
Abstract
Gastric cancer (GC) is the fifth most common malignancy and one of the leading causes of cancer-related death worldwide. Its histological and molecular heterogeneity make it particularly challenging to manage. The Cancer Genome Atlas classifies GC into four molecular subtypes: Epstein–Barr virus-associated GC (EBVaGC), microsatellite instability-high (MSI-H), chromosomal instability (CIN), and genomic stability (GS), each of which has distinct genetic and epigenetic characteristics. Among this biomarker diversity, non-coding RNAs (ncRNAs) such as microRNAs (miRNAs) and long non-coding RNAs play a key role in diagnosis, prognosis, and targeted therapy. For instance, the EBVaGC subtype features PIK3CA mutations and hypermethylation of tumor suppressor genes such as CDKN2A, alongside ncRNAs such as EBV-encoded RNAs and H19 that enhance immunogenicity and response to programmed death-1/programmed death-ligand 1 inhibitors. MSI-H-GC is characterized by high mutational load and DNA mismatch repair defects, and ncRNAs such as MIR99AHG serve as prognostic and immunomodulatory markers. CIN-GC, the most common subtype, is associated with amplification of genes such as ERBB2/HER2 (human epidermal growth factor receptor 2) and ncRNAs such as miR-22 and TERRA, which exacerbate CIN and are linked to a poor prognosis may be amenable to HER2-targeted therapies. GS-GC is characterized by RHOA and CDH1 mutations and epithelial-to-mesenchymal transition (EMT) features, where ncRNAs such as HOX antisense intergenic RNA, ZFAS1, and Linc00152 affect invasion and metastasis by regulating EMT. Through interactions with miRNAs and signaling pathways, these ncRNAs not only influence prognosis but also represent novel therapeutic targets. Integrating multiomics approaches and developing ncRNA-based biomarker panels are essential for advancing precision medicine in GC.
Introduction
Gastric cancer (GC), ranked as the fifth most common cancer, leads to mortality and remained as a public health issue worldwide. The management of GC is challenging due to its histological and molecular heterogeneity, which leads to poor prognosis and clinical outcomes (Thrift et al., 2023). Several risk factors contribute to the development of GC, including infections, lifestyle, and environmental factors. Moreover, inherited genetic factors such as CDH1 mutation are related to GC occurrence (Yusefi et al., 2018). Also, gender association of GC is another factor that showed higher mortality in males (Bray et al., 2024) (Fig. 1).
Summary of GC risk factors. GC, gastric cancer.
Each year, GC affects approximately one million people, and its prevalence, incidence, and mortality rates exhibit significant geographic variation. The highest rates are observed in Asia, parts of Europe, and the Americas (Sitarz et al., 2018; Rawla and Barsouk, 2019) (Fig. 2).
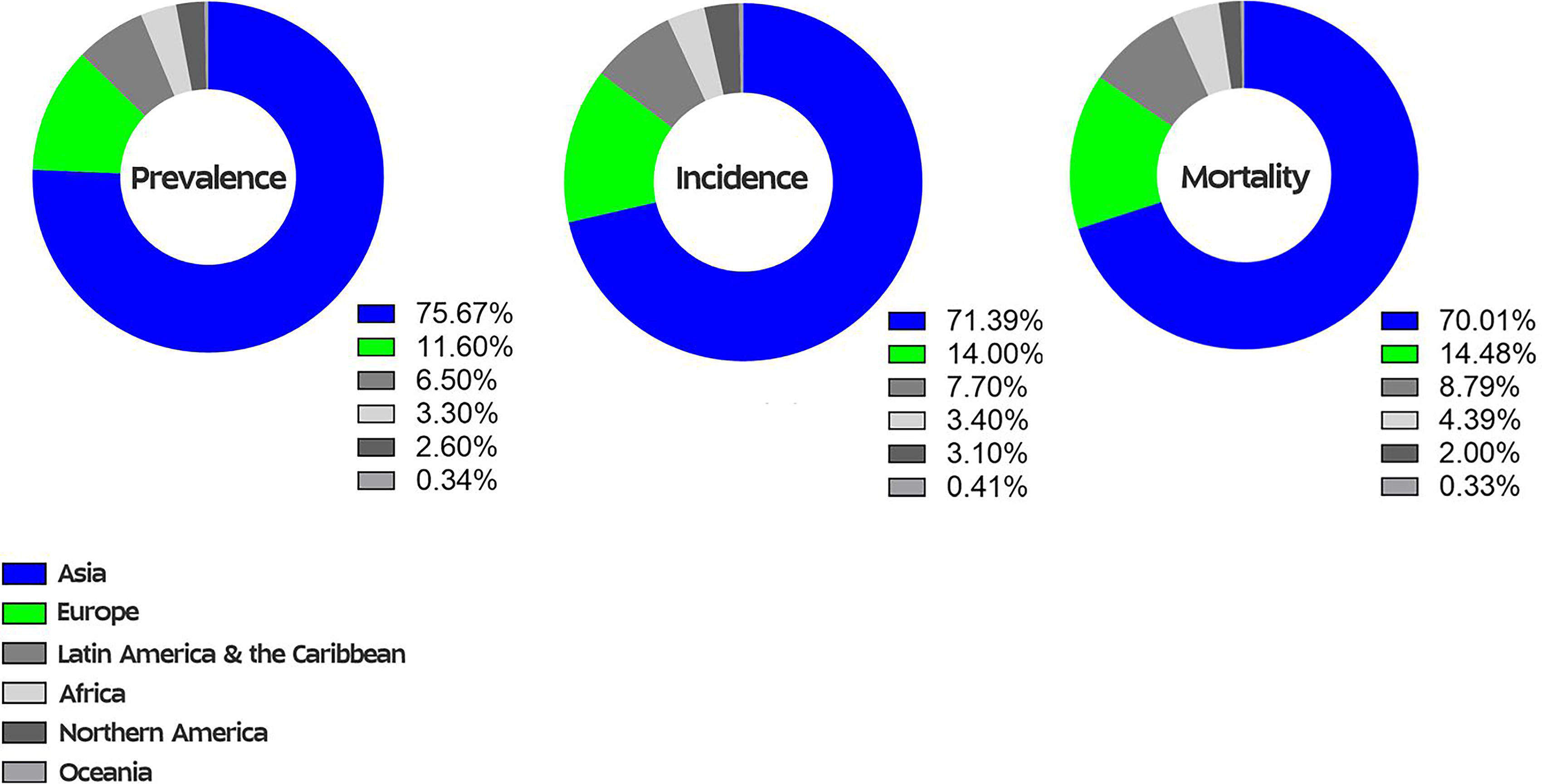
GC prevalence, incidence, and mortality rates worldwide.
Most GC patients are asymptomatic in the early stages, which often results in diagnosis at an advanced stage. The most common clinical symptoms include weight loss, anorexia, dysphagia, dyspepsia, and abdominal pain. Although endoscopy and biopsy are the primary diagnostic approaches, there is a vital need for supplementary methods, such as accurate tumor staging and TNM classification, to determine the most appropriate treatment and intervention (Van Cutsem et al., 2016).
There are two principal methods for classifying GC based on histopathology including the Laurén system and World Health Organization (WHO) classification. The Laurén system discriminates between intestinal and diffuse subtypes, and the WHO classifies GC into papillary, tubular, mucinous, and poorly adherent carcinomas (Chia and Tan, 2016). It is now established that histopathological classification is not sufficient for predicting clinical outcomes and treatment, and there is also a need for molecular subtyping. Advances in genomic and transcriptomic profiling show that these methods are helpful in classifying GC into more detailed subtypes.
The Cancer Genome Atlas (TCGA) is a landmark cancer genomics program that molecularly characterized over 20,000 primary cancer samples and matched normal samples spanning 33 cancer types. This joint effort between the National Cancer Institute and the National Human Genome Research Institute has generated a massive, publicly available dataset of genomic, epigenomic, transcriptomic, and proteomic data. The resulting comprehensive “atlases” of cancer genomes have fundamentally advanced our understanding of cancer biology and are a vital resource for developing new diagnostic and therapeutic strategies (Tomczak et al., 2015).
