
Abstract
Cyclin-dependent kinase 17 (CDK17) is an understudied member of the PCTAIRE family of CDKs, with phosphorylation-guided molecular mechanism being underexplored. In this study, an in-depth mass spectrometry-based phosphoproteomics data integration and harmonization, coupled with replicable statistical analysis, was performed to understand the phosphorylation landscape of CDK17. High-confidence phosphorylation sites of CDK17 were derived from 711 phosphoproteomics profiling studies, where 176 datasets showed differential phosphorylation of CDK17. Among 13 identified phosphorylation sites of CDK17, S180, S137, and S146 were prominently detected in 75% of all the datasets. Notably, sequence conservation of CDK17 (S146, S137, and S180) with CDK16 (S119, S110, and S153) and CDK18 (S98, S89, and S132), respectively, was observed, where CDK16 (S119) is a part of the binding motif for multiple upstream kinases, 14-3-3 protein, and CCNYL1. Furthermore, conserved co-regulatory patterns of other proteins were identified as compared with CDK17 phosphorylation, which revealed 19 upstream kinases, 164 downstream substrates, and several interactors of CDK17, which conserved co-regulatory patterns across diverse biological contexts. Statistical analysis revealed phosphoregulation of CDK17 through other kinases, regulation of CDK17 substrates, protein–protein interactions, and conserved co-differential regulation in multiple datasets. Specifically, this analysis derived through global data integration with a replicable analytical framework lays a groundwork for experimental validation of CDK17 phosphorylation in its functional regulation.
Introduction
Phosphorylation is an important posttranslational modification as it has a central and dynamic role in a wide range of cellular processes, cell signaling, energy regulations, molecular switch function, protein stability-degradation, and therapeutic relevance (Ardito et al., 2017). Mass-spectrometry-based phosphoproteomics has enabled large-scale, unbiased identification and quantification of protein phosphorylation sites across biological samples to understand the cellular signaling and disease mechanisms and identify the potential drug molecule (Wu et al., 2023). Cyclin-dependent kinases (CDKs) are a family of serine/threonine protein kinases, with at least 20 members identified to date. They play essential roles in regulating cell cycle progression, transcription, and splicing (Lukasik et al., 2021). These kinases act within highly coordinated signaling cascades to ensure DNA replication and segregation during cell division. Among these, CDK17, also known as PCTAIRE2, has emerged as a functionally intriguing yet underexplored member of the PCTAIRE subfamily, alongside CDK16 and CDK18 (Dai et al., 2019). PCTAIRE family members are mainly characterized by having a conserved catalytic (CDK) domain flanked by N- and C-terminal extensions involved in cyclin binding (Mikolcevic et al., 2012a).
CDK17 is a 59.5 kDa protein comprising 523 amino acids, which is expressed predominantly in cytoplasmic/membranous structures of normal human tissues, with nuclear translocation observed particularly in cultured cells (Karimbayli et al., 2024). Interestingly, CDK17 is significantly present in the brains of individuals with Alzheimer’s disease and mild cognitive impairment, and it is strongly enriched in terminally differentiated neurons, including those of the olfactory bulbs and hippocampal regions (Hirose et al., 1997). In addition, CDK17 is found to be involved in the glycerophospholipid metabolism pathway, as per a genome-wide association study, indicating its wider involvement in metabolic regulations in the nervous system (Demirkan et al., 2012). Most members of CDKs bind to regulatory cyclins to be structurally activated, but a study states that PCTAIRE family kinases, specifically CDK16 and CDK17, could adopt an active conformation in the absence of cyclin binding and rely on phosphorylation within their N-terminal extensions (Karimbayli et al., 2024). CDK17 differs from other CDKs due to these distinct activation methods, which call for more research into their intricate structural and regulatory mechanisms.
