
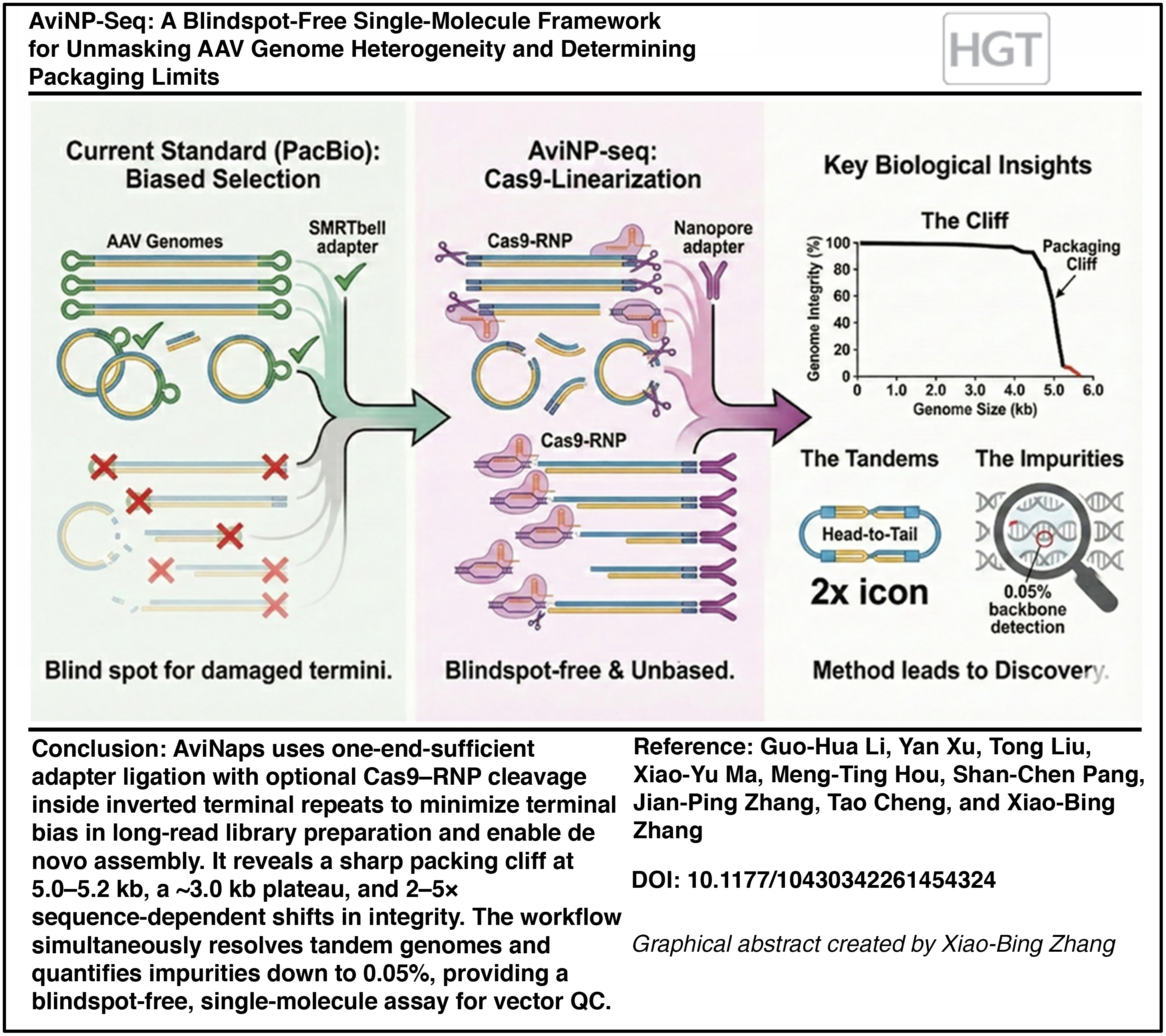
Abstract
Comprehensive recombinant adeno-associated virus characterization is essential for establishing the knowledge base required to ensure clinical safety and efficacy, yet current long-read methods suffer from library preparation biases that obscure genome integrity. We present AviNP-seq, a blindspot-free nanopore sequencing framework utilizing one-end-sufficient ligation and Cas9–ribonucleoprotein (RNP) linearization to minimize terminal selection. Applied to a 1.5–6.5 kb panel, AviNP-seq delineates a sharp packaging cliff at 5.0–5.2 kb and reveals that sequence structure modulates integrity by 2–5× at fixed lengths. It unmasks covalent head-to-tail tandems in sub-3 kb vectors, detecting them with significantly higher sensitivity than PacBio HiFi. The Cas9–RNP step boosts ligation yield ∼7-fold, providing an unbiased assessment of genome integrity (≥95% inverted terminal repeat [ITR]-to-ITR). In addition, the assay quantifies plasmid impurities down to 0.05% with linear response. By integrating integrity mapping, tandem detection, and impurity profiling into a rapid (<36 h), low-input workflow, AviNP-seq provides a robust analytical tool to guide vector design and de-risk early-stage process development.
Keywords
INTRODUCTION
Recombinant adeno-associated virus (rAAV) vectors have become the leading in vivo gene delivery platform, with multiple licensed therapies and a rapidly expanding clinical pipeline.1–4 However, the complex secondary structures of viral genomes pose persistent analytical challenges, complicating library construction and obscuring particle-level heterogeneity.5,6 While regulatory guidance increasingly requires robust comprehensive characterization for genome integrity and residual impurities, existing assays struggle to provide direct, molecule-level resolution. Genomes approaching the ∼5.0–5.2 kb packaging ceiling are prone to premature truncation,7,8 while subcapacity genomes often form heterogeneous or tandemized species.9,10 Conventional methods such as quantitative polymerase chain reaction (qPCR) and droplet digital polymerase chain reaction (ddPCR) are widely used for identity and titer but interrogate only short amplicons and cannot report on full-length structure.11–14 Similarly, short-read sequencing provides base-level accuracy but cannot span inverted terminal repeats (ITRs) or resolve concatemers, leaving standard analytical pipelines blind to structural diversity and packaging heterogeneity.15–19
Incomplete genomes or undetected concatemers can lead to inaccurate viral titer determination and potentially unpredictable pharmacokinetic profiles. Consequently, even emerging long-read methods remain constrained by library designs that impose terminal selection, which can under- or over-represent specific genome states.20,21 Thus, a broadly applicable strategy that minimizes terminal bias and directly profiles genome integrity, tandem packaging, and encapsidated impurities at single-molecule resolution is still lacking.
Several long-read approaches have sought to overcome these limitations. Cas9-guided adapter ligation (nCATS) enables targeted nanopore enrichment, 22 while direct ITR-to-ITR nanopore sequencing profiles capsid-protected genomes end-to-end. 23 PacBio-based thorough molecular configuration analysis-AAV-seq preserves noncanonical AAV configurations, 24 and AAVGP-seq has been applied to vectors packaging clustered regularly interspaced short palindromic repeats (CRISPR) components. 25 These methods have advanced the field by visualizing heterogeneous termini, atypical structures, and complex populations that were previously inaccessible. However, each retains inherent constraints from its library design. SMRTbell circularization requires ligation at both termini and can underrepresent molecules with damaged ends or concatemers.26,27 Furthermore, the requirement for dual-end ligation in circular consensus sequencing (CCS) inherently filters out molecules with noncanonical termini, potentially creating a “survivorship bias” that overestimates vector quality. Conversely, nanopore-based approaches that rely on annealing can inflate apparent integrity by favoring intact molecules. 28 Importantly, no single workflow simultaneously unifies completeness assessment, tandem detection, and impurity quantification nor provides turnaround compatible with rapid process development. The result is a fragmented analytical landscape where complementary assays are needed to piece together genome composition, slowing development and complicating regulatory compliance.
Here, we present AviNP-seq, a one-end-sufficient (motor-anchored) ligation workflow in which ligation-competent ends are generated either by controlled strand annealing or by Cas9–ribonucleoprotein (RNP) cleavage within both ITRs. This design minimizes terminal bias while maintaining throughput and turnaround suitable for iterative process optimization. Applied to a 1.5–6.5 kb AAV panel, AviNP-seq reveals a sharp packaging cliff at 5.0–5.2 kb, a ∼3.0 kb plateau beyond capacity, and strong sequence-encoded effects on integrity at fixed size. At the short end, it resolves covalent tandem genomes, while RNP cleavage boosts usable reads by 7–8× and provides a more unbiased assessment for completeness. In parallel, masked-reference analysis quantifies plasmid and host–cell impurities down to 0.05%. Together, these results establish a blindspot-free, single-molecule framework that generalizes to structured DNA viruses and supports decision-grade analytics for vector engineering and early-stage process characterization.
MATERIALS AND METHODS
Study design and reporting
The objectives of this study were to: (i) map genome integrity across a size spectrum of 1.5–6.5 kb to definitively characterize the 5.0–5.2 kb packaging cliff; (ii) interrogate sequence-intrinsic constraints by comparing matched-length constructs that alter packaging completeness by 2- to 5-fold; (iii) resolve and quantify covalent multicopy species in sub-3 kb vectors; and (iv) establish limits of detection (LODs) for encapsidated impurities, including plasmid backbone, helper sequences, and host–cell DNA (HCD). For each vector lot, sample processing was bifurcated into two parallel library workflows: an annealing + NP protocol optimized for the retention of covalent tandems and a Cas9–RNP + NP protocol designed to provide an unbiased assessment of genome integrity calculations. Biological replicates were defined as independent vector production lots, while technical replicates represented independent library preparations derived from the same DNA eluate. All experiments were conducted in accordance with institutional biosafety level 2 guidelines. No human or animal subjects were utilized in this study.