TCGA has classified GC into four molecular subtypes. Based on this method, GC stratification subtypes are: (1) Epstein–Barr virus-associated GC (EBVaGC), (2) GC with high microsatellite instability (MSI-H-GC), (3) GC driven by chromosomal instability (CIN-GC), and (4) GC with genomic stability (GS-GC) (Rota et al., 2024). This classification facilitates the selection of targeted therapy and prognostic prediction. For instance, PIK3CA mutations, promoter hypermethylation, and programmed death-ligand 1 (PD-L1) overexpression are frequently reported in EBVaGC. Malfunction of mismatch repair (MMR) genes, predisposing patients to high tumor mutational burden (TMB) and increased response to immune checkpoint inhibitors (ICIs) is a hallmark of MSI-H GC. The ERBB2 (HER2) and FGFR2 amplification are commonly reported in CIN-GC subtype and make it a good candidate for HER2-targeted therapy (Rota et al., 2024).
Besides alterations in coding genes, other factors such as non-coding RNAs (ncRNAs), including long non-coding RNAs (lncRNAs) and microRNAs (miRNAs), have also been demonstrated to play key roles in the pathogenesis of GC. While different biomarkers such as proteins and genes have improved GC classification, comprehensive information on the specific distribution of GC biomarkers, mainly based on molecular classifications, is still doubtful.
This review article aims to comprehensively investigate dysregulated ncRNAs specifically lncRNAs and miRNAs in each of the four molecular subtypes of GC based on the TCGA classification.
GC Histopathological and Molecular Classifications
In recent years, in addition to traditional histopathology for GC classifications, molecular classifications have contributed, which provide better insight into tumor biology resulting in better therapeutic and management approaches (Fig. 3) (Baccili Cury Megid et al., 2023).
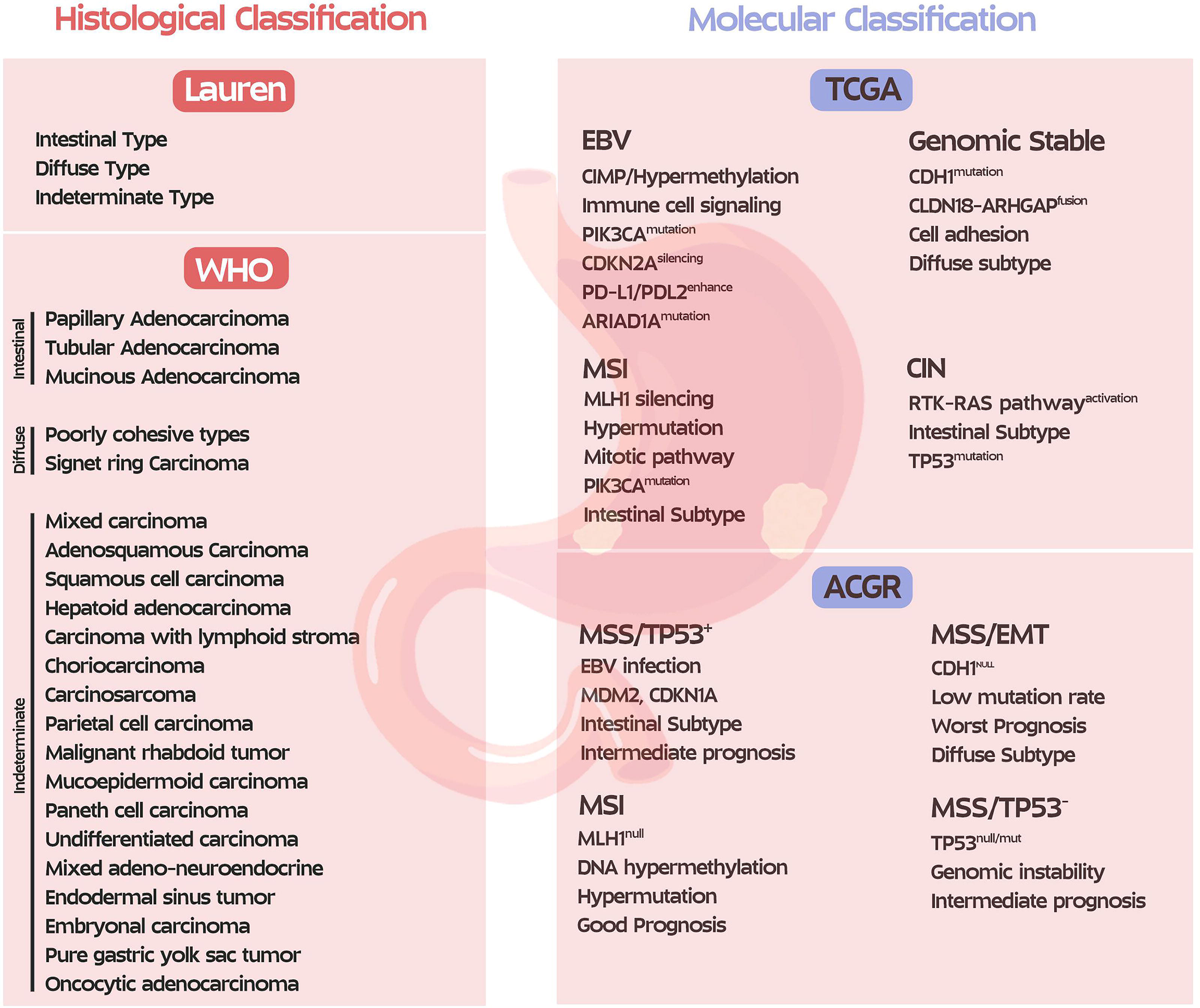
GC histological and molecular classification.
Histopathology remains the preliminary method for GC diagnosis and plays a key role in guiding treatment and prognosis. As depicted in Figure 3, the WHO divides GC adenocarcinoma into papillary, tubular, and mucinous (Baccili Cury Megid et al., 2023). Tubular adenocarcinoma is the most common type of adenocarcinoma, which is more commonly observed in intestinal tumors. Vascular invasion is a common sign of papillary adenocarcinomas. Mucinous adenocarcinomas are characterized by the extensive production of extracellular mucin. In contrast, other types, such as poorly cohesive and signet ring cell carcinomas, typically manifest an infiltrative growth pattern, aggressive clinical behavior, and a frequent association with CDH1 mutations (Lokuhetty et al., 2019).
The Laurén classification categorizes GC into intestinal, indeterminate, and diffuse subtypes (Baccili Cury Megid et al., 2023). Among the fundamental characteristics of intestinal type is glandular differentiation, which is associated with chronic gastritis and intestinal metaplasia and is frequently reported. Other common features of intestinal type are Helicobacter pylori infection, advanced age, TP53 mutations, and CIN. CDH1 gene alteration is a key attribute in diffuse type of GC that is responsible for cell adhesion disruption, tumor invasion, and peritoneal extension. The mixed type integrates intestinal and diffuse features and is clinically intermediate, which is probably a sign of tumor heterogeneity (Chen et al., 2024).
Although histological classifications are the base of GC classification, they have some limitations, including a lack of molecular insight, failure to distinguish between subtypes, intratumoral heterogeneity, and challenges in prognosis (Smyth et al., 2016). Molecular classification systems, such as those from TCGA and the ACRG, help to overcome the aforementioned limitations by providing a more comprehensive understanding of GC (Shi et al., 2024; Baccili Cury Megid et al., 2023).
TCGA classification is primarily for Western populations, but the ACRG identified four subtypes for Asians: MSI, MSS/EMT, MSS/TP53+, and MSS/TP53- (10, 14). Given the widespread use of TCGA, the following discussion will focus on the genetic characteristics of its subtypes (Baccili Cury Megid et al., 2023; Shi et al., 2024), and the following sections will focus on the molecular considerations of each subtype.
Biomarkers in GC: Emerging Perspectives
GC has significant heterogeneity at the molecular and histological levels, which markedly influences prognosis and treatment outcomes. Therefore, identifying additional biomarkers for each subgroup is crucial for addressing these challenges. Accordingly, the following sections will review biomarkers related to the TCGA classification and their roles in diagnosis, prognosis, and targeted therapy.
General biomarkers in EBV-associated GC
Approximately 9% of GC tumors related to EBVaGC and are identified by the presence of EBV in tumor cells, which leads to particular molecular alterations. PIK3CA mutations are a common feature of this subtype (in approximately 80% of cases) and show elevated expression of PD-L1 and PD-L2 (Baccili Cury Megid et al., 2023). These alterations activate the PI3K/AKT signaling pathway, resulting in cell survival and proliferation.
