
Abstract
Adeno-associated virus (AAV) gene therapy is a promising therapeutic approach for both inherited and acquired disorders, yet its effectiveness is often limited by barriers that reduce target cell uptake and transgene expression. While body temperature is known to have a significant impact on viral infection, its controlled regulation for AAV gene therapy has been little explored. This article reviews therapeutic hyperthermia, carefully controlled mild heating, as a complementary strategy to improve delivery and expression. Evidence from cell and animal studies suggests that transient mild heating can increase blood flow and vessel permeability, promoting viral entry from the bloodstream into tissues. It can also trigger cellular stress responses that facilitate entry and downstream processing. Heating can also serve as an external switch when combined with heat-responsive genetic control systems, allowing gene expression to be activated only in the heated region. We summarize clinically used methods for delivering local, regional, and whole-body hyperthermia and discuss how spatially controlled heating could be integrated with AAV gene therapy workflows. We also highlight opportunities for clinical translation, key risks, and open questions, including thermal dose control, potential loss of viral particle stability with excessive heating, and the possibility of increased inflammatory or immune side effects.
BACKGROUND
Adeno-associated virus (AAV) gene therapy has emerged as a transformative tool in modern medicine, providing treatment options for both genetic and acquired diseases by delivering therapeutic genetic materials to target cells. AAV has the ability to transduce both dividing and nondividing cells, 1 and offers long-term expression by forming episomes after reaching the nucleus. It is a safe vector because it cannot replicate without helper viruses, and it is nonpathogenic to humans. These features make AAV particularly well-suited for gene delivery.1,2
Gene therapy offers the possibility of addressing diseases at their molecular cause by restoring, replacing, or editing defective genes, rather than managing downstream pathways. This approach is especially compelling for treating monogenic disorders, in which correction of a single gene can yield substantial clinical benefit. Clinical successes have been reported across diverse tissues and disease areas, including hematological, ocular, and neuromuscular indications, underscoring the versatility of AAV as a therapeutic platform. 3 For instance, in hemophilia B, systemic administration of an AAV vector encoding the high-specific-activity factor IX (FIX) Padua variant (R338L) produced a mean steady-state FIX activity of 33.7 ± 18.5% of the normal level (range = 14–81%). 4 In treated patients, prophylaxis maintenance treatment was discontinued, and the amount of factor use fell markedly (from 2,908 to 49 IU/kg on average). Nine of 10 participants had no bleeding events during follow-up. Similarly, in biallelic RPE65-mediated inherited retinal dystrophy, subretinal delivery of an AAV2.RPE65 vector significantly improved functional vision and light sensitivity in a randomized phase 3 trial, which supported U.S. Food and Drug Administration approval of voretigene neparvovec-rzyl (Luxturna) in December 2017, the first in vivo gene therapy for inherited diseases. 5 Another study showed that in symptomatic infantile-onset spinal muscular atrophy (SMA), a single intravenous dose of AAV9.SMN1 (onasemnogene abeparvovec) improved motor function and survival compared with natural history: 59% of infants achieved independent sitting for ≥30 s by 18 months, and 91% were alive without permanent ventilation at 14 months. 6
AAV transduction processes and barriers
Efficient in vivo delivery of AAV hinges on overcoming route-specific barriers and cell entry steps (Fig. 1). With systemic dosing (e.g., intravenous), vectors must cross the endothelium to access the target-tissue parenchyma and may be intercepted by pre-existing anti-AAV neutralizing antibodies.7,8 Even with engineered capsids that show enhanced barrier crossing in mice, blood–tissue transport (notably across the blood–brain barrier) remains a species-dependent and rate-limiting step.9–11 By contrast, with local administration, such as intramuscular, subretinal, intrathecal, or intraparenchymal injection, the vector is deposited directly into tissue or cerebrospinal fluid and overcomes the endothelial barrier. Therefore, distribution is constrained by injection geometry, extracellular matrix barriers, and pre-existing anti-AAV neutralizing antibodies.7,8,12

Schematic of delivery challenges for systemic AAV gene transfer. After intravenous administration, vectors encounter sequential delivery barriers: (1) intravascular neutralization/clearance by pre-existing neutralizing antibodies, reducing bioavailable dose; (2) off-target sequestration during circulation (e.g., liver uptake), diverting vector away from the intended site; (3) limited transport across the target organ microvascular endothelium to access tissue parenchyma; (4) restricted interstitial transport through extracellular matrix and interstitial fluid to reach target cells; (5) cell-type/serotype-dependent entry limitations, where attachment factors and receptor availability can constrain uptake; (6) post-entry intracellular barriers (inefficient endosomal escape, degradative trafficking, nuclear entry/capsid uncoating, and other processing steps) that limit productive transgene expression. AAV, adeno-associated virus.