Functionally, CDK17 is understudied and thought to have a possible role in key cellular processes, including DNA replication, repair, transcription, and neuronal survival through the phosphorylation of multiple substrate proteins (Karimbayli et al., 2024). Its downregulation, copy number deletions, and missense/frameshift mutations have been associated with low-grade glioma and glioblastoma (GBM), underscoring its potential role as a tumor suppressor in brain malignancies (Karimbayli et al., 2024). However, the structural underpinnings of this selectivity are still unknown, which poses a significant challenge for structure-function research and logical medication development. Interestingly, while CDK16 has been better characterized with respect to phosphorylation at S119 and S153 (sites that create binding motifs for 14-3-3 protein) (Mikolcevic et al., 2012b), the site-specific phosphorylation landscape of CDK17 remains largely unexplored. Upstream kinases are initial stimulus that are responsible for activating or modifying target substrates through phosphorylation events; they transfer phosphate group to target protein. Downstream substrates are the target proteins that are phosphorylated by the upstream kinases; these substrates undergo functional changes such as altered activity, stability, localization, or interaction (Gopalakrishnan et al., 2025; Wang and Gou, 2020). Limited number of studies explored the upstream kinases and downstream substrates of CDK17; given that phosphorylation governs kinase activity, subcellular localization, and substrate interactions, an in-depth phosphoproteomic characterization of CDK17 is crucial. This is particularly significant because atypical activation of the CDK17 mechanism and involvement in both neurodegenerative disease and brain cancer position it as a compelling target for phosphoproteomic studies.
Given the largely unknown role of CDK17 in the atypical CDK family with non-cyclin-dependent activation mechanisms and its connection to neurological disorders, gliomas, and GBMs (Liu et al., 2017), most of the studies to date have focused on its expression patterns, mutational landscape, and inhibitor sensitivity with limited focus toward phosphorylation landscape and its regulatory mechanisms, which are essential for cellular functions. In this study, we aimed to explore global phosphoproteomics landscape of CDK17 by identifying prominent phosphorylation sites, kinase substrate association, protein–protein interaction, and kinases and phosphatases that are coregulated with the prominent phosphorylation sites of CDK17. We believe that this study will be a road map for future studies that focus on biological functions, disease associations, and potential as a therapeutic target.
Methodology
Global assembly of phosphoproteomics datasets identifying phosphopeptides of CDK17
Publicly available human cellular phosphoproteomics data (generated through mass spectrometry platforms) were screened by performing a PubMed search (prompt: “phosphoproteomics” OR “phosphoproteome” NOT “Plant” NOT “Review”). These datasets were manually curated, and Class 1 phosphorylation sites were captured (localization probability ≥75% and/or ambiguity score (A-score) > 13). These datasets were further classified into profile datasets (containing all the detected phosphopeptides of CDK17 from phosphoproteomics profiling) and differential datasets (obtained by comparing specific biological or experimental conditions to their respective control) that show differential abundance of CDK17 phosphopeptides. Study-centric statistical parameters (example: p value < 0.05, fold change ± 1.3) were utilized to catalogue differentially regulated phosphopeptides of CDK17. In addition, co-differentially regulated phosphorylation events of other proteins with CDK17 were also catalogued. Furthermore, each protein was mapped to its corresponding HGNC gene symbol (downloaded on 30.05.2023) (Seal et al., 2023), and individual phosphoproteins were mapped to corresponding UniProt (UniProt, 2023) accessions to ensure uniform mapping (Raghu et al., 2025; Lubaba et al., 2025; Bangera et al., 2026). The annotated biological and experimental conditions were tagged to each dataset in a standard format for efficient categorization by following the previously demonstrated methodology (Sheela et al., 2025; Mahin et al., 2025; Sujina et al., 2025; Fahma et al., 2025; Chakraborty et al., 2025).
Analysis of prominent phosphorylation sites of CDK17
The harmonized phosphoproteomic datasets were compiled and analyzed further to rank the major phosphorylation sites of CDK17. Each of the detected phosphorylation sites was listed and ranked according to the frequency of detection (number of different studies in which the phosphorylation site was detected) in profile and differential datasets. The phosphorylation sites that were detected in >50% of the profiling and differential datasets were considered as prominent phosphorylation sites of CDK17. Importantly, the phosphorylation sites that were detected using mutation-based methods or specific phospho-antibodies, which were not commonly detected or classified as Class 1 in phosphoproteome datasets, were currently not included in this analysis.
Conservation of phosphorylation sites among the PCTAIRE kinases
The CDK family comprises 20 members. To identify conserved phosphorylation sites within this family, we first constructed the phylogenetic tree to understand the closest relatives of CDK17. We used the MUSCLE technique, which is a part of Molecular Evolutionary Genetics Analysis (MEGA version 12) (Kumar et al., 2024), to perform multiple sequence alignment. After the sequences were aligned, a phylogenetic tree was created using the Jones–Taylor–Thornton substitution model and the maximum likelihood technique. Thousand bootstrap repeats were used to assess the tree topology’s stability. The MEGA Tree Explorer was used to visualize and annotate the resultant tree. To further investigate site-specific conservation within CDK16, CDK17, and CDK18, we performed a multiple sequence alignment using Clustal Omega and visualized using ENDscript2 (Robert and Gouet, 2014). This analysis provides insights into understanding the conservation patterns among the PCTAIRE kinases (PCTKs) and their functional relevance.