Increased expression of PD-L1 and PD-L2 is associated with high cytotoxic T lymphocyte (CTL) infiltration, that could be targeted by PD-1/PD-L1 inhibitors such as pembrolizumab. However, one of the key challenges is heterogeneous responses to immunotherapy due to variability in PD-L1 expression and tumor-infiltrating lymphocyte density (Baccili Cury Megid et al., 2023; Gong et al., 2019). Another trait that find in EBVaGC is the hypermethylation of tumor suppressor genes (TSGs) such as CDKN2A (p16) and ARID1A, which are implicated in immune evasion and carcinogenesis (Baccili Cury Megid et al., 2023).
The EBV infection cycle proceeds through latent and lytic phases. During the latent phase, small, non-coding, EBV-encoded RNAs (EBERs) are implicated in cellular transformation and proliferation. Leveraging their stable stem-loop structures, EBERs interact with cellular proteins to promote cell proliferation and immune evasion while inhibiting apoptosis (Ahmed et al., 2021). Identified EBER-interacting targets include TLR3, RPL22, La antigen, RIG-1, PKR, 2′−5′-oligoadenylate synthetase, PAX5, AUF1, IGF-1, IL-6, IL-9, and STAT3 (Ahmed et al., 2021).
EBV carcinogenesis is mediated by viral and host gene expression regulation, and the pattern of altered gene expression is determined by the latent (0, I, II, III) or lytic cycle (Ren et al., 2012). In most of the EBVaGC tumors gene expression alteration that associated with latency phases I and II, including EBER1, EBER2, and BamHI-A rightward transcripts (BARTs) (Sugiura et al., 1996). Also, some studies reported the expression of latency phase II genes such as BARF1, LMP-1, LMP-2A, and LMP-2B in EBVaGC, with EBER, EBNA1, and BARTs showing the highest expression levels during latency (Luo et al., 2005). Among the EBERs, EBER1 serves as a great target for diagnosis due to its stable and highly expression (Ahmed et al., 2021; Ahmed and Khan, 2014).
Aberrant methylation is another characteristic of EBVaGC. A significant association has been reported between EBVaGC and the CpG island methylator (CIMP) phenotype (Stanland and Luftig, 2020). Hypermethylation of p14ARF, p16INK4A (CDKN2A), and RCOR2, in the absence of concurrent MLH1 methylation, is a key differentiator between EBVaGC and MSI (Morales-Sanchez and Fuentes-Panana, 2017).
Although many gene mutations are involved in EBVaGC, certain mutations are more prevalent. For instance, mutations in PIK3CA, ARID1A, BCOR, TP53, JAK2, PD-L1, and PD-L2 are frequently reported (Yang et al., 2020). These mutations can also occur in other GC subtypes, and their prevalence is significantly higher in EBVaGC, approximately 80% for PIK3CA, 47–55% for ARID1A, and 23% for BCOR (Cancer Genome Atlas Research Network, 2014). Interestingly, TP53 mutations are relatively rare in EBVaGC, which could be due to EBV-mediated stabilization of the p53 protein (Cancer Genome Atlas Research Network, 2014).
Gene deletions also have been investigated in EBVaGC, including WWOX (16q23.1), MACROD2 (20p12.1), PTEN (10q23.31), IMMP2L (7q31.1), FAM190A (4q22.1), and PTPRD (9p24.1) (Cancer Genome Atlas Research Network, 2014; Gulley, 2015). Moreover, other less-specific alterations, such as amplification of CD44 (11p13) and HER2 (17q12), have been reported (Gulley, 2015). According to TCGA analyses, there is a substantial association between EBVaGC and upregulation of interleukin-12 (IL-12) signaling and downregulation of hypoxia-inducible factor 1-alpha (HIF-1α) transcription (Cancer Genome Atlas Research Network, 2014).
The therapeutic landscape for EBVaGC is multifaceted, incorporating conventional chemotherapy, immunotherapy, and targeted agents. While some in vitro studies suggest chemoresistance, retrospective clinical data indicate that EBVaGC patients achieve high disease control rates (90.3–100%) and better survival with first-line platinum and fluoropyrimidine-based regimens (Corallo et al., 2020; Qiu et al., 2020; Corallo et al., 2024). Immunotherapy represents a particularly compelling strategy, underpinned by the characteristic dense lymphocyte infiltrate and frequent amplification of immune checkpoint molecules PD-L1 and PD-L2 in these tumors. Despite this strong rationale, clinical outcomes for ICIs have been variable, with overall response rates ranging from 0% to 100%. The unique genetic profile of EBVaGC also presents opportunities for targeted therapies; the high prevalence of activating PIK3CA mutations (∼80%) supports the investigation of PI3K inhibitors, while the frequent inactivation of the ARID1A tumor suppressor gene has prompted research into synthetic lethality strategies using EZH2 and PARP inhibitors (Corallo et al., 2024).
The prognostic significance of EBV positivity in GC remains a subject of discussion, though a significant body of evidence suggests a more favorable outcome. Multiple large-scale studies, including an international pooled analysis, have associated the EBV-positive subtype with improved survival and lower mortality compared with EBV-negative cases, even after adjusting for factors such as disease stage. Although some conflicting reports find no significant survival difference, a consistent observation is that within the EBVaGC cohort, the lymphoepithelioma-like carcinoma histological subtype is associated with the best prognosis (Corallo et al., 2024).
ncRNA biomarkers in EBV-GC
miRNAs, short ncRNAs (18–24 nucleotides), regulate gene expression by interacting with the 3′ untranslated region (3′ UTR) of mRNAs and causing mRNA degradation or translation inhibition. Based on the studies, EBVaGC can dysregulating miRNAs, such as downregulating pri-miR-200 that leads to impairment of E-cadherin and cell adhesion (Shinozaki et al., 2010). Also, increased expression of EBV-miR-BART14-3p showed in EBVaGC, which induce the host DNA damage response inefficiency by inhibiting the ATM protein (Lung et al., 2018).
As depicted in Figure 4, EBV-derived miRNAs play crucial roles in various oncogenic processes. These include suppressing pattern recognition receptors, modulating host cell antigen expression, inhibiting apoptosis, maintaining viral latency, and promoting cell proliferation, invasion, and metastasis (Zebardast et al., 2021). High-throughput analyses have identified several host miRNAs (hsa-miR-5787, hsa-miR-4446-3p, hsa-miR-335-3p, hsa-miR-6877-3p, and hsa-miR-1915-3p) as most involved miRNAs in EBVaGC (Jing et al., 2018). It has been suggested that hsa-miR-5787 inhibits cell proliferation by targeting eIF5; however, carcinogenesis or tumor suppression of this miRNA is not fully understood (Yoo et al., 2011). Another investigation shows that hsa-miR-5787 enhances glycolysis and chemotherapy resistance by mitochondrial oxidative phosphorylation impairment (Chen et al., 2019).

Mechanistic pathways of EBV-miRNAs in GC development. APC, adenomatous polyposis coli; BAD, the BCL2 associated agonist of cell death; BALF5, DNA polymerase catalytic subunit; BID, subgroup of the BCL‐2 family; BRUCE, baculovirus inhibitor of apoptosis repeat‐containing ubiquitin‐conjugating enzyme; BZLF1, BamHI Z fragment leftward open reading frame 1; Caprin, cytoplasmic activation/proliferation‐associated protein; DAB2, disabled‐2; EBF1, early B‐cell factor‐1; FOXP1, forkhead box protein P1; IP07, importin‐7; KLF2, Kruppel‐like factor 2; LMP, latent membrane protein; MAP3K2, mitogen‐activated protein kinase kinase; MICB, MHC class I chain‐related protein B; NDRG1, N‐myc downstream‐regulated gene 1; NF‐Κb, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; NKG2D, natural killer group 2D; NLK, Nemo‐like kinase (Serine/threonine‐protein kinase); NLRP, NOD‐like receptor protein; PDL1, programmed death‐ligand 1; PTEN, phosphatase and tensin homolog; PUMA, the p53 up-regulated modulator of apoptosis; RIG, retinoic acid‐inducible gene; TOM, translocase of the outer mitochondrial membrane; TP53, tumor protein 53.