Systemic AAV dosing also yields serotype-dependent biodistribution, resulting in off-target distribution, often with predominant liver uptake that can divert the dose away from the target organ.13–15 These patterns are governed by two separable determinants: (1) capsid–receptor interactions and (2) organ-specific microvascular exposure. For example, AAV9’s recognition of terminal N-linked galactose defines which cells it can bind, whereas the liver’s fenestrated sinusoids provide ready vector access to hepatocytes.16,17 Clinically, dose-related hepatic aminotransferase elevations have been observed in high-dose AAV-treated cohorts, underscoring that liver-biased exposure can constrain the practical dose window.18,19 Additionally, systemic exposure at very high AAV doses can trigger complement-associated thrombotic microangiopathy, an endothelial injury syndrome that often presents with renal microvascular damage and acute kidney injury, further narrowing the safe dosing margin beyond hepatotoxicity.20,21
After reaching target cells, productive AAV entry typically begins with attachment to cell-surface glycans. For example, AAV2 can bind heparan sulfate proteoglycans, 22 and AAV5 relies on sialic acid binding for efficient transduction. 23 This attachment is followed, for many serotypes, by engagement of the AAV receptor (AAVR/KIAA0319L), which promotes endocytosis and intracellular trafficking. 24 Once internalized, only a fraction of particles escape the endosomal system and reach the nucleus,25,26 whereas many are routed to late endosomes/lysosomes and to proteasome-dependent degradation. 27
THERAPEUTIC HYPERTHERMIA
Why temperature matters for viral gene delivery
Fever and controlled thermal stress shape host–virus biology and the tissue microenvironment in ways that are directly relevant to vector delivery. At fever-range temperatures (39–40°C), high endothelial venules upregulate homing cues and adhesion molecules, most notably intercellular adhesion molecule 1 (ICAM-1) and C-C motif chemokine ligand 21 (CCL21) via Interleukin-6 (IL-6) trans-signaling, thereby increasing lymphocyte trafficking and dynamically modulating the vascular interface. 28 In T cells, a heat-sensing pathway in which heat-shock protein (HSP) 90 activates α4-integrins enhances adhesion and transmigration during fever, illustrating that modest temperature elevations rapidly tune cell–surface interactions at the blood–tissue interface. 