Plasmids and vector panel
Cis-acting plasmid constructs harboring AAV2 ITRs were engineered with transgene inserts ranging from 1.5 to 6.5 kb. To decouple sequence context from physical length, we generated isogenic pairs or trios of vectors within the critical 5.0–5.2 kb window, varying only in segment order, guanine-cytosine (GC) content, or predicted thermodynamic stability. Additional comparative pairs were designed for the short-genome regime (1.5–2.7 kb). All plasmids were assembled using established molecular cloning techniques. 29
Cell culture, AAV production, and purification
Recombinant AAV vectors were produced via triple-plasmid transient transfection of adherent HEK293T cells. Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C in 5% CO2 and transfected at ∼70–80% confluency using polyethylenimine (PEI) at a DNA:PEI ratio of approximately 1:2 (w/w). The transfection mix contained the AAV2 ITR-flanked vector plasmid, an AAV2 Rep/Cap plasmid (serotype as indicated), and an adenoviral helper plasmid at a 1:1:2 ratio. To minimize nonencapsidated DNA background, cultures were treated with Benzonase (20 U/mL; Santa Cruz Biotechnology) 96 h post-transfection. Vectors were harvested at 120 h, and supernatants were clarified by centrifugation (3,000 g, 15 min). Purification was performed via iodixanol density gradient ultracentrifugation (15/25/40/60% steps) at 350,000 g for ∼2 h. The 40% fraction was extracted and buffer-exchanged into phosphate-buffered saline containing 0.001–0.01% Pluronic F-68 using 100 kDa molecular weight cut-off centrifugal filters. Purified aliquots were stored at −80°C. Detailed production protocols follow previously described methods. 30
Viral DNA extraction and cleanup
To release encapsidated genomes, DNase-treated AAV preparations were incubated with genomic DNA extraction buffer (100 mM NaCl, 10 mM Tris–HCl pH 8.0, 1 mM ethylenediaminetetraacetic acid (EDTA), and 0.5% Tween-20) supplemented with 1% proteinase K (10 mg/mL) at 56°C for 1 h. Lysates were subsequently heat-inactivated at 95°C for 5 min. Viral DNA was purified using 1.0× solid-phase reversible immobilization (SPRI) paramagnetic beads (Zymo Research, D4084) and eluted in elution buffer or 10 mM Tris–HCl (pH 8.5). DNA concentration was quantified via Qubit fluorometry or Nanodrop spectrophotometry. Input material was normalized to 50–100 ng for RNP + NP libraries and 0.5 µg for annealing + NP libraries.
Neutral and alkaline agarose gel electrophoresis
Electrophoretic characterization was performed under both neutral and alkaline conditions. For neutral analysis, samples were combined with loading dye and resolved on 1% agarose gels in Tris-acetate-EDTA buffer (40 mM Tris-acetate, 1 mM EDTA, and pH 8.0), alongside a molecular weight marker (ABM, G248) for size referencing. For alkaline denaturation analysis, 31 samples were denatured and loaded onto 1% agarose gels prepared and electrophoresed in alkaline running buffer (50 mM NaOH and 1 mM EDTA) using a horizontal apparatus pre-equilibrated with the same buffer. Following electrophoresis, gels were neutralized in 0.1 M Tris–HCl (pH 8.5), stained with GelRed (Biotium), and imaged.
Cas9–RNP design, assembly, and ITR cleavage (RNP16/RNP34)
The Cas9–RNP system was deployed in two distinct experimental contexts: (i) the linearization of pAAV plasmids to isolate pure ITR–cassette–ITR reference fragments and (ii) the generation of short, ligation-competent termini within the ITRs for nanopore library construction. RNP complexes were reconstituted under RNase-free conditions by incubating Alt-R SpCas9 with sgRNA at a 1:2 molar ratio for 10 min at room temperature.22,32 A typical 10 µL reaction consisted of 5 µL RNP complex, ∼200 ng DNA substrate, and 1 µL 10× reaction buffer. Digestions were incubated for 2–4 h at 37°C, quenched with Proteinase K (∼1% v/v, 30 min, 37°C), and purified via 1.0× SPRI bead cleanup. In the RNP + NP workflow, cleavage at both ITRs exposes two ligatable duplex termini; sequencing initiates provided at least one terminus successfully ligates to the motor-bearing adapter.
Library preparation for nanopore sequencing
Library construction utilized ONT Ligation Sequencing Kit V14 (SQK-LSK114) employing a “one-end-sufficient” (motor-anchored) ligation strategy, which permits sequencing initiation as long as one terminus acquires a functional motor adapter. The protocol bifurcated based on the method of terminus exposure. In the annealing + NP workflow, 0.5 µg of AAV DNA was thermally denatured at 95°C for 5 min and allowed to gradually cool to room temperature to facilitate strand annealing. These samples subsequently underwent end-repair, dA-tailing, and adapter ligation per manufacturer instructions; this mode is optimized for the detection of covalent tandem species in sub-3 kb vectors. In the RNP + NP workflow, 100 ng of AAV DNA underwent targeted cleavage at both ITRs (using RNP16 or RNP34) to generate ligation-competent duplex ends prior to end-repair and ligation. This mode maximizes usable read counts and establishes an unbiased baseline for genome integrity assessment. For multiplexed runs, Native Barcoding Kit V14 was applied prior to pooling in equimolar ratios.
Nanopore sequencing (devices, chemistry, and run quality control)
Sequencing was conducted over a 24-h period on R10.4.1 flow cells (FLO-MIN114) utilizing either PromethION or MinION Mk1C platforms (Oxford Nanopore Technologies), adhering to manufacturer recommendations and established protocols.33–35 High-throughput PromethION runs were executed at Novogene (Tianjin, China), while MinION Mk1C runs were performed at GenoSTAR. Real-time run quality control (QC) monitored active pore count, total yield, read N50, and mean Q-score, with standardized start-up diagnostics and barcode cross-talk assessments performed for every acquisition.
PacBio HiFi library preparation and sequencing (cross-platform control)
To establish a cross-platform benchmark, SMRTbell libraries were constructed from native AAV DNA using the SMRTbell Prep Kit 3.0, adhering to the manufacturer’s protocol without shearing. Hairpin adapters were ligated to both termini to generate circularized templates, followed by exonuclease treatment to remove linear byproducts. Sequencing was performed on a Revio system (SMRT Cell 25M, 30-h movie) at Novogene (Tianjin, China). High-fidelity CCS reads generated from these runs served as orthogonal controls for validating genome integrity findings.
Validation of sequencing readthrough using linearized plasmid templates
To rigorously test the hypothesis that ITRs inherently impede nanopore readthrough, pAAV plasmids were restriction-digested to excise the bacterial backbone and isolate the full-length ITR–insert–ITR cassette. The resulting fragments (1.5–6.4 kb) were isolated via agarose gel electrophoresis and subsequently recovered using a Universal DNA Purification Kit (Tiangen, Cat. #DP219-03) according to the manufacturer’s instructions. The purified DNA was then used for direct sequencing library preparation. These reference fragments exhibited uniform coverage across the entire insert length, confirming that the terminal drop-offs observed in encapsidated vector genomes arise from biological packaging constraints rather than sequencing artifacts.
Spike-in control design and impurity classification pipeline
To enable absolute quantification and LOD assessment, a 4.75 kb PCR amplicon was spiked into libraries at 0.1% (w/w) to serve as a 100% ligation efficiency standard. For impurity profiling, reads were aligned to a masked composite reference to classify sequences as vector genome, plasmid backbone, adenoviral helper, or HCD. Reads mapping exclusively to masked regions were excluded from impurity calculations to prevent ambiguity. HCD reads aligning to the human reference (GRCh38) were characterized via length histograms and chromosome-normalized density distributions. Vector-assigned reads were excluded from impurity fractions to ensure independence of variables.
Basecalling and demultiplexing
Raw signal data (POD5) were basecalled directly within the MinKNOW software environment (v24.11.8; core v6.2.6, Bream v8.2.5, Script Config v6.2.12) using Dorado v7.6.7 to generate FASTQ.gz output. Processing utilized the Kit 14 chemistry model (SQK-NEB114-96) with super-accuracy (SUP) settings, providing Q10 + simplex accuracy with optional duplex support. Following basecalling, reads were demultiplexed based on the extended barcode set, 36 yielding per-barcode files and an unclassified bin. Adapter and barcode trimming were subsequently performed using Porechop with default parameters to eliminate residual synthetic sequences (e.g., porechop -i input.fastq -o output.fastq). All analyses were strictly standardized to Kit 14 chemistry.
Composite references and masking
For each experimental batch, a composite FASTA reference was assembled containing (i) the specific ITR–cassette–ITR sequence of the AAV vector; (ii) the corresponding plasmid backbone; (iii) the adenoviral helper sequence; and (iv) the human genome (GRCh38) for HCD classification. To minimize ambiguous mapping, shared homologous elements present across multiple plasmids (e.g., antibiotic resistance markers, origins of replication, common promoters) were computationally masked by replacing these regions with Ns. Human reference sequences were derived from NCBI GRCh38.p14 (downloaded March 2024, accession GCA_000001405.29).