The role of hsa-miR-4446-3p in tumor biology is not well defined; however, high expression of hsa-miR-4446-3p is associated with tumor progression and targets oncogenes and TSGs such as PAX5, KMT2A, RUNX1, N-RAS, IDH2, and CEBPA (Han et al., 2023; Gaur et al., 2020). hsa-miR-335-3p is reported to have a considerable role in promoting GC proliferation, migration, invasion, and lymph node metastasis (Ma et al., 2022). The role of hsa-miR-6877-3p in tumorigenesis is unclear, but its decreased expression is associated with ovarian cancer recurrence (Sato et al., 2020). hsa-miR-1915-3p suppresses DUSP3, activating ERK1/2 signaling, and increasing cell proliferation and migration in GC (Guo et al., 2018). It is also involved in the pathogenesis of GC by suppressing antiapoptotic factor BCL-2 (Cui et al., 2019). circRNA-ABCB10 may function as a molecular sponge for hsa-miR-1915-3p, thereby promoting gastric carcinogenesis through the Rac1 axis (Liu et al., 2022a).
LncRNAs by interacting with DNA, RNA, and proteins could play a regulatory role in tumor initiation, progression, and metastasis (Xie et al., 2019). Abnormal expression of lncRNAs in EBV-infected cells is linked to the pathogenesis of EBVaGC (Zhang et al., 2020). For instance, SNHG8 is significantly upregulated in EBV-positive compared with EBV-negative GC cell lines and tissues (Huang et al., 2016). SNHG8 regulates various genes (e.g., BNLF2a, LF3, BHRF1, and BHLF1) and host targets (EIF4A2, TRIM28, PLD3, NAP1L1, TRPM7, and RPL18A), collectively resulting in oncogenesis (Huang et al., 2016). Silencing SNHG8 with shRNA in vitro and in vivo arrests the G0/G1 phase of the cell cycle, which indicates its oncogenic role and highlights its potential as a therapeutic target (Liu et al., 2018).
RNU12, a host-derived lncRNA, is involved in tumor suppression, and EBV infection modulates its expression in EBVaGC (Huang et al., 2016). Although the precise mechanism remains unclear, it may inhibit tumor progression by silencing miR-575 and regulating the proapoptotic factor BLID (Wang et al., 2023).
Upregulation of lncRNA H19 in EBVaGC is associated with increased tumor proliferation (Yang et al., 2012). H19, which is activated by c-Myc, suppresses the TP53/BAX/BCL2 apoptotic pathway and induces tumor progression (Liu et al., 2016). Decreasing plasma level of H19 after tumor resection shows a considerable biomarker potential for EBVaGC diagnosis (Yörüker et al., 2018). The role of H19 in EMT, metastasis, and poor prognosis make it a more interesting biomarker for GC (Li et al., 2019; Liu et al., 2022b).
MIR143HG is another lncRNA that although its role in EBVaGC is not clear, emerging evidence suggests its association with gastric tumorigenesis. MIR143HG probably acts as a tumor suppressor by inhibiting the Wnt/β-catenin and MAPK pathways and modulating HOXB7 methylation, which its expression alters in EBVaGC (Gou et al., 2023; Zhu et al., 2024; Nishikawa et al., 2017).
General biomarkers in MSI-GC
Microsatellites are short, repetitive DNA sequences found throughout the coding and noncoding regions of the genome. Their repetitive structure makes them prone to replication errors, such as insertions and deletions. These errors are normally corrected by the DNA MMR system, which is essential for maintaining GS (Puliga et al., 2021).
MSI-H-GC (about 22% of cases) have a favorable prognosis, especially in early stages. MSI-H-GC produce high levels of TMB and neoantigen due to MMR deficiency, which enhances immunogenicity and improves response to ICIs such as pembrolizumab. Hence, these tumors have good potential for immunotherapy (Ajani and Pedersen, 2024).
The relationship between clinical features and MSI-GC is still undefined; however, research is ongoing to identify prognostic and predictive markers to better classify MSI-GC cases. MMR gene mutations frequently result in missense or frameshift mutations affecting critical cell cycle regulators, apoptosis, genomic integrity, and mitotic progression, as illustrated in Figure 5 (Hudler, 2012; Cancer Genome Atlas Research Network, 2014; Cancer Genome Atlas Research Network, 2014; Puliga et al., 2021). The altered expression of MMR proteins (hMLH1, hPMS2, hMSH2, hMSH6), often due to promoter hypermethylation or coding region mutations, causes MMR deficiency (Ma et al., 2005). Compared with MSS tumors, MSI is a significant risk factor in GC and can increase mutation rates up to 1000-fold (Dudley et al., 2016).
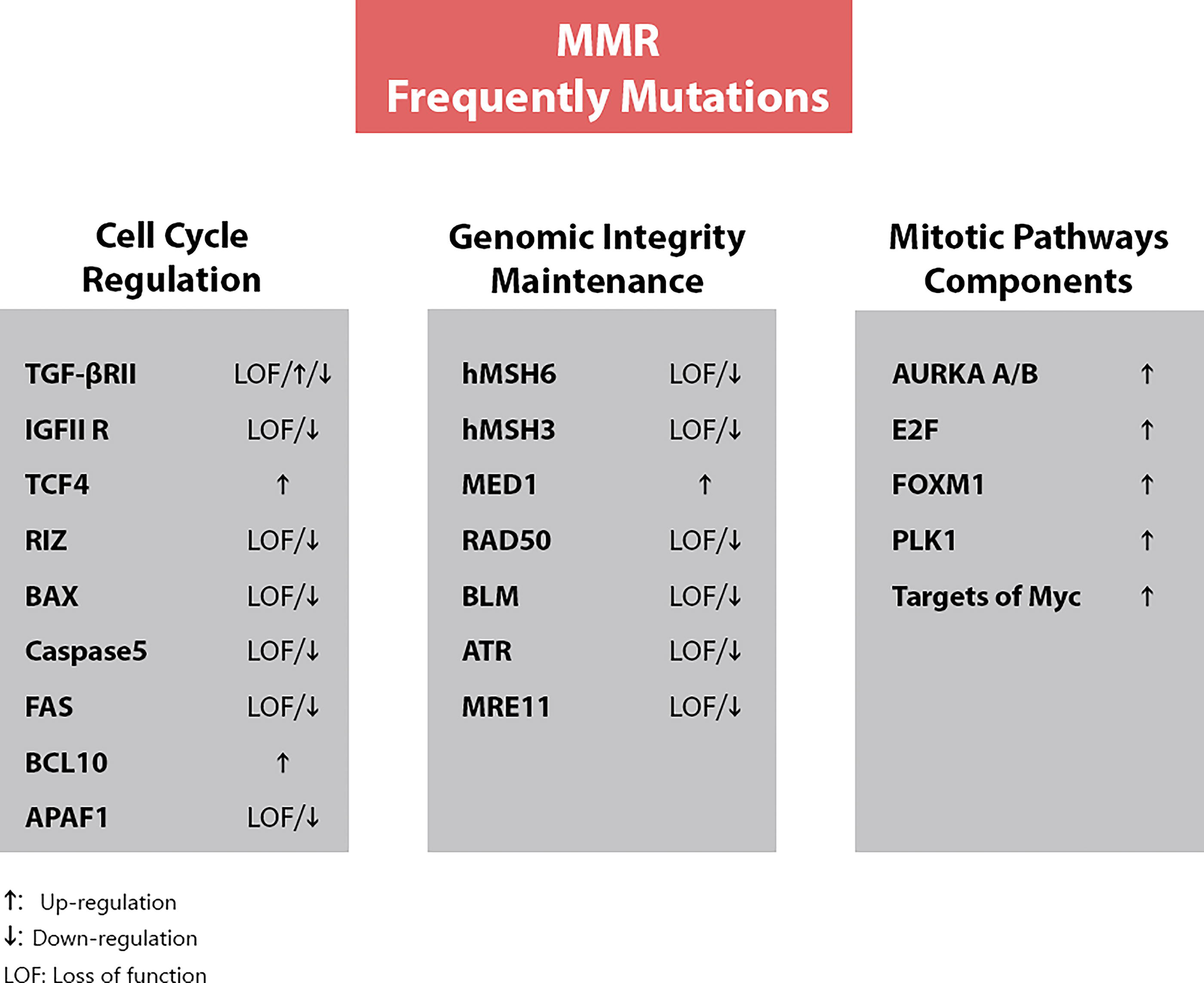
Functional loss of MMR proteins causes mutations in oncogenes and tumor suppressor genes. MMR, malfunction of mismatch repair.