29 Beyond adhesion signal modifications, hyperthermia increases vascular permeability and facilitates macromolecule/nanoparticle extravasation in vivo, providing a parallel mechanism for improving vector access to parenchyma.30,31 Together, these temperature-responsive changes provide a biological rationale for testing hyperthermia as a means to improve AAV uptake in targets by modulating access and adhesion-dependent interactions and by aiding transvascular transport that precedes productive entry.
Combining hyperthermia with AAV gene therapy
There is evidence indicating that controlled heating can increase AAV transduction (Table 1). In colorectal cancer cell lines, 43°C × 1 h heating enhanced AAV transduction, and this was accompanied by heat-shock-linked shifts in AAV receptor and chaperone gene expression. 36 In melanoma cell lines, the same 43°C × 1 h regimen improved recombinant AAV-DJ gene transduction, with increased AAVR (KIAA0319L) expression across all studied cell lines. Heparan sulfate proteoglycans (HSPG1/HSPG2) were upregulated in a subset (A375/G-361 for HSPG1; G-361 for HSPG2) of cell lines, alongside the induction of HSP70/HSP90 family members. 37 Meanwhile, earlier mechanistic work with AAV2 showed that heat-shock treatment can raise transduction independently of HSP90, suggesting multiple, capsid- and context-dependent routes by which temperature facilitates entry or post-entry steps. 33 Although studies remain limited and largely preclinical, the direction of effect is consistent: mild hyperthermia improves AAV gene transfer under regimens such as 43°C × 1 h that are practical using standard clinical or laboratory equipment.36,37
Past Studies Investigating Hyperthermia’s Effect on Adeno-Associated Virus Transduction Efficiency
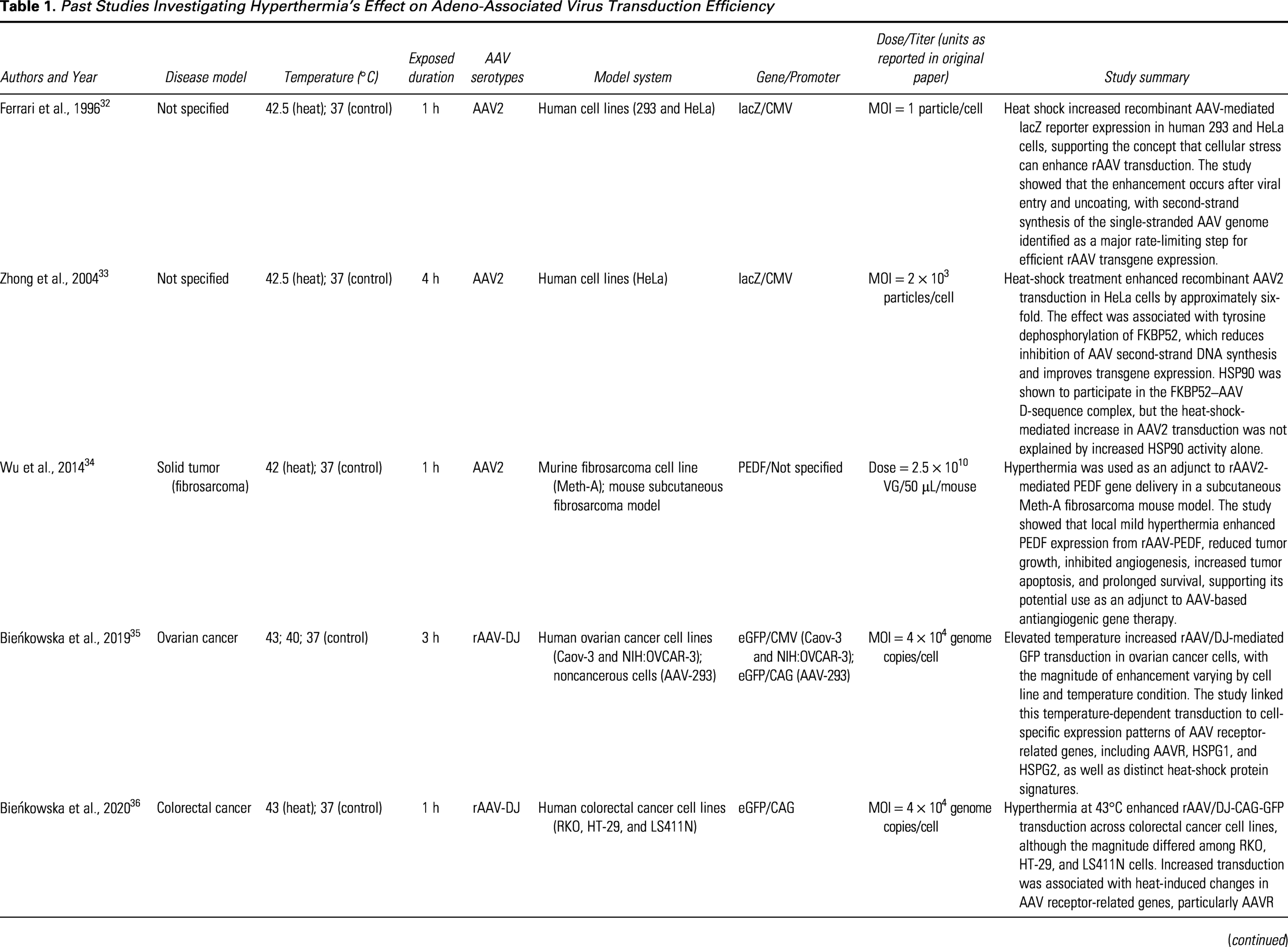
AAV, adeno-associated virus; AAVR, adeno-associated virus receptor; CAG, CMV early enhancer/chicken β-actin promoter; CMV, cytomegalovirus immediate-early promoter; eGFP, enhanced green fluorescent protein; FKBP52, FK506-binding protein 52; GFP, green fluorescent protein; HSP90, heat-shock protein 90; HSPG, heparan sulfate proteoglycan; lacZ, β-galactosidase reporter gene; LS411N, human colorectal adenocarcinoma cell line LS411N; MOI, multiplicity of infection; PEDF, pigment epithelium-derived factor; rAAV, recombinant adeno-associated virus; VG, vector genome.