Mapping and orientation
Read alignment was executed in a two-stage process. Stage 1 (taxonomic classification): To maximize specificity, reads were assigned to reference bins using a custom alignment workflow rather than standard preset parameters. Each read was queried against the composite reference (comprising ITR–cassette–ITR, plasmid backbone, helper plasmid, and GRCh38), and assignments were made based on the highest alignment identity score, provided the score exceeded 95%. Reads failing to meet this stringency threshold were sequestered into an unclassified bin. Stage 2 (per-vector structural analysis): Reads assigned to the vector bin underwent fine-grained structural characterization. Alignments were parsed to evaluate high-scoring segment pairs (HSPs) against two specific reference panels: (i) the full-length cassette spanning the ITRs and (ii) the terminal ITR motifs enumerating all alternative flip/flop conformations. For each read, HSP coordinates, percent identity, and query coverage were analyzed to (a) verify vector integrity, defined as continuous high-identity coverage across the cassette with correctly positioned ITR boundaries and (b) detect concatemerization or chimeric ligation events, evidenced by multiple nonoverlapping HSPs or cross-target matches. ITR orientation (flip vs. flop) was determined based on the highest-scoring alignment to specific ITR seed sequences relative to the cassette body.
Strand normalization and Integrative Genomics Viewer (IGV) sessions
To account for the stochastic packaging of positive and negative strands in AAV, alignments were computationally postprocessed to orient all reads in a canonical 5′→3′ direction relative to the reference. This normalization ensures a consistent coordinate system for 5′-end capacity analyses. For visualization, bedGraph coverage tracks were generated, with reads grouped by strand and soft-clips preserved to highlight terminal variability. For figure generation, datasets were randomly subsampled to 200 reads per condition to ensure visual comparability. The alignment and structural features of the AAV genome were visualized and analyzed using the IGV (Broad Institute), a high-performance interactive tool designed for the visual exploration of large genomic datasets. To ensure consistency and reproducibility in data interpretation, all viewing parameters (including track scales, color-coding for chimeric reads, and grouping criteria) were standardized and recorded via IGV session XML files using IGV software (version 2.19.1). 37
Definition of integrity and “raw versus unique”
Two complementary metrics were established to characterize the library. The Raw Mapping Length Distribution encompasses all aligned molecules—including fragments, partial reads, and concatemers—to provide a global view of the read population. Genome Integrity (Unique/Intact) was strictly defined as the fraction of molecules whose primary and supplementary alignments collectively span ≥95% of the designed ITR–cassette–ITR reference, extending from the 5′ ITR boundary to the 3′ ITR boundary. Molecules failing to meet this criterion were classified as truncated or partial. This ≥95% ITR→ITR definition was applied consistently across all analyses and sequencing platforms to ensure cross-comparability.
Per-read structure typing (0×/1×/2×/3×)
Single-molecule topology and copy number were assigned based on alignment (HSP) architecture relative to the cassette reference. The classification criteria were as follows:
1× (Monomer): A single contiguous alignment spanning ≥95% of the reference insert with exactly two distinct ITR contacts at the termini. 2× (Dimer): Two consecutive, head-to-tail alignments abutting at a central ITR junction (within a ≤50 bp tolerance), collectively covering approximately twice the unit insert length. 3× (Trimer): Three serial head-to-tail alignments collectively covering approximately three times the unit insert length. 0× (Partial/Null): No contiguous full-length insert detected.
Terminal orientation (flip/flop) was recorded based on alignment directionality near the D-sequence. Notably, in RNP-treated libraries, calls for concatemer species were abolished due to the cleavage of ITR junctions, confirming that the multicopy signals observed in annealing-based libraries represent genuine covalent head-to-tail linkages rather than library artifacts.
Terminology note
Throughout this article, the term “one-end-sufficient (motor-anchored) adapter ligation” denotes ONT ligation libraries in which sequencing initiates provided at least one ligatable terminus carries a functional motor adapter; the opposing terminus need not be ligated. This mechanism is distinct from PacBio SMRTbell libraries, which strictly require successful hairpin ligation at both termini to generate a circular template. We emphasize that this terminology refers specifically to in vitro library construction requirements not to the biological multiplicity of ITRs on the viral genome.
Statistical analysis
Biological replicates were defined as independent vector production lots, while technical replicates represented independently prepared libraries derived from the same DNA eluate. All statistical analyses were conducted using GraphPad Prism v9.
For comparisons between two groups, paired or unpaired two-sided Student’s t-tests were applied as appropriate based on experimental design. Normality of data distributions was assessed using the Shapiro–Wilk test, and homogeneity of variance was evaluated via F-test or Levene’s test. Pearson’s correlation coefficients and simple linear regression models were calculated where indicated.
Data are presented as mean ± standard deviation. The specific number of replicates (n), statistical tests employed, and exact probability values are specified in the figure legends. Significance thresholds are annotated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; “ns” indicates not significant (p > 0.05).
RESULTS
A distinct 5.1–5.2 kb packaging cliff and ∼3 kb plateau constrain rAAV cargo capacity
To systematically map the physical packaging limits of the AAV8 capsid, we profiled a panel of vectors ranging from 1.5 to 6.5 kb using single-molecule nanopore sequencing and gel electrophoresis (Supplementary Fig. S1). On neutral and alkaline gels, vectors ≤4.8 kb resolved as discrete monomer bands, whereas designs ≥5.0 kb exhibited significant trailing and smearing under both conditions, consistent with increased truncation and heterogeneous packaging (Fig. 1A). Notably, short genomes (<2.6 kb) frequently produced ≈2× bands, indicative of tandem species packaging. While these gel patterns recapitulate the size-dependent heterogeneity long observed in rAAV biology,3,38–40 they lack the resolution to quantify the exact boundaries of genome completeness.
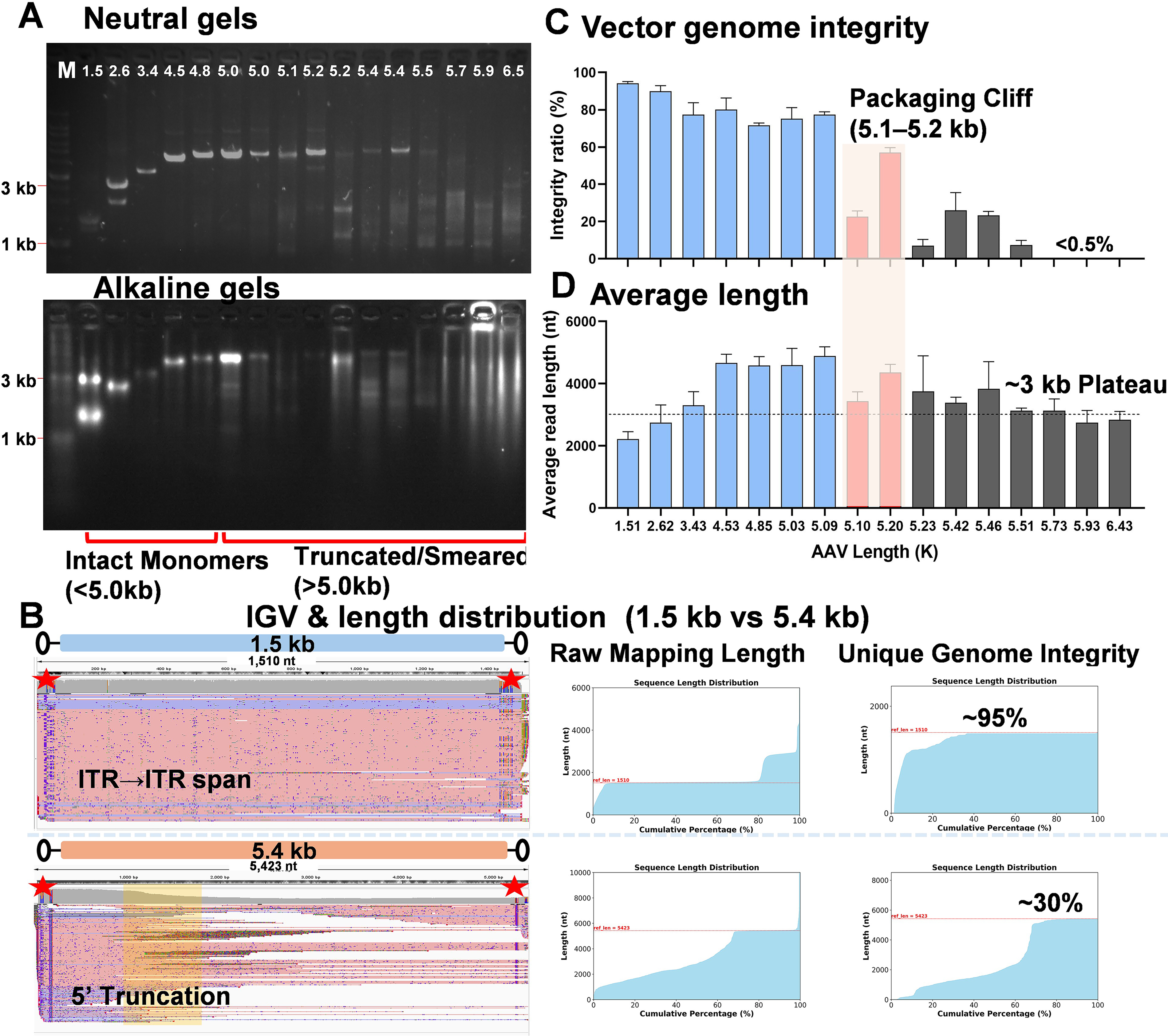
AAV cargo capacity is strictly constrained by a 5.0–5.2 kb packaging cliff and a ∼3.0 kb truncation plateau.