The assessment of MSI is thorough comparing microsatellite sequences in normal and tumor tissues. Five loci including BAT-25, BAT-26, D17S250, D5S346, and D2S123 by The Bethesda panel analyzes and based on that samples divides into MSI-H (instability at ≥2 loci), MSI-L (instability at 1 locus), or MSS (no change) (Murphy et al., 2006; Corso et al., 2011; Smith et al., 2023).
“Hotspots” are specific genomic regions with a high frequency of mutations, recombination, or structural rearrangements, playing a vital role in gene regulation and disease pathogenesis (Edsinger and Moroz, 2024). EGFR, ERBB2, ERBB3, and PIK3CA have been identified as MSI-associated hotspots in cancers and especially GC (Ghojazadeh et al., 2022). A meta-analysis also showed that HER2 expression is absent in more than 97% of benign MSI-H tumors (Zhang et al., 2024). Notably, mutations in tumor suppressor genes such as TP53, which are common in many cancers, are less frequent in MSI-H tumors. Unlike microsatellite stability (MSS) tumors, MSI tumors exhibit high expression of immune ligands such as CTLA-4, IDO, LAG-3, and PD-L1 (Llosa et al., 2015).
Gene expression profiling has revealed MSI-GC markers, such as HLA-DRA, HLA-DRB5, HLA-DQA1, HLA-DMA, FAS, JAK2, and CASP8, which are implicated in the prognosis and mechanism of the disease (Hang et al., 2018). The retinoic acid receptor beta gene is associated with good prognosis due to its inverse relationship with MSI and immune infiltration, and could be a biomarker and therapeutic target (Cai et al., 2024). This role is maintained despite its association with proliferation, invasion, and EMT.
ncRNA profile in MSI-GC
Regardless of extensive research on MSI-GC, its association with ncRNAs is still not clear. According to the ceRNA hypothesis, lncRNAs such as RP11-999E24.3, LINC00106, HULC, and RP11-314B1.2 could affecting the prognosis of GC through cross-regulated gene expression by competing for shared miRNAs (Qi et al., 2020).
A comprehensive analysis of the ceRNA network in MSI-GC identified 60 lncRNAs. Of these, 43 were found to be associated with MSI and were subsequently investigated for their prognostic value.MIR99AHG was recognized as the center of a subnetwork in which let-7f-5p played a protective role and miR-125a-5p played a risky role (Zhang et al., 2023b).
MIR99AHG shows a remarkable correlation with immune markers within the tumor microenvironment (TME) of MSI-GC, suggesting its potential as a biomarker for diagnosis, targeted therapy, and immunomodulation (Zhang et al., 2023b).
The heterogeneity of ncRNAs in MSI-H tumors implies the role of additional molecular factors in immune evasion. Even though MSI-H tumors have high immunogenicity, the response to ICIs varies across patients; thus, there is a need for complementary markers such as T-cell receptor (TCR) diversity, TME composition, and cytokine signatures to predict prognosis and treatment outcomes (Zhang et al., 2023a; Cancer Genome Atlas Research Network, 2014).
General biomarkers in CIN-GC
The CIN is commonly observed in various cancers and generally characterized by the frequent gain or loss of whole chromosomes or large chromosomal segments (Maleki and Rocken, 2017). CIN triggers carcinogenesis through inducing loss of heterozygosity or amplification of oncogenes, resulting in marked tumor heterogeneity (Maleki and Rocken, 2017; Bakhoum and Landau, 2017). This subtype is the most common molecular variant in GC (about 50% of cases) and is associated with considerable copy number alterations in genes such as ERBB2, FGFR2, MET, and KRAS, all of which serve as potential therapeutic targets (Danishevich et al., 2023).
Despite extensive research, the precise cause of CIN remains unknown. Some hypotheses suggest that it is owing to defects in oncogenes such as RAS and TSGs such as TP53; however; mutations in these genes have further been reported in tumors with stable karyotypes (Maleki and Rocken, 2017).
TP53, a key regulator of DNA damage and cell cycle, is predominantly mutated in the CIN subtype and related to drug resistance and poor prognosis (Nemtsova et al., 2023). Based on TCGA data, 71% of CIN tumors contain TP53 mutations, regularly linked to an intestinal phenotype (Zhang et al., 2022). These mutations typically occur in the DNA-binding domain, interfere with the regulation of target genes, and sometimes enhance carcinogenesis through a dominant-negative effect. TP53 mutations also prevent its degradation by ubiquitin ligases, leading to the accumulation of the mutant protein and increasing tumorigenesis (Nemtsova et al., 2023).
CCNB1 (Cyclin B1), an essential mediator of the G2/M transition in the cell cycle, is commonly overexpressed in the CIN-GC. Its presence in the nucleus and cytoplasm is contributed to CIN, poor prognosis, and reduced survival, establishing it as a potential therapeutic target (Nemtsova et al., 2023).
CDK1, vital for entry into mitosis, is overexpressed in CIN-GC and is related to tumor progression. It is implicated in advanced cancer by phosphorylating hTERT. Inhibition of CDK1 causes cell cycle arrest and reduced proliferation, particularly in cells with CDKN2A mutations. CDKN2A inhibitors such as PD-0332991 and dinaciclib have exhibited favorable antitumor effects (Nemtsova et al., 2023).
Several genes regulating cell division exert essential influences on CIN-GC. AURKA, a mitosis-essential kinase at 20q13, contributes to CIN by interfering with mitotic mechanisms (Mesic et al., 2016). This enzyme is involved in GC progression by phosphorylating targets such as histone H3, NDEL1, TACC3, and TPX2, and interacting with β-catenin and TP53 (Nemtsova et al., 2023).
AURKB, a crucial mitotic kinase, is implicated in CIN-GC by regulating chromosome condensation, the mitotic spindle, and cytokinesis. Its abnormal expression stimulates the G1/S transition by activating CCND1 via histone H3 phosphorylation (Nemtsova et al., 2023). Inhibition of AURKB reduces CCND1 levels, suppresses proliferation, and induces G2/M arrest (Wang et al., 2020).
HER2, located on chromosome 17, is overexpressed in 7–34% of GC cases, particularly in the CIN subtype. This increased expression is associated with activation of the PI3K and MAPK pathways, tumor growth, a poor prognosis, and metastasis. Drugs such as trastuzumab, which target HER2, improve clinical response and survival in HER2-positive cases (Kanayama et al., 2018; Shitara et al., 2020).
Despite advances, the genes that drive CIN-GC remain unclear, as many mutations are linked to other subtypes. CDH1, CTNNA1, and RHOA mutations are more common in GS-GC, and KRAS, PIK3CA, and ARID1A are more common in CIN-GC or EBVaGC (Cancer Genome Atlas Research Network, 2014). Current investigations have linked copy number changes and aneuploidy to particular mutations and clinical factors such as metastasis and treatment efficacy (Smith et al., 2023).
The clinical features of CIN-GC are well understood, allowing for the prediction of tumor progression and the selection of more aggressive treatments. This subtype is associated with a poor prognosis and shorter survival (Díaz del Arco et al., 2018; Tsai et al., 2020), and more than 40% of cases have high PD-L1 expression (Kim et al., 2016). Checkpoint inhibitors such as avelumab, nivolumab, and pembrolizumab have shown promising results in treating CIN-GC (Kang et al., 2017).
In the CIN subtype, defined by genomic disruption, aneuploidy, and recurrent mutations in the TP53 gene, there is no distinct signature of promoter hypermethylation of tumor suppressor genes. This subtype falls into the CIMP-low category (low methylation phenotype), and its carcinogenesis pathway is driven primarily by genetic events. In fact, the main epigenetic event associated with this subtype is its opposite phenomenon, global hypomethylation. This overall decrease in methylation, particularly in repetitive regions of the genome, can promote structural instability of chromosomes and directly contribute to the CIN that is the main characteristic of this subtype (Zeng et al., 2017; Toshima et al., 2023; Usui et al., 2021).