Hyperthermia can also be used to control post-transduction gene expression using heat-responsive promoters. For this application, localized heating induces heat-induced promoter activity (e.g., Hsp70.CreERT2-driven gene expression can be activated selectively in heated tissues), allowing thermally regulated gene expression. 38 Earlier preclinical tumor studies used heat-shock promoter constructs to confine expression of potent cytokines or suicide genes to heated tumors and reduce systemic exposure.39,40 This strategy is especially relevant when the therapeutic payload has a narrow safety window, as illustrated by heat-regulated gene-therapy studies that used hyperthermia to spatially restrict expression of potent therapeutic genes,39–42 or when systemic AAV biodistribution is broader than the desired expression field because of serotype-dependent tropism and liver-biased exposure.13–17 In practice, an HSP promoter could be used alone for strong thermal responsiveness38,39,41,42 or combined with tissue-selective regulatory elements, such as tissue-selective promoters or microRNA-de-targeting sequences, to create a two-layer control system in which expression requires both the correct cell type and the correct thermal stimulus.43,44
Clinical application of hyperthermia
Therapeutic hyperthermia (typically 39–45°C) is most established clinically as an adjunct to radiotherapy and select chemotherapies, whereas higher temperatures (>50°C) are used for thermal ablation. 45 In oncology, mild heating can sensitize tumors to radiation and drugs through combined effects on tumor physiology (e.g., perfusion/oxygenation) and cellular stress responses. 45 Because these heating modalities can be applied with spatial precision, they also provide a practical control input for thermally gated gene-expression strategies, although this application remains largely preclinical.
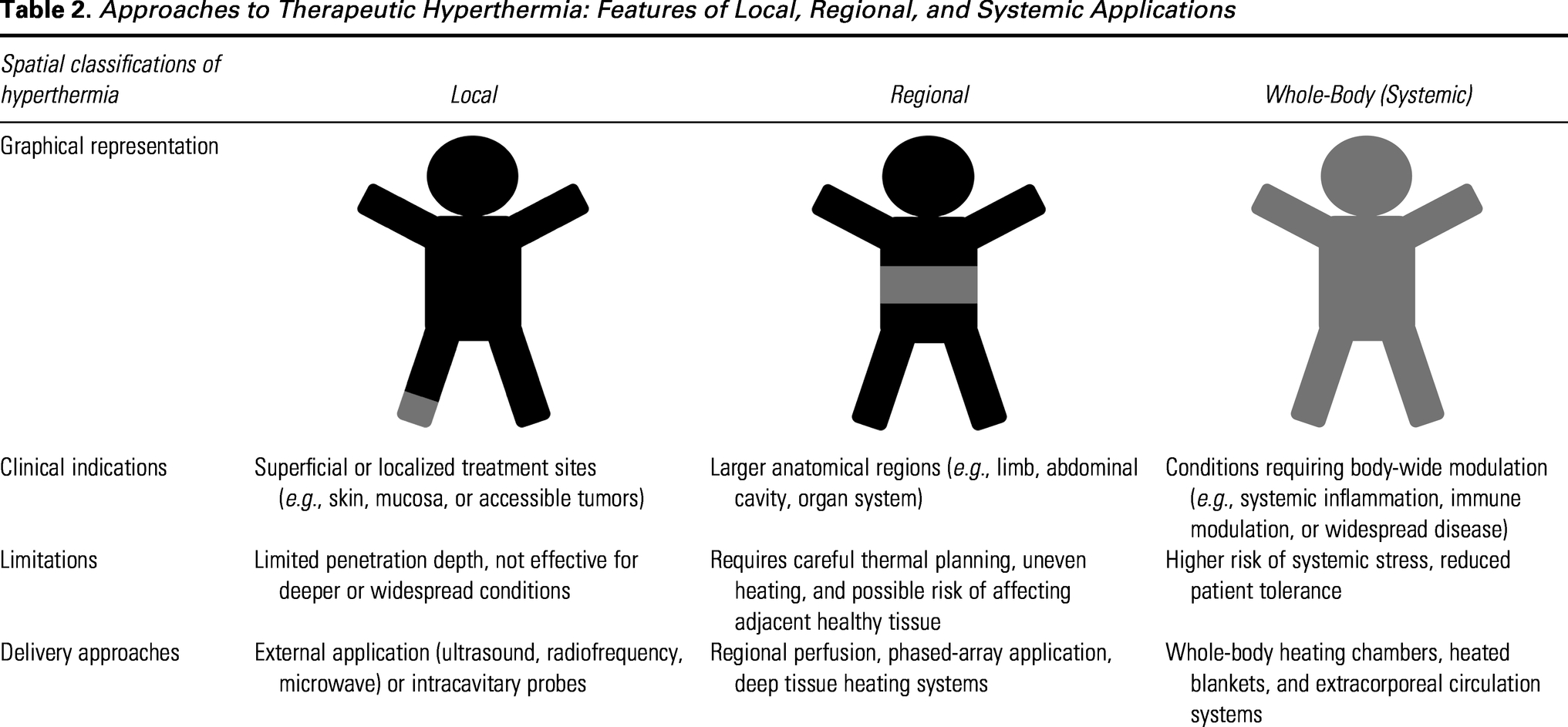
Three principal clinical heating approaches are used (Table 2): local, regional, and whole-body hyperthermia. 45 Local hyperthermia targets superficial or otherwise easily-accessible lesions using surface, interstitial, intraluminal, or intracavitary applicators, including focused ultrasound (FUS) and superficial electromagnetic systems, making it well-suited to confined disease sites. 45 Regional hyperthermia treats larger and deeper anatomical volumes (e.g., pelvis, abdomen, limbs) and commonly relies on phased-array or capacitive electromagnetic applicators. Whole-body hyperthermia elevates core body temperature and has mainly been studied as an adjunct in metastatic or advanced diseases; however, clinical evidence remains limited, and grade 3/4 toxicities (often myelosuppression), as well as occasional treatment-related mortality, have been reported in whole-body hyperthermia and chemotherapy combinations. 46
Approaches to Therapeutic Hyperthermia: Features of Local, Regional, and Systemic Applications
Methods of delivering hyperthermia