At the single-molecule level, IGV visualizations of 200 subsampled reads revealed uniform ITR→ITR coverage for a 1.5 kb cassette, whereas a 5.4 kb construct displayed a pronounced 5′ drop-off, confirming preferential 3′ packaging (Fig. 1B; additional examples in Supplementary Fig. S2A). Thus, molecule-resolved maps definitively distinguish between fully packaged genomes and the truncated species that accumulate near the capacity limit.
Quantitatively, genome integrity—rigorously defined as reads spanning ≥95% of the ITR–cassette–ITR reference—declined progressively with increasing size before hitting a sharp “packaging cliff” in the 5.1–5.2 kb window (Fig. 1C). Furthermore, while mean aligned read length tracked with insert size up to ∼4.5 kb, it plateaued at ∼3.0–3.3 kb once designs exceeded 5.0 kb (Fig. 1D). These data delineate a strict manufacturing design window: near 5.1 kb, integrity falls precipitously, and beyond this physical limit, capsids preferentially enclose ∼3 kb fragments rather than random truncations. Furthermore, the integrity metric proved robust and threshold-insensitive: analyses at 90%, 95%, and 98% coverage yielded indistinguishable values (paired two-sided t-tests, all ns), indicating that the 5.1–5.2 kb cliff and ∼3 kb plateau represent intrinsic biophysical constraints rather than artifacts of the integrity definition (Supplementary Fig. S2B).
Sequence-encoded thermodynamic structure modulates packaging success and offers a rescue strategy
Having established the length-dependent limits, we next investigated whether primary sequence composition modulates packaging outcomes at a fixed size. We hypothesized that regions of high thermodynamic stability (high GC content/negative ΔG) might impede the packaging motor, creating localized truncation hotspots.
Testing this hypothesis on a pair of equal-length ∼5 kb vectors (P2340 vs. P2341), we observed a distinct 3.5–4.0 kb truncation hotspot in P2340 that perfectly coincided with a region of highly negative ΔG and elevated GC content (Fig. 2A; Supplementary Table S1). Consequently, genome integrity in this thermodynamically stable vector was reduced ∼2-fold compared with its partner (Fig. 2D; Supplementary Fig. S3A), demonstrating that even within the permissive size range, sequence barriers can severely compromise packaging success.
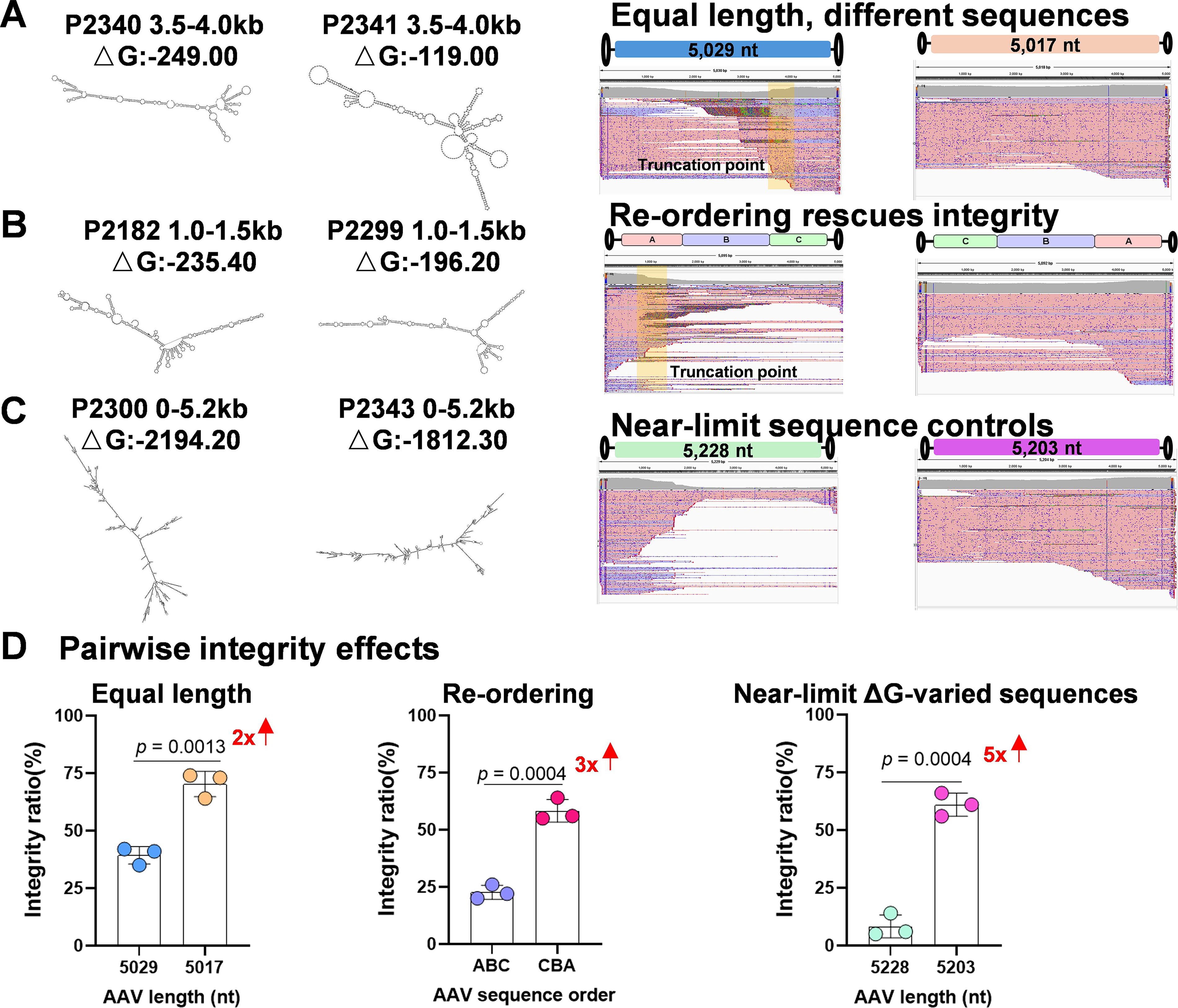
Sequence-encoded structural constraints modulate genome integrity independent of physical length.
Significantly, we found that cassette architecture can be engineered to overcome these barriers. By simply reordering two 1.55 kb segments (P2182→P2299) to lower the local ΔG and GC content in the critical 1.0–1.5 kb window, we successfully tripled the genome integrity (Fig. 2B,D; Supplementary Table S1; Supplementary Fig. S3B). This finding provides a vital design strategy: rational segment reordering can rescue otherwise unstable vector designs without altering the genetic cargo.
The impact of sequence composition became most pronounced near the packaging limit. In two designs pushing the ∼5.2 kb ceiling (P2300 vs. P2343), we observed a striking ∼5-fold gap in integrity, with the more stable, GC-rich construct significantly outperforming its less stable counterpart (Fig. 2C, D; Supplementary Table S1; Supplementary Fig. S3C). This highlights that sequence-encoded effects are nonlinear and intensify dramatically at the edge of capsid capacity. Across all matched pairs, sequence-encoded stability shifted integrity by 2–5×, indicating that the effective packaging ceiling is not a static number but a dynamic boundary governed by the interplay of genome length and thermodynamic structure.
RNP-mediated verification to distinguish biological truncations from sequencing artifacts
We evaluated library designs based on their distinct terminal requirements. While PacBio SMRTbell libraries necessitate hairpin ligation at both ends to form circular templates—a requirement that can inherently underrepresent molecules with atypical or damaged termini26,41,42—AviNP-seq employs a “one-end-sufficient” adapter ligation strategy following strand annealing or Cas9–RNP cleavage within the ITRs. By avoiding circularization and minimizing terminal bias (Fig. 3A), this design ensures that truncated or aberrant molecules are not systematically excluded during library construction.