The key genomic regions of interest include (1) long interspersed nuclear elements (LINE-1), which comprise approximately 17% of the human genome. Due to their abundance, LINE-1 methylation serves as a reliable surrogate for global DNA methylation, and studies have shown that LINE-1 hypomethylation in GC is associated with poorer prognosis and increased genomic instability. (2) Alu elements are among the most prevalent repetitive sequences in the genome, and their hypomethylation can disrupt heterochromatin organization, promote chromatin relaxation, and compromise genome stability by increasing DNA vulnerability and impairing repair efficiency. (3) Satellite DNA consists of highly repetitive sequences concentrated in centromeric and pericentromeric regions, with satellite alpha (Satα) being one of the most important types. Since centromeres play a crucial role in accurate chromosome segregation, hypomethylation in these regions can impair centromere function, increase segregation errors, and ultimately lead to aneuploidy, which is a defining feature of the CIN subtype (Ichida et al., 2018; Abe et al., 2022).
ncRNA profile in CIN-GC
There is a lack of significant evidence for the exact role of ncRNAs in CIN-GC, but some studies have suggested their involvement in the development of CIN. miR-22 (Jafarzadeh-Samani et al., 2017), miR-26a (Qiu et al., 2017), miR-28 (Jeddi et al., 2019), and miR-186 (Cao et al., 2016) have been identified in GC, but their precise role in CIN-GC remains undefined. miR-22 inhibits MDC1 and PTEN at the S-phase checkpoint, resulting in chromosomal repair impairment and CIN (Lou et al., 2006). Also, in colon cancer, HER2 activates AKT and miR-22 leads to aneuploidy and CIN by reducing homologous recombination. miR-22 directly suppresses MDC1, disrupting DNA repair and exacerbating CIN. The transcription factor AP-4 regulates this pathway by activating MDC1 and repressing miR-22; consequently, the loss of its function promotes defective DNA repair and increased CIN (Mohapatra et al., 2023). miR-26a has a dual role in cancer. Overexpression of miR-26a causes centrosome and multipolar spindle defects, inducing CIN (Mohapatra et al., 2023; Castellano et al., 2017). This miRNA impairs mitotic checkpoints by repressing CHFR and YWHAE, and restoration of these genes corrects the defects (Mohapatra et al., 2023). miR-28 promotes CIN by inhibiting the translation of MAD2, a crucial mitotic checkpoint protein. miR-28 amplification is associated with loss of the Von Hippel-Lindau (VHL) tumor suppressor, and anti-miR-28 therapy could be beneficial in VHL-deficient cancers. However, due to the sensitivity of MAD2 function, therapeutic targeting requires high precision (Mohapatra et al., 2023). miR-186 amplification is associated with increased CIN, particularly in the presence of arsenite, and disrupts cell division and DNA repair. In HaCaT cells, increased miR-186 induces bipartite, dicentric, and ring chromosomes (Sage et al., 2017). Although the possibility of BUB1 repression by miR-186 is has been suggested, direct evidence is lacking, and the role of BUB1 in CIN remains complex and controversial (Fujibayashi et al., 2020; Wu et al., 2019).
In addition to miRNAs, some lncRNAs such as TERRA, cenRNA, enhancer RNAs, PCAT2, GUARDIN, Ginir, CCAT2, and NORAD are involved in the progression of CIN and various cancers (Mohapatra et al., 2023); however, their exact role in CIN-GC is still not elucidated.
TERRA maintains chromosomal integrity by regulating telomere dynamics. This lncRNA regulates chromatin structure by suppressing telomerase activity and interacting with telomere proteins. Excessive overexpression or depletion of TERRA disrupts telomere homeostasis and induces CIN; overexpression produces telomere shortening and DNA damage, while depletion promotes telomere lengthening and tumorigenesis (Yang et al., 2022; Sagie et al., 2017; Mohapatra et al., 2023). Hence, TERRA is a potential therapeutic target that requires fine-tuning.
CenRNAs constitute a subset of lncRNAs that are characterized by interaction with centromeric proteins such as CENP-A. Elimination of CenRNAs leads to loss of CENP-A and chromosome segregation instability, contributing to CIN. Furthermore, amplification of CENP-A in cancer cells results in the aberrant localization of centromeric proteins and exacerbates genomic instability (Mohapatra et al., 2023; Shrestha et al., 2017).
Ginir is an lncRNA that induces CIN via promoting DNA breaks, mitotic abnormalities, and centrosome duplication. This outcome is mediated via disrupting the interaction between the proteins Cep112 and BRCA1, which modulates centrosome function. The role of Ginir in promoting genomic instability reveals its carcinogenic properties (Panda et al., 2018).
GUARDIN is a p53-induced lncRNA that supports telomere stability and DNA repair through the preservation of TRF2 expression by inhibiting miR-23a and interacting with BRCA1/BARD1. GUARDIN suppression disrupts associated pathways, promotes chromosomal fusion, and impairs DNA repair, resulting in CIN (Yoshino et al., 2021; Hu et al., 2018).
Colon cancer-associated transcript 2 (CCAT2) transcript (at 8q24) is overexpressed in GI cancers, particularly colon cancer, and is associated with poor prognosis (Deng et al., 2023). This lncRNA contributes to tumor growth, metaphase defects, and polyploidy through the upregulation of MYC and regulation of BOP1 (Ling et al., 2013). It also disrupts chromosome-microtubule associations by activating AURKB, thereby enhancing CIN and tumorigenesis (Chen et al., 2020).
NORAD is another lncRNA that is activated following DNA damage and is a vital factor in maintaining GS. The deletion of NORAD triggers mitotic errors such as anaphase bridges and mitotic slippage, consequently resulting in CIN. NORAD is implicated in the maintenance of DNA repair through inhibiting PUMILIO proteins (particularly PUM2). The loss of function in NORAD causes chromosome segregation defects; however, this dysfunction can be corrected by circular RNA containing PUMILIO response elements (Lee et al., 2016; Elguindy and Mendell, 2021).
Some investigations indicate the involvement of NORAD in GC, showing that its inhibition inducing apoptosis and repressing cell proliferation (Raei et al., 2022). Additionally, another study has associated six lncRNAs, including LINC02678, HOXA10-AS, RHOXF1-AS1, AC010789.1, LINC01150, and TGFB2-AS1, with genomic instability in GC (Sun et al., 2021). Despite the potential role of these ncRNAs in the development of CIN, further research is required to elucidate their exact role in CIN-GC.
General biomarkers in GS-GC
GS-GC encompasses approximately 20% of GC cases and is predominantly marked by diffuse, poorly adherent carcinomas. Another feature of GS-GC is mutations in EMT-related genes, such as RHOA and CDH1 mutated forms, which are linked to tumor invasion and peritoneal dissemination (Cancer Genome Atlas Research Network, 2014). RHOA mutations promote cell migration, cytoskeletal remodeling, and therapy resistance. Furthermore, rearrangements involving genes such as CLDN18 with ARHGAP26 or ARHGAP6 are common in this type of tumor, consistent with the diffuse phenotype according to the Lauren classification (Röcken, 2023).
In diffuse-type of GS-GC, overexpression of CLDN18.2 is linked to invasion and increased lymphatic metastasis (Rohde et al., 2019). These changes reflect EMT features, such as decreased cell adhesion in GS-GC, and may facilitate peritoneal metastasis. The reduced expression of CLDN18.2 in HER2-overexpressing tumors suggests a cross-regulation between these two pathways (Moentenich et al., 2020). Targeted therapies such as zolbetuximab in combination with chemotherapy have shown promising results in improving survival (Sahin et al., 2021), highlighting CLDN18.2 as a potential therapeutic target.
Bioinformatics analyses and gene expression studies have suggested crucial signaling pathways in GS-GC development, including H19-mediated Rb-E2F1 signaling and CDK-β-catenin activity. These pathways are activated in the GS subtype, indicating their role in oncogenesis and progression. (Ling et al., 2020). CDH1, CTNNA1, HIST1H1C, RHOA, and TGFBR2 are among the most frequently altered genes in GS-GC patients.