Conserved co-differential regulation among phosphorylation sites of other proteins as compared with prominent phosphorylation sites of CDK17
To identify the protein phosphorylation sites that are either positively (similarly) or negatively (oppositely) co-regulated with the prominent phosphorylation sites of CDK17, differential datasets for each prominent phosphorylation site were classified separately. Due to considerable heterogeneity across datasets, including differences in experimental conditions, biological systems, and analytical methods, it was not practical to conduct a comprehensive reanalysis of raw data. The differential datasets were categorized based on the regulation status of prominent phosphorylation sites of CDK17 (denoted as “c”) and the associated co-regulated phosphorylation sites on the other proteins (denoted as “p”). Datasets where the CDK17 sites were upregulated were labeled as (Uc), while those where they were downregulated were labeled as (Dc). Within these, we identified phosphorylation sites that were upregulated or downregulated in association with CDK17 upregulation (UcUp and UcDp) and with CDK17 downregulation (DcUp and DcDp). Furthermore, the differential datasets that showed the above pattern of expression were quantified. The combination of expressions falling into the UcUp and DcDp groups was further considered positively co-regulated with CDK17, while those in the UcDp and DcUp groups were considered negatively co-regulated. Based on this, combinations of phosphorylation sites were reclassified into two categories for each CDK17 site: UcUpDcDp (UUDD) for positive co-regulation and UcDpDcUp (UDDU) for negative co-regulation. Also, instances in datasets where either or neither of the pair of co-regulated phosphorylation sites were differentially regulated were also catalogued as an additional layer to co-regulation interpretation. This calculation resulted in a 4-value contingency table that was used to perform a differential Fisher’s exact test (FET) to assess the likelihood of co-regulation between prominent sites of CDK17 and the co-regulating phosphorylation sites. This approach intentionally excluded adjustments for potential confounders like rare phosphorylation site occurrences or dataset redundancy for the same stimulus. Subsequently, combinations of phosphorylation sites with a p value < 0.05 in these UUDD and UDDU groups were regarded as statistically significant (therefore conserved across multiple experimental conditions) and were screened for subsequent analysis.
Additional filtering was done based on the ratio of the total number of occurrences in (UcUp and DcDp) compared with (UcDp and DcUp) for positively co-regulated phosphorylation sites, and vice versa for negatively co-regulated ones. Only those phosphorylation site combinations where this ratio represented at least 10% of the total occurrence frequency of the prominent CDK17 phosphorylation sites were considered significant. Finally, to enhance confidence, additional criteria were applied: selected phosphorylation sites needed to show co-regulation in at least three independent studies (with distinct PubMed IDs ≥3) and under a minimum of three different experimental conditions (code confidence ≥3). Phosphorylation sites meeting all four of these criteria were defined as high-confidence co-regulated phosphorylation sites on other proteins. These were subsequently examined for their involvement in biological processes, upstream kinase associations, and protein–protein interaction networks related to prominent phosphorylation sites of CDK17 (Priyanka et al., 2024). Similarly, co-occurrence analysis of CDK17 phosphorylation sites among each other was explored to distinguish mutual occurrence among different datasets. For each pair, we calculated UU, UD, DD, and DU frequencies and their positive co-regulation pattern using ∑ (nUU + nDD)/∑ (nUD + nDU) and negative co-regulation pattern using ∑ (nUD + nDU)/∑ (nUU + nDD) was identified. The co-occurrence of the CDK17 phosphorylation sites was carefully analyzed to exclude interpretation of the phosphorylation sites harbored on the same tryptic peptides (as they will be invariably co-regulated in multiple studies).
Protein and phosphorylation site-specific CDK17 interactors extraction
The experimentally known protein–protein interactors of CDK17 were extracted from HPRD (Keshava Prasad et al., 2009), BIND (Bader et al., 2003), BioGRID (Oughtred et al., 2021), and ConsensusPathDb release 35 (downloaded on 22.05.2023) (Kamburov and Herwig, 2022), CORUM (downloaded on 03.03.2023) (Tsitsiridis et al., 2023), and RegPhos 2.0 (downloaded on 24.05.2023) (Huang et al., 2014).