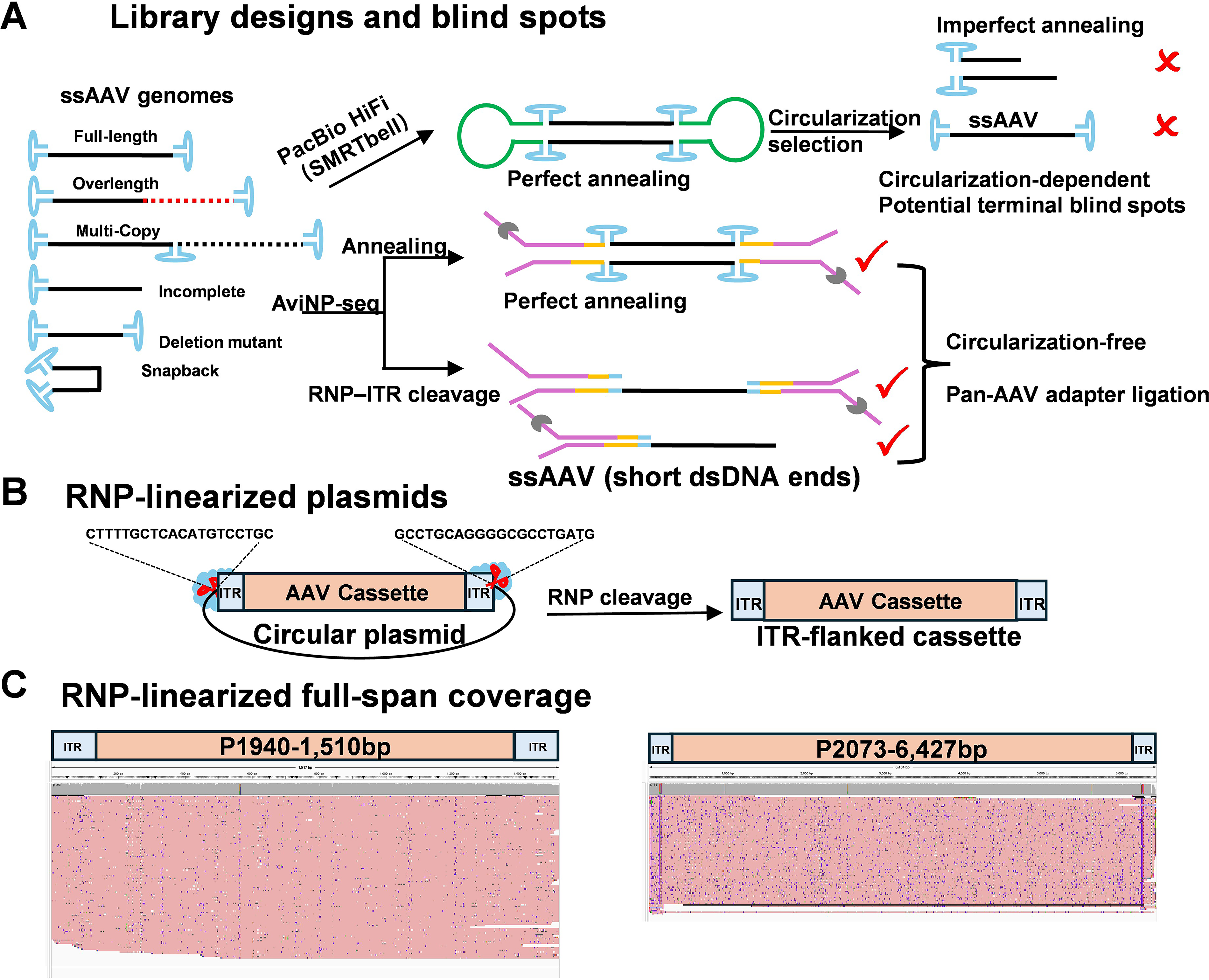
A blindspot-free library design to exclude the possibility of nonbiological truncation artifacts.
To determine whether ITR sequences intrinsically impede nanopore readthrough, we excised ITR–cassette–ITR fragments from plasmids using Cas9–RNP and sequenced them directly (Fig. 3B). Across inserts ranging from 1.5 to 6.4 kb, IGV traces demonstrated uniform coverage with no systematic drop-offs at either ITR (Fig. 3C; Supplementary Fig. S4). These findings disprove the hypothesis that terminal loss is a sequencing artifact, thereby confirming that the drop-offs observed in packaged vectors are biological in origin.
Cas9–RNP ITR cleavage enhances yield while providing an unbiased integrity baseline
To improve ligation efficiency for ssAAV genomes, AviNP-seq employs a one-end-sufficient adapter ligation strategy, performed either after strand annealing or following Cas9–RNP cleavage within the ITRs. By generating short, ligation-competent double-stranded termini without requiring circularization (Fig. 4A; Supplementary Fig. S5A), this design minimizes the terminal bias inherent in SMRTbell libraries, which necessitate dual-end hairpin ligation.
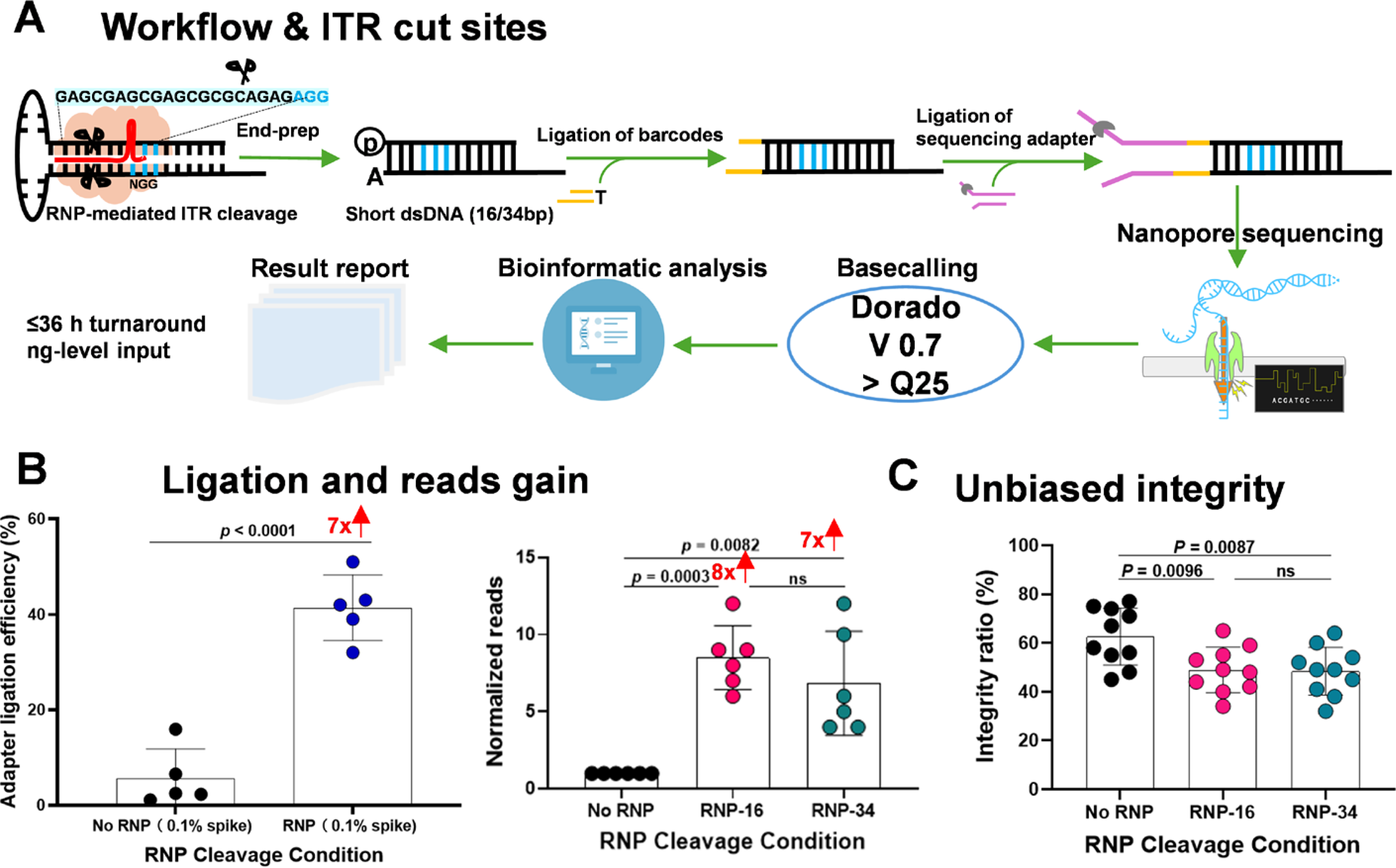
Cas9–RNP cleavage enhances ligation yield and establishes an unbiased integrity baseline.
Across vectors ranging from 1.5 to 5.5 kb, Cas9–RNP treatment achieved a ∼7-fold increase in adapter ligation efficiency relative to a 0.1% dsPCR spike-in control, translating to a ∼7–8-fold rise in usable read counts compared with libraries prepared without RNP (Fig. 4B). IGV visualization confirmed precise excision at the programmed ITR cut sites (Supplementary Fig. S5B), demonstrating that RNP cleavage provides a reproducible yield advantage independent of genome size.
Importantly, this enhancement in yield did not artificially inflate integrity estimates. Using the ≥95% ITR→ITR definition, paired analyses of 4.5–5.5 kb vectors revealed significantly higher apparent integrity in non-RNP samples compared with those treated with RNP16 (p = 0.0096) or RNP34 (p = 0.0087), with no significant difference observed between the two RNP conditions (Fig. 4C). The modestly lower integrity observed following RNP treatment reflects the removal of annealing-dependent selection bias, thereby establishing a truer baseline. In effect, RNP libraries capture molecular species that annealing protocols tend to exclude, yielding a more accurate calculation of the intact fraction.
Together, these results establish Cas9–RNP cleavage as a robust, low-bias strategy that enhances ligation yield while delivering an unbiased assessment of genome completeness. This combination renders the approach particularly well-suited for vector design and process development.