In the GS subtype, epigenetic changes occur in a targeted and focal manner. The most prominent and characteristic event is hypermethylation of the promoter of the tumor suppressor gene CDH1, which encodes the protein E-cadherin. This hypermethylation often acts as a “second hit” alongside genetic mutations in the same gene, leading to its complete silencing, disruption of cell integrity, and promotion of an aggressive phenotype. In contrast, there is evidence of hypomethylation of genes related to the Wnt signaling pathway in this subtype. This suggests that the epigenetic landscape of GS is a combination of targeted silencing of tumor suppressor pathways through hypermethylation and activation of oncogenic pathways through hypomethylation (Toshima et al., 2023; Usui et al., 2021; Cancer Genome Atlas Research Network, 2014).
A study of methylation status across 23 genetic loci in 98 GC samples showed that GS tumors have a distinct epigenetic profile. Unsupervised clustering placed most GS tumors in a group with lower overall methylation levels, and inter-variable clustering identified Wnt-associated loci with the lowest mean methylation score in GS tumors. In particular, SFRP2 and APC were significantly hypomethylated, with a mean score of 1.3 compared with MSI (2.7), EBV (2.1), and CIN (2.4) tumors. This pattern was most pronounced in signet-ring cell carcinoma, which is enriched in the GS subtype (Canale et al., 2020; Toshima et al., 2023). While hypomethylation of proto-oncogenes such as c-MYC and HRAS occurs in gastric carcinogenesis more broadly, the specific hypomethylation of SFRP2 and APC defines the GS subtype and suggests Wnt pathway dysregulation through a distinct epigenetic mechanism.
Among the key genes mentioned above, Claudin 18.2 (CLDN18.2) has been extensively studied as a crucial therapeutic target for GS-GC. The primary treatment targeting CLDN18.2 is the chimeric monoclonal antibody zolbetuximab, which was evaluated in the Phase II FAST trial. This study demonstrated that adding zolbetuximab to first-line EOX chemotherapy, consisting of epirubicin, oxaliplatin, and capecitabine, significantly improved both progression-free survival (hazard ratio [HR] 0.44) and overall survival (HR 0.55) in patients with advanced esophagogastric cancer expressing moderate to strong levels of CLDN18.2. The survival benefit was even greater in the subgroup with the highest CLDN18.2 expression (Röcken, 2023).
Despite conflicting findings on association of CLDN18.2 with clinical features and survival, its restricted expression in normal tissue and consistent tumor presence make it a specific therapeutic target. The FAST trial’s positive results with zolbetuximab confirm CLDN18.2’s potential, with efforts underway to standardize its detection and scoring (Sahin et al., 2021).
ncRNA profile in GS-GC
One of the main features of GS-GC is the EMT process, and ncRNAs associated with EMT could serve a critical role in clarifying its molecular mechanism. For instance, specific miRNAs, such as miR-26a, suppress EZH2, increase E-cadherin, and prevent EMT and invasion (Ma et al., 2016). In contrast, downregulation of miR-200c in GC elevates ZEB1 expression, leading to E-cadherin suppression and promotion of EMT (Kurata et al., 2018). Loss of miR-101 also disrupts E-cadherin function by upregulating EZH2 (Carvalho et al., 2012). However, the effects of miRNAs vary depending on the tumor type; for example, miR-9 enhances EMT by suppressing E-cadherin in esophageal cancer (Song et al., 2014); however, in melanoma, it increases E-cadherin by suppressing SNAIL1 (Liu et al., 2012).
Studies have suggested that reduction in expression of miR-128 and miR-148a in GC cells is mediated by H. pylori, which also increases MMP-3 and MMP-7 (SGC-7901). These enzymes degrade E-cadherin, thereby promoting cancer cell migration and invasion (Costa et al., 2016). Activation of the MMP/E-cadherin pathway in H. pylori-infected cells is linked to downregulation of these miRNAs (Yang et al., 2018).
MiR-148a reduces the invasive properties of GC cells through inhibiting DNMT1 and targeting SMAD2, thereby inhibiting EMT (Wang et al., 2013). Similarly, miR-29b/c, by suppressing DNMT3A, causes CDH1 silencing and promotes metastasis. Downregulation of miR-204 in GC reportedly results in E-cadherin levels elevation and a decrease in EMT markers such as N-cadherin, vimentin, and TWIST through inhibiting TGFBR2 and modulating the TGF-β pathway (Bure et al., 2019).
MiR-5003-3p, miR-217, and miR-9, targeting the 3′ UTR of the CDH1 gene, reduce E-cadherin expression and thereby increase migration, invasion, and EMT in GC. Additionally, miR-544a, by reducing CDH1 and AXIN2, induces EMT through activation of the Wnt pathway and the translocation of β-catenin into the nucleus; this miRNA is proposed as a prognostic marker and candidate as a therapeutic target in metastatic GC (Bure et al., 2019; Yanaka et al., 2015).
Among the miRNAs regulating EMT in GC, miR-376a is associated with advanced disease stages and poor prognosis. miR-381 acts as a tumor suppressor, which inhibits TMEM16A and the TGF-β pathway. miR-128 is downregulated in paclitaxel-resistant cells. Conversely, expression of miRNAs that increase E-cadherin can reverse EMT, reduce invasion, and increase sensitivity to apoptosis, suggesting their vital role in overcoming drug resistance (Bure et al., 2019).
HOX antisense intergenic RNA (HOTAIR) is one of the first lncRNAs identified as an inducer of cancer progression, including GC. By engaging the PRC2 complex, HOTAIR induces epigenetic silencing of miR-34, which activates HGF/c-MET/SNAIL cascades and drives EMT (Liu et al., 2015). Furthermore, HOTAIR promotes the methylation of histone H3K27 in the E-cadherin promoter region, thereby reducing its expression and promoting tumor invasion. Knockdown of HOTAIR reduces PRC2 activity, elevates acetylation, reduces H3K27 methylation, and suppresses EMT (Song et al., 2019).
The lncRNA XLOC_010235 is significantly upregulated in GC tissues and affects EMT and tumor progression by inhibiting SNAIL1. Amplification of XLOC_010235 leads to the elevation of N-cadherin, vimentin, and MMP2/MMP9, while decreasing E-cadherin levels (Bure et al., 2019). Similarly, the lncRNA ZFAS1 is upregulated in tissue, serum, and exosomes of GC patients and promotes EMT through ZEB1 activation and ZEB2 stabilization. Exosomes containing ZFAS1 contribute to metastasis. Inhibition of ZFAS1 reduces invasive markers and elevates E-cadherin levels (Pan et al., 2017; Zhou et al., 2016).
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is an oncogenic lncRNA, and its amplification in GC leads to the upregulation of E-cadherin and EMT modulation through N-cadherin, ZEB1, and SNAIL. FRLnc1 also promotes cell migration by activating FOXM1 and EMT genes such as TGFβ−1 and TWIST (Bure et al., 2019).
LINC00978 overexpression in GC correlates with cancer progression by promoting EMT. Inhibition of LINC00978 results in increased E-cadherin, suppression of EMT factors such as SLUG and TWIST1, decreased TGF-β/SMAD2, and decreased cell invasion and migration (Fu et al., 2018).
Urothelial carcinoma-associated 1 (UCA1) is amplified in GC and is associated with invasion and metastasis. UCA1 upregulation is observed in response to TGFβ−1. Suppression of UCA1 increases the levels of E-cadherin and ZO-1 and reduces vimentin and SNAIL levels, suggesting its role in promoting EMT (Zhang and Huang, 2018; Ramli et al., 2021).
Taurine upregulated gene 1 (TUG1) overexpression in GC is linked to tumor growth. This lncRNA interacts with the PRC2 complex and represses cell cycle inhibitors such as P21, P15, P16, and P57, leading to increased cell proliferation. TUG1 inhibition reduces GC cell proliferation in vitro and in vivo (Zhang et al., 2016).
LncRNAs commonly modulate this process through sponging EMT-regulating miRNAs. For example, Linc00152, by sponging miR-193b-3p and increasing ETS1 expression, inhibits cell proliferation, migration, and invasion in GC. Also, the lncRNA XIST, as a miR-101 sponge, modulates EZH2 expression in GC cells (Bure et al., 2019).