Within the local, regional, and whole-body framework, clinically used thermal therapies span several energy-delivery families: electromagnetic and ultrasonic heating, hyperthermic perfusion, and conductive heating. They can be tuned for mild hyperthermia (39–45°C) or for ablation (>50°C), depending on the clinical objective. 45 FUS, including magnetic resonance (MR)-guided FUS, is best positioned as a local hyperthermia modality because it can deposit energy noninvasively into a confined focal volume with image guidance and real-time thermometry. FUS is explicitly listed among clinically used local heating techniques. 45 In preclinical oncology, FUS-induced mild hyperthermia (42°C for 20 min) improved tumor response when combined with radiation in a subcutaneous glioma model. 47 Complementary work shows that microbubble-assisted MR-guided FUS can maintain stable mild hyperthermia at reduced acoustic power levels in vivo. 48 Ultrasound-mediated heating has also been used to activate heat-shock promoter–regulated gene expression in solid tumors, offering proof of principle for spatiotemporal control of transgene expression via local thermal dosing. 41
Magnetic nanoparticle hyperthermia likewise falls under local heating and uses iron oxide nanoparticles activated by alternating magnetic fields to generate controlled, localized hyperthermia. 45 Clinically, intratumoral thermotherapy using magnetic nanoparticles combined with external-beam radiotherapy has been evaluated in patients with recurrent glioblastoma, demonstrating the feasibility of this platform in humans. 49 Preclinical implementation and dosimetry workflows for magnetic nanoparticle hyperthermia have also been described in methodological studies.
For regional hyperthermia, larger anatomical fields are commonly heated using phased-array or capacitive electromagnetic systems and, in selected indications, isolated/hyperthermic perfusion approaches. These strategies typically require treatment planning plus multipoint thermometry to manage heterogeneous tissues and avoid temperature-limiting hot spots.45,50
Whole-body hyperthermia is generally achieved by limiting heat loss and adding systemic energy deposition (e.g., infrared-based radiant heating) or by heating blood in an extracorporeal circuit, but it carries a higher physiological burden and has shown substantial grade 3/4 toxicities in oncology combination studies, so integration with systemic gene-therapy dosing would require careful safety evaluation.45,46
MECHANISMS OF HYPERTHERMIA—INDUCED IMPROVEMENT IN GENE THERAPY
Mild therapeutic hyperthermia can enhance viral gene delivery through coordinated effects at tissue, cellular, and molecular scales. First, controlled heating transiently increases perfusion and vascular permeability, creating a time-limited window during which vectors extravasate more readily from the microcirculation into parenchyma. In temperature-controlled tumor models, localized mild hyperthermia in the 39.5–40.5°C range for 30–60 min produces a measurable and transient rise in tumor blood flow. For example, water-filtered infrared-A heating increased tumor blood flow by about 30–80% above baseline during the heating period, with improvements typically no longer evident after 1 h post-heating. 51 Intravital microscopy studies further show that localized hyperthermia can increase tumor-selective vascular transport of macromolecules and nanoparticles, including liposomes as large as around 400 nm in tumor chamber models. 30 In this localized hyperthermia setting, permeability improvements persisted for up to 24 h and returned to baseline by 36 h. 52 Together, these vascular effects plausibly increase the number of AAV particles reaching the interstitium for successful cell entry.