Covalent tandem genomes occupy spare capacity in sub-3 kb vectors
Neutral agarose gel electrophoresis of 1.5–2.7 kb vectors revealed prominent bands migrating at approximately twice the unit length (Fig. 5A). These species persisted under alkaline denaturation, confirming that they represent covalently linked concatemers rather than hydrogen-bonded annealed duplexes (Fig. 5B). Thus, vectors with substantial spare packaging capacity frequently form stable tandem copies.
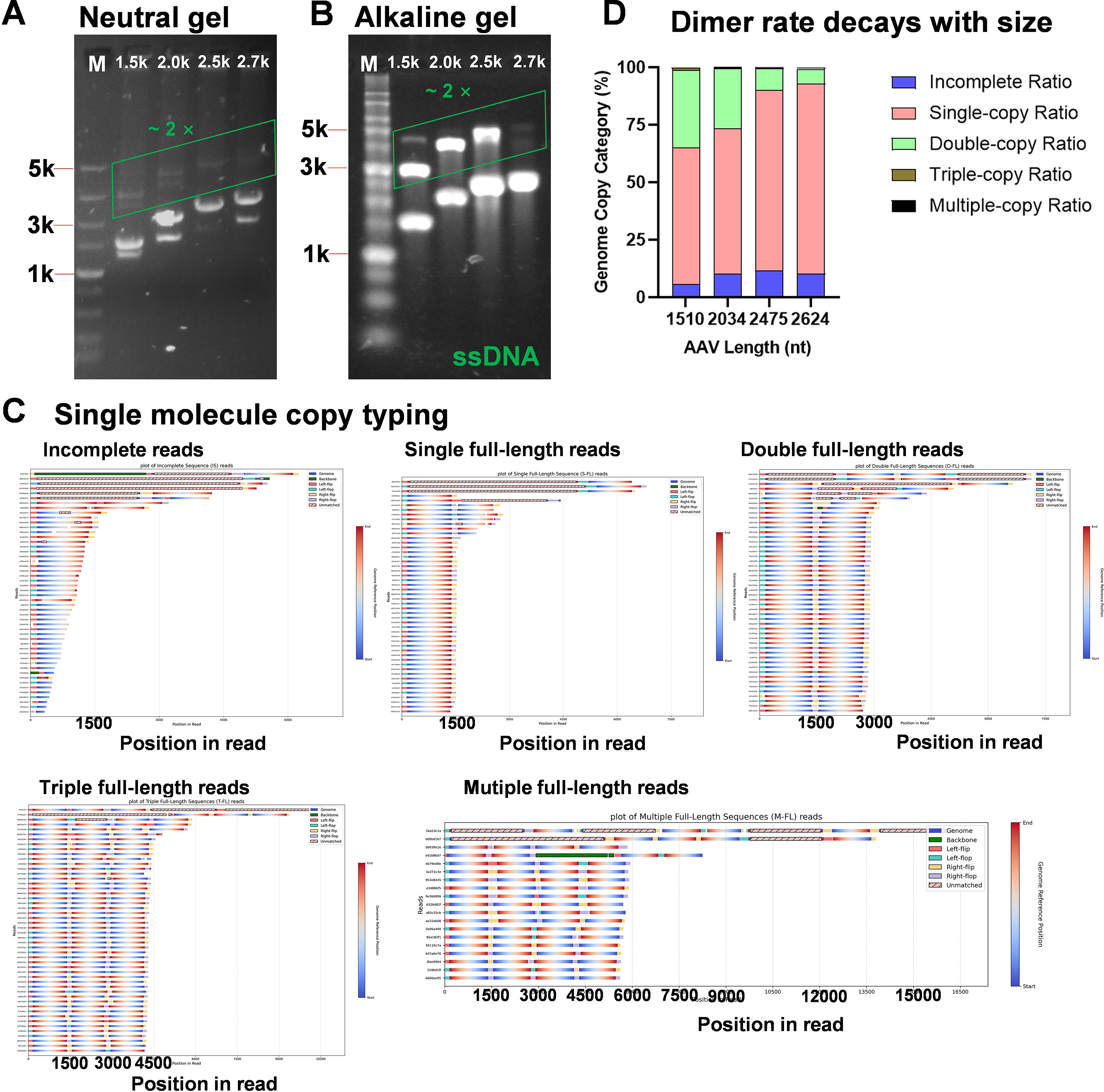
Covalent tandem genomes occupy spare packaging capacity in sub-3 kb vectors.
Single-molecule typing categorized reads by multiplicity (0×, 1×, 2×, or 3×) based on ITR junctions (Fig. 5C). The 2× fraction was prevalent at 1.5 kb but showed an inverse correlation with vector size, decreasing markedly at 2.0–2.5 kb and becoming negligible by 2.6–2.7 kb (Fig. 5D). This length-dependent decay supports a volumetric packaging model in which dimers of ∼2.6 kb approximate the physical capacity limit of a canonical ∼5 kb monomer.
Analysis of ITR orientations in single-copy genomes revealed an even distribution across eight flip/flop configurations, indicating an absence of terminal bias (Supplementary Fig. S6A–B). When both ITRs were targeted for cleavage by Cas9–RNP, these tandem signals were abolished (Supplementary Fig. S6C), definitively proving that these species exist as covalent head-to-tail concatemers. Together, these results demonstrate that AviNP-seq can resolve and validate multicopy genomes at single-molecule resolution, a capability that is critical for characterizing vectors below the standard capacity.
Validation of genome integrity across serotypes and benchmarking against PacBio HiFi sequencing
Across serotypes AAV8, AAV9, and AAVDJ, alkaline agarose gel electrophoresis displayed characteristic single-stranded bands alongside distinct species corresponding to approximately twice the unit length in 1.5 kb samples (Fig. 6A; see neutral gels in Supplementary Fig. S7A). When integrity values were normalized to the non-RNP control (set to 100%), both RNP16 and RNP34 treatments consistently yielded lower apparent completeness scores across all serotypes (Fig. 6B; Supplementary Table S2). This reduction reflects the elimination of annealing-dependent selection bias, thereby providing a more rigorous and unbiased baseline for calculating genome integrity.
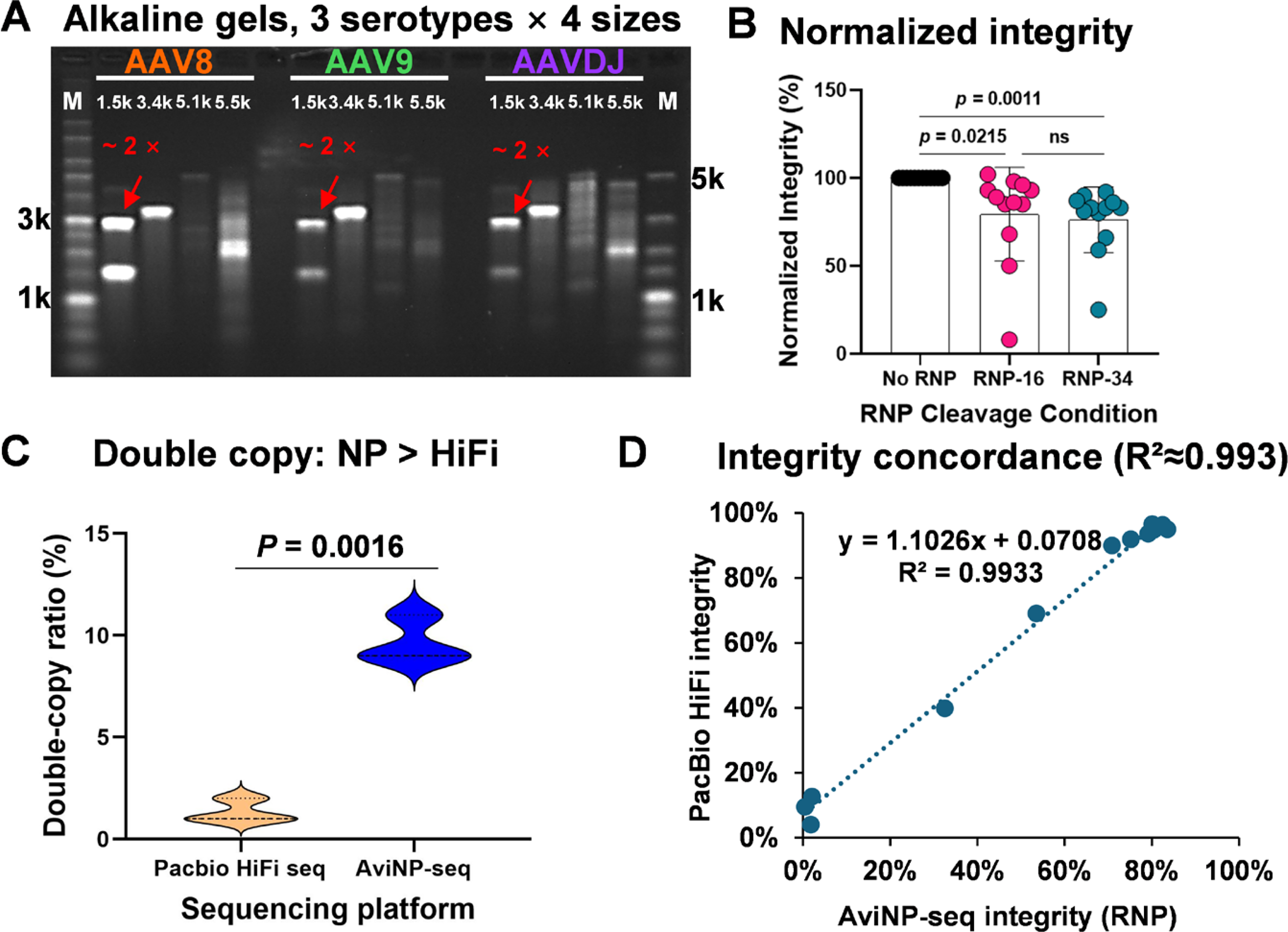
Serotype-independent validation and superior sensitivity to tandems compared with PacBio HiFi.