LncRNA-ATB is implicated in the EMT of GC via the TGF-β/miR-200/ZEB axis. Increased expression of lncRNA-ATB upregulates ZEB1, suppresses miR-200, and induces EMT. Downregulation of lncRNA-ATB upregulates E-cadherin and downregulates mesenchymal markers (ZEB1 and N-cadherin). LncRNA SNHG6 is also upregulated in GC and leads to invasion and metastasis. SNHG6 inactivation increases p21, inhibiting EMT by downregulating EZH2 and activating the c-Jun N-terminal kinases (JNK) pathway. Acting as a ceRNA, SNHG6 sponges miR-101-3p and enhances ZEB1 to increase invasive properties (Bure et al., 2019).
Microbiome Associations in GC According to TCGA
The understanding of the role of gastric microbiome in GC has evolved significantly. Initial research focused on markers such as the Boas–Oppler bacillus (Lactobacillus) and the nitrosamine hypothesis of the 1970s, which suggested that bacteria could convert nitrates into carcinogens. This concept was integrated into the Correa cascade model. However, the discovery of H. pylori in the 1980s fundamentally reshaped the field, establishing it as the primary driver of chronic inflammation and carcinogenesis that can lead to GC, largely superseding earlier theories (Engstrand and Graham, 2020).
While H. pylori is a key initiator, advanced culture-independent sequencing techniques have revealed that the stomach harbors a complex microbial community beyond a single pathogen. In healthy individuals, the stomach contains a relatively low microbial load dominated by phyla such as Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria. However, GC is associated with a profound dysbiosis. This state is often characterized by a marked reduction in overall microbial diversity and the enrichment of specific bacterial genera, including Clostridium, Fusobacterium, Lactobacillus, Prevotella, Streptococcus, and Veillonella, as well as oral taxa such as Dialister and Peptostreptococcus in the TME (Chattopadhyay et al., 2023).
A comprehensive study leveraging data from TCGA and Memorial Sloan Kettering Cancer Center cohorts provided significant insights into how these microbial alterations correlate with GC molecular subtypes. Microbial DNA reads were extracted from next-generation sequencing data and classified using Nucleotide Basic Local Alignment Search Tool (BLASTn). The analysis confirmed that GC tumors exhibit significantly lower microbial diversity compared with adjacent nonmalignant tissues. Across both cohorts, five bacterial genera including Helicobacter, Lactobacillus, Streptococcus, Prevotella, and Bacteroides were consistently enriched in tumor samples (Abate et al., 2022).
Strikingly, Abate et al. revealed distinct microbial signatures among the TCGA subtypes. The MSI-H subtype was characterized by a significant enrichment of Lactobacillus, with other genera such as Fusobacterium and Streptococcus also showing notable enrichment. This unique microbial pattern is thought to be linked to the distinct immune microenvironment of MSI-H tumors. In contrast, the GS and CIN subtypes did not show a clear or consistent microbial profile. The EBV-positive subtype was excluded from detailed analysis due to limited sample sizes (Abate et al., 2022).
The enrichment of specific bacteria, particularly lactic acid bacteria such as Lactobacillus and Streptococcus, points to their potential role in promoting carcinogenesis. Their protumorigenic mechanisms are believed to be multifaceted and include production of reactive oxygen species, which can lead to host DNA damage, reduction of dietary nitrate to mutagenic nitrite, a process that can foster angiogenesis, and secretion of lactate, which serves as an alternative energy source for tumor cells and may confer chemoresistance (Chattopadhyay et al., 2023).
Despite these compelling findings, the field faces significant methodological challenges that limit the comparability and interpretation of studies. Inconsistencies in sample collection, DNA extraction protocols, and bioinformatic analyses can lead to conflicting results. Some researchers argue that due to these limitations, there is not yet definitive evidence for the role of the non-H. pylori microbiome in carcinogenesis beyond the inflammatory effects of H. pylori itself (Engstrand and Graham, 2020). Therefore, standardizing methodologies is crucial for future research to build upon the groundwork laid by TCGA and other large-scale studies.
Finally, the paradigm has shifted from a singular focus on H. pylori to recognizing a complex dysbiosis in the GC TME. Large-scale analyses have identified reduced microbial diversity and the enrichment of specific genera, with a particularly unique microbial signature in the MSI-H subtype. While significant methodological hurdles remain, these findings highlight the potential of the non-H. pylori microbiome as a factor in carcinogenesis and lay the foundation for future research into its role across different GC subtypes.
Conclusion
The molecular classification provided by TCGA, which divides GC into four distinct subtypes, including EBVaGC, MSI-H, CIN, and GS, has offered significant insights into tumor biology and provided new strategies for precision medicine. Each subtype is characterized by unique genetic and epigenetic aspects, presents great opportunities and challenges for diagnosis, prognosis, and targeted therapy. Within these subtypes, ncRNAs, including miRNAs and lncRNAs, are emerging as crucial biomarkers and regulators in the pathogenesis of GC. In addition to influencing prognosis by regulating EMT, proliferation, invasion, and metastasis, ncRNAs also provide novel therapeutic targets. ncRNAs related to EBVaGC, such as EBERs and H19, promote tumor immunogenicity and improve response to immune checkpoint inhibitors (ICIs) such as pembrolizumab by modulating immune signaling and hypermethylation of TSGs. However, the variability in PD-L1 expression and lymphocyte infiltration indicates the need for complementary markers to refine therapeutic response prediction. In MSI-H-GC, ncRNAs such as MIR99AHG participate in ceRNA networks serving as prognostic and immunomodulatory markers through the regulation of immune-related gene expression. This subgroup shows a remarkable response to immunotherapy due to its high mutational burden; however, the heterogeneity of responses supports the demand for more comprehensive biomarker panels. CIN-GC, as the most common subtype, is characterized by ncRNAs such as miR-22 and TERRA, which exacerbate CIN and result in a poor prognosis. Nevertheless, overexpression of genes such as ERBB2 offers opportunities for targeted therapies such as trastuzumab. Finally, GS-GC is characterized by EMT features and ncRNAs such as HOTAIR, ZFAS1, and Linc00152, which enhance invasion and metastasis by the modulation of miRNAs involved in EMT. By engaging Transforming growth factor β (TGF-β) and Wnt pathways, these ncRNAs emerge as potential therapeutic targets to inhibit disease progression.
Recent progress in multi-omics technologies, including high-throughput sequencing and transcriptomic analysis, has facilitated the identification and validation of subtype-specific ncRNAs. Paired with bioinformatic analyses, these help develop robust biomarker panels that can improve early diagnosis, allow for more accurate subtype classification, and predict treatment response. However, challenges such as biomarker diversity, intratumoral heterogeneity, and the lack of standardization for ncRNAs remain barriers to clinical application. To overcome these barriers, future investigations should focus on unifying multi-omics data, validating biomarkers in diverse populations, and developing noninvasive diagnostic methods such as liquid biopsy. In addition, the involvement of ncRNAs in modulating drug resistance and the TME demonstrates their potential as innovative therapeutic targets. For instance, inhibition of lncRNAs such as HOTAIR or SNHG6 has the potential to reverse EMT and increase sensitivity to current therapies. Targeting oncogenic ncRNAs via gene editing technologies such as CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats) could also introduce novel therapeutic approaches. However, the intricate ncRNA regulatory networks and the possibility of off-target effects underscore the need for extensive preclinical studies to ensure the safety and efficacy of these interventions. The molecular classification by TCGA and the identification of subtype-specific ncRNAs have established a new paradigm in managing GC. These advances pave the way for developing personalized diagnostic and therapeutic strategies. Through the continued evolution of research and the integration of new technologies, ncRNAs are expected to become fundamental components of precision medicine in GC, helping to improve the prognosis and quality of life of patients. Investment in collaborative multicenter studies and international cooperation to standardize and validate these markers is vital to realizing this potential.
Authors’ Contributions
M.S. and A.S. contributed to the conception and design of the study; M.S., S.S., and O.D. contributed significantly to screening and data collection; M.S., F.B., O.D., and B.S. contributed significantly to article preparation; M.T. and A.S. contributed to supervising and final approval of the article. All authors read and approved the final article.
Footnotes
Funding Information
The authors did not receive a specific grant for this research from any funding agency in the public, commercial, or not-for-profit sectors.
Disclosure Statement
The authors declare that they have no conflicts of interest.
Confirmation Statement
All authors hereby affirm that the research presented in this article was supported by an institution that is exclusively involved in educational and research activities.