Beyond vascular transport, hyperthermia may also modulate extracellular matrix (ECM) remodeling pathways by inducing matrix metalloproteinases (MMPs). MMPs are proteases that cleave structural ECM components (e.g., collagens and proteoglycans), thereby altering matrix architecture and stiffness and potentially increasing interstitial transport by reducing steric and adhesive constraints with the stromal meshwork. 53 In an in vitro model, heat shock upregulates MMP-1 and MMP-3 in human dermal fibroblasts via extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling, as well as an autocrine IL-6 loop, 54 although whether comparable MMP induction occurs in vivo in tumor stroma, and whether it is sufficient to measurably alter transport barriers, requires further investigation.
At the cellular level, brief exposures to mild heat increase the abundance or availability of AAV-relevant attachment and entry factors, leading to elevated transduction yield. Across cancer cell models, heating at 43°C for 1 h consistently increased AAV transduction and was accompanied by heat-shock–linked remodeling of entry determinants. Most notable was increased AAVR (KIAA0319L), and in a cell-dependent manner, heparan sulfate proteoglycans (e.g., HSPG1/HSPG2) were also upregulated together with induction of HSP70/HSP90 chaperone networks that may support post-entry trafficking and processing.36,37 Because AAV entry routes vary by serotype and cell type, these receptor-involvement and uptake effects likely contribute in a context-dependent manner rather than via a single universal endocytic program. 55
After cell entry, heat-shock programs can relieve intracellular bottlenecks that limit productive gene expression. With AAV2, heat shock increases transduction, and this gain is not abrogated by HSP90 inhibition, implicating additional chaperone/co-chaperone axes. 33 A body of work identifies FKBP52, an HSP90-associated immunophilin, as a regulator of post-entry nuclear processing of AAV2 genomes. In particular, altering FKBP52 abundance or phosphorylation state can relieve inhibition on second-strand DNA synthesis and thereby increase AAV2 transgene expression.56,57 Collectively, these data support the view that heat-triggered proteostasis programs can shift intracellular AAV fate decisions toward more productive nuclear processing and expression of AAV genomes.
Overall, controlled mild hyperthermia can plausibly enhance AAV gene transfer through (1) improved vascular delivery and extravasation,30,51,52 (2) potential loosening of extracellular transport constraints,53,54 and (3) increased cellular permissiveness for AAV entry trafficking and genome processing.33,36,37,55–57 The relative contribution of each mechanism is expected to depend on tissue context, heating modality, dose, and AAV serotypes.
CHALLENGES AND LIMITATIONS
Applying hyperthermia to augment AAV delivery introduces safety and control considerations at both the vector and host levels. From a biophysical standpoint, excessive heating can destabilize AAV capsids and promote genome release. Thermal profiling (differential scanning fluorimetry) and single-particle atomic force microscopy analyses show that AAV capsids undergo temperature-dependent structural transitions, with DNA ejection and subunit dissociation occurring as temperatures climb well above physiological ranges.58,59 Although most gene delivery studies that report delivery benefit use mild hyperthermia, local “hot spots” or prolonged heat exposures thus can increase the risk of capsid damage and reduce infectivity. On the host side, systemic overheating poses organ-level toxicities. The heat-stroke literature in animals and humans documents hepatic injury driven by mitochondrial dysfunction, oxidative stress, coagulopathy, and inflammatory cascades when core temperature is sustained near or above 40°C. 60 These data underscore the importance of precise thermal dosimetry and real-time thermometry to stay within a therapeutic window.