Assembly of experimentally validated and predicted kinases and substrates of CDK17
A collection of experimentally validated kinases targeting the prominent phosphorylation sites of CDK17, as well as the known downstream substrates of CDK17, along with the specific phosphorylation site they phosphorylate, was catalogued from databases including PhosphoSitePlus (downloaded on 22.05.2023) (Hornbeck et al., 2015), Phospho.ELM 9.0 (downloaded on 24.05.2023) (Dinkel et al., 2011), and RegPhos 2.0 (downloaded on 24.05.2023) (Huang et al., 2014). In addition, we retrieved predicted kinases and substrates of the specific CDK17 from multiple computational tools such as NetworKIN (downloaded on 04.01.2023) (Linding et al., 2008) and AKID (downloaded on 24.05.2023) (Parca et al., 2019), as well as from high-throughput kinase–substrate interaction data available in iKiP-DB (Mari et al., 2022). Since most of these tools require individual protein sequences as input, we used the RefSeq protein sequences for our queries, submitted via their respective APIs. Moreover, we also included all CDK17 substrates and kinases categorized by Johnson et al. (Yaron-Barir et al., 2024) identified through synthetic peptide library screening for kinome-wide substrate specificity profiling, using a stringent cutoff at the 90th percentile. These datasets were compiled for further analysis and compared with the co-regulation datasets to fetch corresponding (co-regulating) kinase–substrate interactions.
Gene enrichment analysis of conserved co-regulation events and data representation
Gene ontology (GO) and pathway enrichment analyses representing co-regulated phosphorylation events were conducted using g:Profiler to understand associated biological functions and pathways (Raudvere et al., 2019). The R/Bioconductor package trackViewer (10.18129/B9.bioc.trackViewer) was used to generate lollipop plots for data visualization. Cytoscape (version 3.10) (Shannon et al., 2003) and PathVisio 3 (Kutmon et al., 2015) were employed for interaction map visualization. Clinical significance of the phosphorylation sites was analyzed using the cProsite database (Wang et al., 2023). RAWGraph 2.0 was used to create dendrograms and bar charts generated using R packages such as dplyr, ggplot2, pheatmap, and tidyr for representing GO enrichment results. ENDscript (Robert and Gouet, 2014) server was used to represent the conservation of phosphorylation sites. The sequence coverage visualizer (Shao et al., 2023) tool was used to spot the prominent phosphorylation sites and their motif in the Alphafold (AF-Q00537-F1-v6) model of CDK17.
Results
Assembly of phosphoproteomics datasets exhibiting CDK17 phosphorylation and prominent phosphorylation sites of CDK17
A total of 3,825 global phosphoproteomics data sets generated from human cell lines were curated and assembled to study the phosphorylation landscape of CDK17. Subsequently, 711 profiling and 176 differential datasets, consisting of class 1 phosphorylation sites on CDK17 phosphopeptides, were extracted (Supplementary Tables S1 and S2). Profiling datasets analysis identified frequently occurring phosphorylation sites across diverse experimental and biological conditions. The phosphorylation sites detected in both profile datasets and differential datasets are shown in Figure 1A. Further attempt to identify key/prominent phosphorylation sites of CDK17 from profile and differential datasets by ranking the frequency of detection resulted in the following. In the profile datasets, among 26 phosphorylation sites detected, S137, S180, and S146 were detected with the frequency of 564, 530, and 352, respectively. Similarly, in differential datasets, S180, S137, and S146 phosphorylation sites were detected with a frequency of 86, 62, and 58, respectively. These sites are not localized on the protein kinase domain but are located in the close vicinity of it. Hence, it can be speculated that they are potentially involved in regulatory mechanisms. Although the complete tertiary structure of CDK17 has not yet been determined experimentally, comprehending its molecular mechanism is still a top research objective (Karimbayli et al., 2024). The AlphaFold predicted structural model (AF-Q00537-F1-v6) Figure 1B of CDK17 showed around 30% sequence coverage achieved through mass spectrometry-based phosphoproteomics experiments, along with the predicted aligned error plot to show the confidence of the model. The mean pLDDT score of 69.94, which is greater than the 70% confidence score. It is important to note here that a major part of the protein kinase domain is still uncharacterized through mass spectrometry, opening further avenues for targeted approaches that may uncover other underexplored phosphorylation events that represent prominent phosphoregulation of CDK17.
