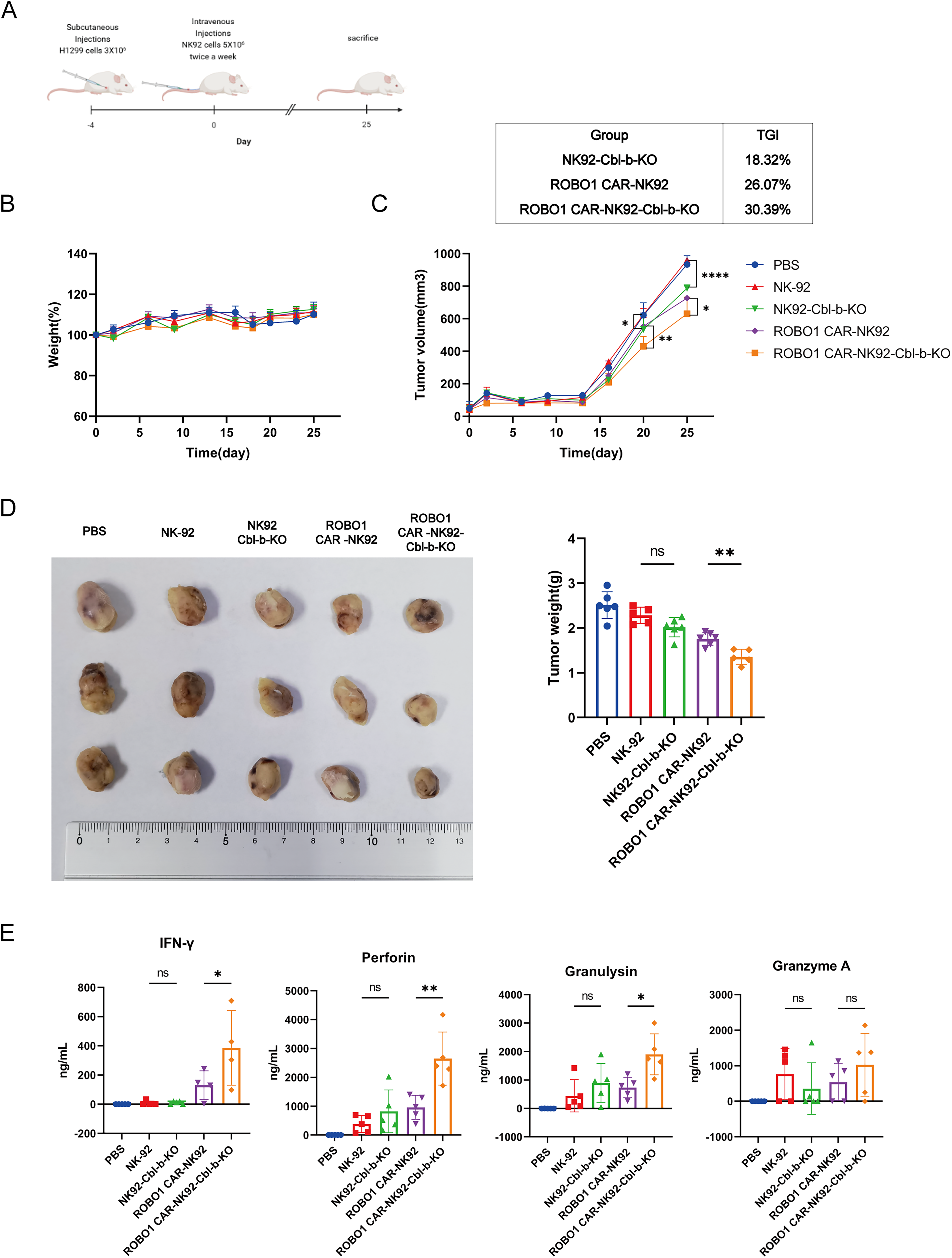
Therapeutic efficacy evaluation of H1299 subcutaneous tumor models in NCG mice.
Serum effector molecule levels across all groups were quantified using magnetic bead-based assays (Fig. 3E). Quantification revealed significantly higher concentrations of IFN-γ, perforin, and granulysin in the ROBO1 CAR-NK92-Cbl-b-KO group compared with PBS controls, parental NK-92, NK92-Cbl-b-KO, and ROBO1 CAR-NK92 groups. Granzyme A levels in the ROBO1 CAR-NK92-Cbl-b-KO group trended higher than those in the ROBO1 CAR-NK92 group, though this difference did not reach statistical significance.
Collectively, these results validate that Cbl-b knockout synergizes with ROBO1 CAR engineering to augment antitumor efficacy in vivo, primarily by enhancing effector molecule secretion and maintaining cytotoxic superiority under physiological effector cell constraints.