To benchmark performance across sequencing platforms, we compared AviNP-seq with PacBio HiFi using identical DNA preparations. In 1.5 kb vectors, AviNP-seq detected a significantly higher fraction of double-copy genomes compared with HiFi (p = 0.0016; Fig. 6C), a finding that corroborates the prominent ∼2× bands observed in gel electrophoresis. Consequently, AviNP-seq demonstrates superior sensitivity for detecting tandem species in short-genome vectors.
Conversely, estimates of genome integrity were virtually indistinguishable between the two platforms. Linear regression analysis of AviNP-seq (RNP-treated) versus HiFi data yielded an R2 of approximately 0.9933 with a slope near unity (Fig. 6D). These findings establish robust cross-platform concordance for assessing genome completeness, while simultaneously highlighting the specific advantage of AviNP-seq in identifying concatemer populations.
High-sensitivity impurity quantification at single-molecule resolution
Reads were classified as vector, plasmid backbone, helper, or HCD using a masked composite reference alignment strategy (Fig. 7A). In a representative 1.5 kb production lot, >90% of reads mapped to the vector genome. Backbone and helper sequences contributed at subpercent frequencies, while HCD fragments were characteristically short and uniformly distributed throughout the host genome (Fig. 7B; Supplementary Fig. S8). These data indicate that residual impurities persist at trace levels without evidence of site-specific enrichment.
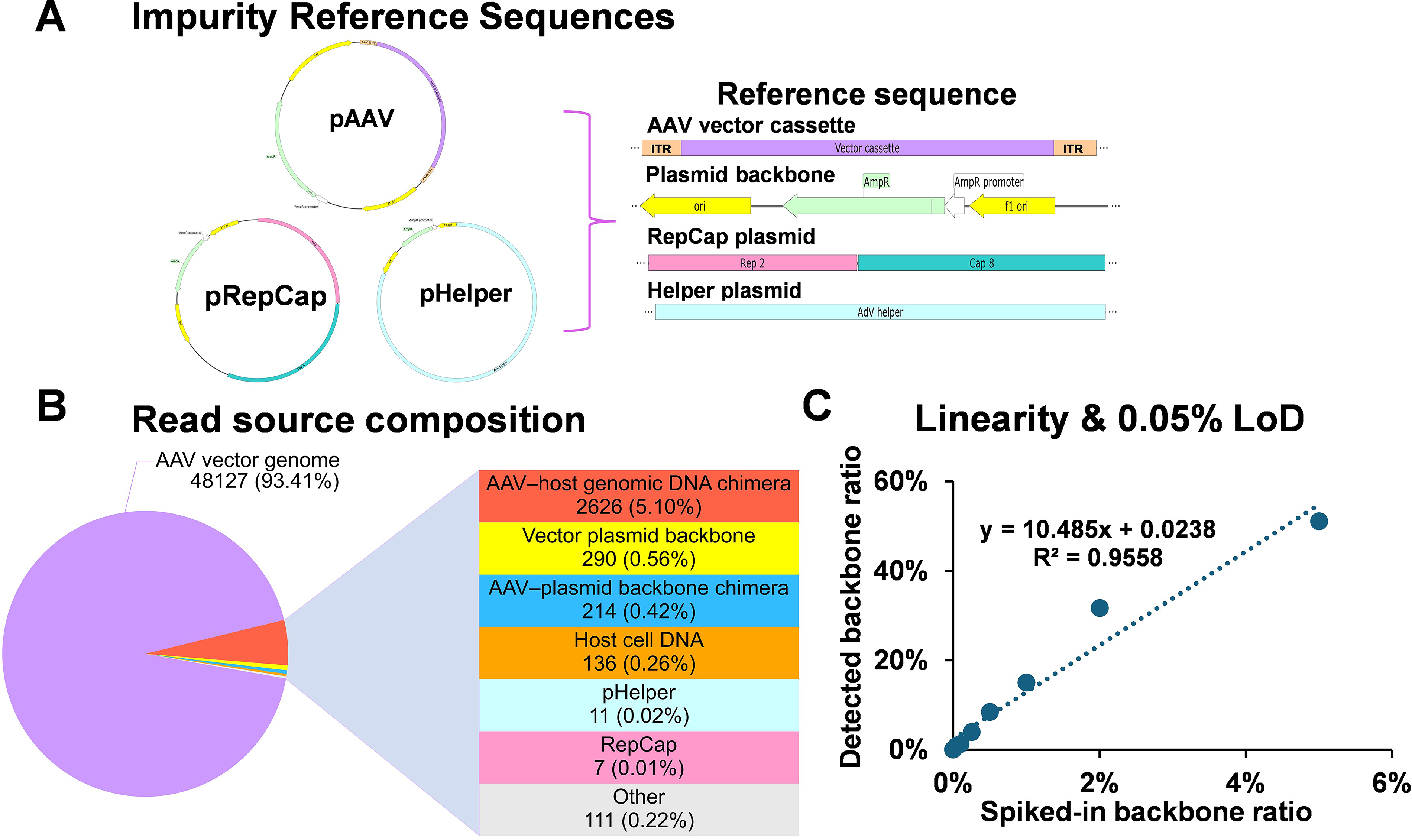
High-sensitivity impurity profiling and limit-of-detection (LOD) validation.
Analytical sensitivity was validated using a backbone spike-in titration ranging from 0% to 5% (w/w). Linearity was maintained across this range (R2 ≈ 0.956), with an LOD confirmed at 0.05% (corresponding to 59 backbone-derived reads at standard sequencing depth; Fig. 7C). These findings establish a robust quantitative framework for impurity trending, providing defined detection limits appropriate for vector characterization and process optimization.
A decision framework linking single-molecule readouts to process-relevant actions
We synthesized our findings into a comprehensive decision matrix designed to guide process development and vector characterization (Fig. 8). For vectors approaching the capsid capacity limit (5.0–5.2 kb), the framework mandates the use of RNP + NP libraries. This strategy circumvents terminal bias and enables precise quantification of completeness based on the ≥95% ITR→ITR criterion, ensuring accurate integrity estimates at the upper bounds of packaging.
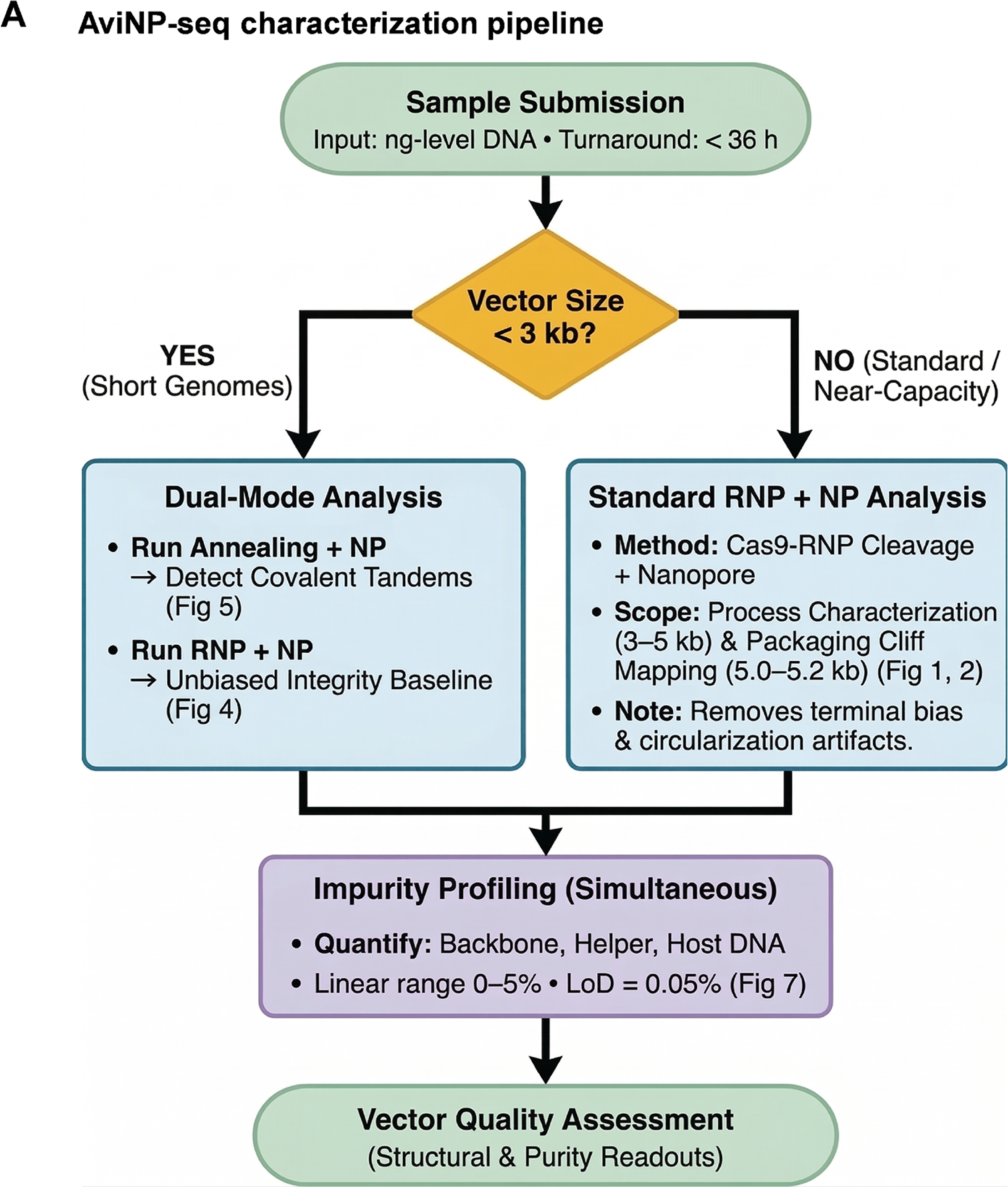
A comprehensive analytical framework for AAV vector characterization and process optimization.