A second challenge is the immune risk under heat. As discussed above, fever-range hyperthermia can acutely increase leukocyte trafficking at the vascular interface and enhance antigen-presenting cell activity.28,29,61,62 While these temperature-responsive programs provide a mechanistic rationale for improving tissue access, they might also heighten anti-AAV immune responses and inflammatory toxicities when heat is combined with vector delivery. Against this backdrop, AAV is intrinsically immunogenic, although less than many other vectors: pre-existing neutralizing antibodies, capsid-specific CD8 T cell responses, innate sensing, like TLR9, and treatment-emergent transaminitis remain common challenges in humans.63,64 Complement activation is also an important toxicity mechanism with high-dose systemic delivery and has been implicated in thrombotic microangiopathy in clinical reports and mechanistic studies. Accordingly, combining heat with AAV might increase the probability or severity of these events. Practical guardrails include patient selection and timing, predefined monitoring, and protocolized immunomodulation where indicated.
A third challenge concerns parameter optimization. Beneficial effects on vascular permeability and cell entry are typically achieved with mild hyperthermia (41–43°C for 20–60 min).47,48,65 Because biological effects scale with both temperature and exposure time, contemporary practice expresses thermal dose as cumulative equivalent minutes at 43°C to guide dosing and minimize unintended tissue injury. 66 In preclinical AAV work, brief heating at 43°C for 1 h increased AAV transduction in vitro in colorectal cancer and melanoma cell lines.36,37 In vivo, combining AAV with local hyperthermia improved outcomes in a murine fibrosarcoma model. 34 By contrast, escalating thermal dose (higher temperatures and/or longer exposure) is well established to steeply increase normal tissue injury risk based on thermal dosimetry frameworks and compiled tissue damage thresholds, motivating time- and temperature-limited “mild hyperthermia” regimens unless ablation is intended.65,67,68
For clinical translation, protocols should prespecify thermal-dose targets, map intra-target temperature distributions, and incorporate safety cutoffs to prevent focal overheating. Coordination of treatment sequence and timing is also critical. In a murine Meth-A fibrosarcoma model using intratumoral AAV2.PEDF combined with hyperthermia, applying heat at 42 ± 0.5°C for 1 h on days 3 and 10 post-injection, resulted in higher PEDF protein expression in tumor tissue and serum. 34 This suggests that hyperthermia can also augment the post-cell entry process of AAV-mediated gene expression.
Finally, the genetic control layer adds both opportunity and constraint. Heat-shock promoters can provide spatial and temporal control of transgene expression in response to localized heating with short thermal exposures.39,42 However, HSP promoters are stress-responsive and may exhibit low-level basal or nonthermal activation under certain conditions, which can reduce treatment specificity. 38 Promoter selection should therefore consider baseline activity in the target tissue and any co-treatments that might inadvertently engage the heat-shock response.
TRANSLATIONAL APPLICATION AND FUTURE PERSPECTIVE
Because studies combining hyperthermia with AAV gene therapy remain limited, the immediate priority is to determine whether heating should serve as a delivery enhancer (thermal priming), an expression switch (thermal gating), or a synergistic combination of both. Thermal priming applies mild heating to improve tissue access and cellular permissiveness during administration, whereas thermal gating utilizes heat-responsive promoters to restrict transgene activation to the heated volume.
For systemic AAV delivery, a potential use of hyperthermia would be to create a transient organ- or region-specific window of increased vector access without increasing the total vector dose. However, this concept remains largely hypothetical and will require direct testing in animal models. For local delivery, such as intramuscular, intraparenchymal, subretinal, or intracardiac administration, heating may be more practical because the thermal field can be aligned with the injection field. In this setting, local or regional hyperthermia could be used to improve vector spread, tissue penetration, or post-entry expression while limiting systemic physiological stress. This rationale is strongest for superficial or anatomically confined targets where established local hyperthermia approaches can be applied with spatial control. 45
The heart illustrates both the potential and the technical hurdles of this strategy. Clinical and translational studies have shown that AAV-mediated myocardial gene transfer is feasible,69,70 but inefficient myocardial uptake remains a major barrier. 71 Conceptually, mild image-guided heating could be paired with intracoronary, retrograde coronary venous, intramyocardial, or pericardial vector delivery to improve regional vascular transport and cellular permissiveness in a defined myocardial territory. Alternatively, a heat-responsive promoter could be used to restrict expression to heated myocardium after broader vascular exposure.38,42 However, cardiac application would require strict safeguards because increased oxygen demand, cardiac dysfunction, ischemic vulnerability, and arrhythmia risk could narrow the therapeutic window. Therefore, future cardiac studies would need real-time thermometry, electrocardiographic monitoring, conservative thermal-dose limits, and direct measurement of both vector biodistribution and myocardial transgene expression.