DISCUSSION
In this study, we successfully generated ROBO1 CAR-NK92-Cbl-b-KO and NK92-Cbl-b-KO polyclonal populations via CRISPR/Cas9-mediated gene editing, followed by isolation of fully edited monoclonal cell lines through limiting dilution and flow cytometry sorting. RTCA revealed enhanced cytotoxicity of NK92-Cbl-b-KO cells compared with parental NK-92 cells, directly demonstrating that Cbl-b ablation potentiates NK cell killing capacity. Under physiologically relevant low effector-to-target (E:T) ratios (≤0.25:1), a hierarchical cytotoxic potency was established: ROBO1 CAR-NK92-Cbl-b-KO > ROBO1 CAR-NK92 > NK92-Cbl-b-KO > NK-92, highlighting the context-dependent additional benefit of Cbl-b knockout in ROBO1 CAR-engineered NK92 cells. In vivo, ROBO1 CAR-NK92-Cbl-b-KO cells exhibited superior tumor suppression and induced higher secretion of effector molecules (e.g., IFN-γ, Perforin) compared with ROBO1 CAR-NK92, supporting their enhanced therapeutic efficacy. Collectively, these findings provide a foundation for further optimization of CAR-NK cell engineering strategies and suggest that dual-targeting approaches (CAR + Cbl-b knockout) may offer a promising avenue for improving adoptive immunotherapy against solid tumors.
However, several critical observations emerged from this work that warrant further investigation.
Context-dependent efficacy of Cbl-b knockout
At a 1:1 E:T ratio, ROBO1 CAR-NK92-Cbl-b-KO and ROBO1 CAR-NK92 achieved comparable cytotoxicity (>90% target cell lysis within 2 h) against H1299 cells, indicating that saturating CAR-mediated activation is sufficient to drive maximal target cell killing irrespective of Cbl-b expression status. Notably, under these saturating conditions, ROBO1 CAR-NK92-Cbl-b-KO cells secreted significantly less TNF-α and IFN-γ than ROBO1 CAR-NK92 cells. This paradoxical reduction may reflect the engagement of Cbl-b-independent negative feedback mechanisms in Cbl-b-deficient NK cells—including upregulation of inhibitory receptors (e.g., PD-1) 11 and induction of activation-induced cell death (AICD) 12 —which serve to constrain excessive inflammatory responses and mitigate NK cell exhaustion following hyperactivation. This interpretation is consistent with the established role of Cbl-b in setting signaling thresholds; 1 in its absence, the cellular machinery may trigger compensatory regulatory circuits to maintain homeostasis. In contrast, under limiting E:T conditions (0.1:1 and 0.05:1), ROBO1 CAR-NK92-Cbl-b-KO exhibited significantly enhanced cytotoxicity and cytokine production relative to ROBO1 CAR-NK92 control, demonstrating its functional superiority in resource-constrained microenvironments. This context-dependent efficacy highlights the physiological relevance of Cbl-b deletion: by lowering the activation threshold for NK cells, Cbl-b ablation enables robust effector responses even when CAR engagement or co-stimulatory signaling is limiting. Therefore, the observed benefit at low E:T ratios is not merely a quantitative difference but a fundamental rewiring of NK cell sensitivity that is directly relevant to the therapeutic challenge of eradicating solid tumors, where effector cell infiltration is often sparse and target antigen density may be heterogeneous.
Model optimization for translational relevance
While H1299 subcutaneous xenografts (cell line-derived models, CDX) enable rapid tumorigenesis, 13 patient-derived xenograft (PDX) models better recapitulate human tumor biology and drug responses. 14 Future investigations may focus on: (1) evaluating ROBO1 CAR-NK92-Cbl-b-KO efficacy in PDX models of colorectal carcinoma, hepatocellular carcinoma, and glioblastoma; (2) establishing humanized immune-reconstituted PDX platforms to evaluate tumor-immune interactions; and (3) leveraging tumor organoids for preclinical assessment of toxicity and efficacy to further define clinical translatability.
Addressing modest in vivo efficacy and future multiplexed editing
While the ROBO1 CAR-NK92-Cbl-b-KO cells demonstrated statistically significant reductions in tumor burden and elevations in systemic pro-cytolytic molecule levels in vivo, the absolute magnitude of these effects remains modest. This limited therapeutic impact in current murine models likely reflects suboptimal experimental conditions, such as excessive tumor burden or insufficient effector cell numbers; therefore, protocol refinement—reducing tumor inoculum size and escalating therapeutic doses—may enhance treatment efficacy. More importantly, however, this modest efficacy suggests that single-gene ablation is insufficient to fully overcome the immunosuppressive tumor microenvironment. Consequently, future studies should employ multiplexed knockout of additional immune modulatory signal transduction molecules. Multiplexing Cbl-b ablation with disruption of complementary inhibitory pathways may be required to unleash the full, durable cytolytic capacity of CAR-NK therapies in vivo.
AUTHORS’ CONTRIBUTIONS
J.J.H., J.Z., H.S.L., and J.P.Z. conceptualized and designed this study. J.J.H. performed most of the experiments, and J.Z. performed partial experiments. Y.S. and Y.Y. executed the acquisition and analysis of data. J.J.H. and J.Z. prepared figures, performed statistical analysis, and wrote the article. J.J.H., J.Z., H.S.L., and J.P.Z. provided administrative, technical, or material support. All authors read and approved the final article.
Footnotes
ACKNOWLEDGMENTS
The authors thank the Institutes of Biology and Medical Sciences, Soochow University, for providing the experimental mice and related animal facility support. They are grateful to Ascle Therapeutics for their expertise and technical assistance in gene editing and cell cytotoxicity assays.
AUTHOR DISCLOSURE
The authors declare no competing financial interests.
FUNDING INFORMATION
This work received funding from