In the case of sub-3 kb genomes, the framework recommends a dual-mode operational strategy: RNP + NP is utilized to establish unbiased integrity baselines, while annealing + NP is employed for the high-sensitivity detection of covalent concatemers that frequently occupy spare packaging capacity (Figs. 5–6). This bifurcated approach comprehensively characterizes both genome integrity and multicopy species within truncated constructs.
Simultaneously, impurity profiles are generated from the same dataset via masked reference classification (classifying vector, backbone, helper, and HCD), demonstrating linear quantification from 0% to 5% with an LOD at 0.05% (Fig. 7C). Supported by run-level QC metrics (Supplementary Fig. S9) and a cross-platform benchmarking matrix (Supplementary Table S5), this unified decision card offers a rapidly deployable solution. By integrating readouts for integrity, tandem packaging, and impurities within 36 h using nanogram-scale inputs, the framework effectively translates complex sequencing data into actionable quality metrics tailored for preclinical development and process optimization.
DISCUSSION
This study establishes a blindspot-free, single-molecule framework that integrates genome integrity, tandem packaging, and impurity profiling into a unified assay requiring nanogram-level input and less than 36 h turnaround. By coupling one-end-sufficient adapter ligation with Cas9–RNP cleavage within the ITRs, the workflow circumvents the circularization requirements that inherently bias SMRTbell libraries. This methodological innovation not only yields reproducible baselines across diverse serotypes but also enables a rigorous exclusion of technical artifacts, revealing new biological insights into the constraints of AAV packaging.
Mechanistic constraints: the packaging cliff and sequence composition
Across a systematic size series, we mapped a sharp packaging cliff at 5.0–5.2 kb, refining classical approximations of the AAV capacity limit. Importantly, matched-length comparisons demonstrated that thermodynamic stability (ΔG) and GC-rich hotspots significantly exacerbate truncation, while segment reordering can rescue integrity by 2–5-fold. These findings elucidate that the effective packaging ceiling is governed not solely by physical length but by the interplay between sequence composition and capsid volume. This precise mapping provides a definitive guide for vector design, cautioning that “overstuffing” vectors beyond the 5.0–5.2 kb threshold precipitates heterogeneous truncation events rather than a linear loss of yield.
Volumetric constraints: the prevalence of covalent tandems
At the lower end of the size spectrum (<3 kb), our data challenge the canonical “one genome per capsid” dogma.43,44 We observed that short genomes frequently package as covalent head-to-tail concatemers, evidenced by the persistence of ∼2× species under alkaline denaturation and the length-dependent decay of tandem frequencies. These results support a volumetric model where undersized genomes form multimers to approximate the steric volume of a full-length (∼5 kb) monomer. Notably, AviNP-seq detected these species with superior sensitivity compared with PacBio HiFi. This discrepancy highlights how the dual-hairpin requirement of SMRTbell libraries acts as a filter, blinding the analysis to odd-numbered or atypical termini, whereas the linear, one-end-sufficient design of AviNP-seq captures the true biological heterogeneity of the population.
Translational and regulatory implications
From a translational perspective, the resolution of covalent tandems has profound implications. Clinically, the presence of multimeric genomes necessitates a re-evaluation of dosing calculations, which typically assume monomeric payloads. Furthermore, concatemers may exhibit distinct transduction kinetics or recombination risks compared with monomers, mandating their rigorous quantification during process development.
Simultaneously, this framework advances impurity profiling by quantifying residual HCD, helper, and backbone sequences within the same dataset. As detailed in Supplementary Table S4, AviNP-seq demonstrates high concordance with PacBio HiFi in identifying major contaminants across AAV8, AAV9, and AAVDJ serotypes. Notably, unlike qPCR or short-read Illumina sequencing—which rely on short amplicons and provide only mass-based estimates—AviNP-seq characterizes the full-length distribution and genomic origin of residual DNA. Our cross-platform analysis (Supplementary Table S4) reveals that AviNP-seq is particularly sensitive in identifying structural impurities, such as AAV-host genomic DNA chimeras (up to 2.52%), which are often underrepresented in other libraries. This single-molecule resolution enables a more comprehensive assessment of the vector’s genetic landscape beyond simple concentration metrics. This capability is increasingly critical for regulatory risk assessments, particularly regarding the oncogenic potential of long, promoter-containing HCD fragments.45–47 By aligning these high-sensitivity readouts with established guidelines (e.g., ICH Q5A), the assay consolidates multiple critical quality attributes into a streamlined workflow.
Advancing AAV analytics: cross-platform validation and the future landscape of AviNP-seq
Despite foundational differences in library chemistry, AviNP-seq and PacBio HiFi demonstrated excellent concordance for genome integrity (R2 ≈ 0.9933) (Fig. 6D; Supplementary Table S3). However, the platform-specific divergence in concatemer detection reinforces the necessity of “blindspot-free” library designs for comprehensive characterization. The resulting decision framework (Fig. 8) transforms these complex multiparametric datasets into actionable analytical criteria suitable for preclinical development and process optimization.
While AviNP-seq offers distinct advantages for macrostructural mapping and concatemer detection, it is important to acknowledge the inherent trade-offs of the platform. The single-pass nature of our one-end-sufficient ligation minimizes terminal selection bias, but it inherently lacks the ultra-high single-molecule consensus accuracy achieved by PacBio’s CCS. Furthermore, the intrinsically higher base-calling error rate of the nanopore platform makes it less optimal for detecting rare point mutations or small indels within the vector payload. Consequently, AviNP-seq and PacBio HiFi should be viewed as complementary tools: the former excelling in unbiased structural profiling, and the latter providing superior sequence-level fidelity.
Looking forward, the principles of Cas9–RNP-mediated terminus exposure and one-end-sufficient ligation may extend to other complex DNA viruses, such as parvoviruses or circoviruses, where strong secondary structures complicate analysis. By systematically mapping tandem packaging grammars and structural hotspots, this approach serves as both a tool for mechanistic discovery and a robust platform for the QC of next-generation gene therapies.
AUTHORS’ CONTRIBUTIONS
X.Z. and G.L. conceived the study and designed the experiments. G.L. performed the experiments and acquired data. Y.X., X.M., and S.P. developed algorithms and conducted data QC. G.L., Y.X., X.M., T.L., and X.Z. performed data analysis and interpretation. G.L. conducted statistical analyses. X.Z. and G.L. wrote and revised the article. X.Z., T.C., and J.Z. provided administrative support and supervision. All authors reviewed and approved the final article for submission.
DATA AVAILABILITY
Basecalled reads (FASTQ) have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession PRJNA1327863. The complete analysis pipeline, including all custom scripts and command-line workflows, is archived in a version-controlled repository (https://github.com/Mxy0331/AviNP-seq). Exact software versions and parameter configurations are detailed in the Methods Source Data.
Footnotes
AUTHOR DISCLOSURE STATEMENT
None declared.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82402188, 92368202, 92568302, 82570286, 81870149, 82070115, 81890990, and 81730006), the National Key Research and Development Program of China (Grant Nos. 2019YFA0110803, 2019YFA0110204, and 2021YFA1100900), the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (Grant Nos. 2024-I2M-3-018, 2025-I2M-XHZY-040, 2024-I2M-ZH-015, 2023-I2M-2-007, 2022-I2M-2-003, 2022-I2M-2-001, 2021-I2M-1-041, 2021-I2M-1-040, and 2021-I2M-1-001), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Grant No. 2020-PT310-011), the CAMS Fundamental Research Funds for Central Research Institutes (Grant No. 3332021093), the Tianjin Natural Science Foundation Project (Grant No. S25YB0754), the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (Grant No. TSBICIP-KJGG-017), the Haihe Laboratory of Cell Ecosystem Innovation Fund (Grant No. 24HHXBSS00005 and HH22KYZX0022), China Foundation For Youth Entrepreneurship and Employment-Incaier Public Welfare Fund (HH25KYHX0009), Fundamental Research Funds for the Central Universities (Grant No. 3332024074), and Postdoctoral Fellowship Program of CPSF (Grant No. GZC20240142).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.