Skeletal muscle and limb disorders may represent another practical application. Regional limb hyperthermia or isolated limb perfusion could, in principle, be combined with intravascular or intramuscular AAV delivery to enhance transduction across a muscle compartment while limiting whole-body exposure. This approach may be relevant to diseases in which a large tissue mass and high vector dose requirements limit efficacy. However, significant barriers remain, including heterogeneous heating across different tissue types (fat vs. muscle) and the risk that heat-induced leukocyte trafficking28,29,61,62 might exacerbate anti-capsid immune responses.63,64
Other organs may require different strategies. In the liver, hyperthermia may be less useful as a simple uptake-enhancing method because many AAV serotypes already show liver-biased exposure after systemic administration.13–17 However, thermal gating could be useful if expression needs to be restricted to a heated liver segment, lesion, or regional treatment field. In ocular or central nervous system applications, local heating or MR-guided FUS could theoretically support barrier modulation or regional promoter activation. However, the low tolerance for thermal injury in these tissues necessitates extreme precision. 66 In accessible solid tissues or tumors, the rationale is currently strongest because local heating is already clinically established 45 and can be aligned with intratumoral, regional, or systemic vector delivery. This setting also has the strongest precedent for heat-inducible gene expression strategies and AAV–hyperthermia combination therapy. 34
Heat-responsive promoter systems may be particularly useful because they decouple vector distribution from transgene activation. For example, an AAV vector carrying a therapeutic gene under an Hsp70-derived or related heat-responsive promoter could be administered systemically or regionally, followed by focal heating of the intended target.38–42 This design may be especially relevant for immunomodulators, genome editors, or angiogenic/anti-angiogenic genes where unrestricted expression could be harmful. The main limitation is specificity: heat-shock promoters are stress responsive rather than disease specific, so promoter leakiness, activation by nonthermal stress, and variable induction across tissues must be measured carefully.38,42
In addition, patient stratification will be critical. Selection paradigms should integrate anti-AAV neutralizing antibody titers, tissue-level perfusion characteristics, and host genetics relevant to thermal stress responses to personalize both heating and vector choice.64,72,73 Computational modeling of heat distribution and vector pharmacokinetics could then enable patient-specific optimization of thermal dose and dosing schedules. Despite the substantial work required to enable clinical translation, improvement in gene transduction efficiency remains an urgent priority across AAV gene therapy applications, in light of serious adverse events recently highlighted in clinical trials. Heat application is thus a promising adjunctive strategy that can synergistically integrate molecular and physiological therapeutic mechanisms. Further rigorous studies are critically needed to define its utility, optimize its implementation, and evaluate its safety for clinical AAV gene therapy.
CONCLUSION
Hyperthermia-enhanced AAV gene therapy is a promising direction in genetic medicine. By leveraging the biological effects of mild heat, such as transient increases in vascular permeability and extravasation, context-dependent upregulation of entry factors, and activation of heat-shock programs, controlled hyperthermia can increase vector access to target cells and augment AAV transduction. Across in vitro systems and selected in vivo models, pairing mild hyperthermia with an AAV vector has improved reporter or therapeutic gene expression compared with AAV delivery alone.
Translational success will depend on careful parameter optimization and risk management. Clinical protocols should (1) define thermal-dose targets and use real-time thermometry to remain the temperature within a therapeutic window; (2) integrate heating with vector dosing, immunoprophylaxis, and monitoring to mitigate risks such as tissue injury, capsid destabilization, complement activation, and transaminitis; and (3) account for patient-specific factors including neutralizing antibody titers, tissue perfusion, and variation in heat-stress pathways.
With continued progress in image-guided heating modalities, AAV capsid engineering, manufacturing, and patient stratification, hyperthermia-assisted AAV delivery could expand treatment options and enable more effective, personalized gene therapies. Achieving this vision will require coordinated preclinical and clinical studies that couple rigorous dosimetry with mechanistic endpoints and safety frameworks.
AUTHORS’ CONTRIBUTIONS
N.-H.L. drafted the article and figures. N.-H.L., S.A.M., and K.I. made critical edits to the article. All authors approved the final version of the article.
Footnotes
AUTHOR DISCLOSURE
K.I. serves as the principal investigator on the grant from ABIOMED Inc. K.I. received honorarium from Abiomed Inc. and served as a consultant for Pfizer Inc., Gordian Biotechnology, and Lexeo Therapeutics.
FUNDING INFORMATION
K.I. was supported by National Institutes of Health grants