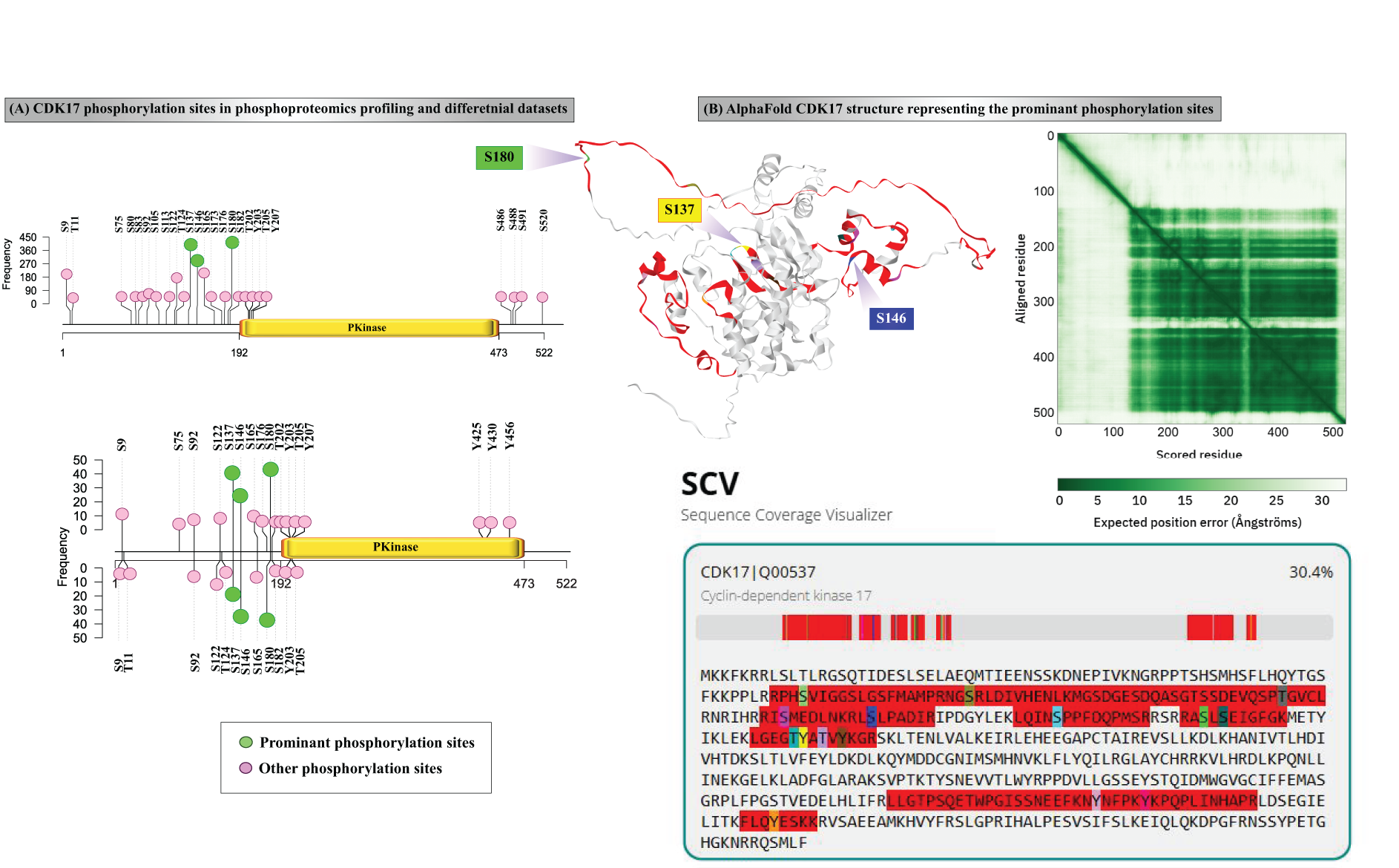
Phosphorylation landscape of CDK17.
Sequence and phosphorylation conservation among PCTKs
To identify conserved phosphorylation sites within the PCTAIRE family, we generated a phylogenetic tree through MEGA 12 tool. The tree shows the close relation of CDK16, CDK17, and CDK18 as they are the closest members within the PCTAIRE subfamily (Fig. 2A). Therefore, to further investigate on site-specific conservation within this group, we performed a multiple sequence alignment using Clustal Omega tool (Sievers and Higgins, 2021). The results showed all prominent phosphorylation sites of CDK17 (S146, S137, and S180) were conserved in CDK16 (S119, S110, and S153) and CDK18 (S98, S89, and S132), respectively (Fig. 2B). However, the site-specific functionality of S146, S137, and S180 of CDK17 is currently not well characterized. Interestingly, other phosphorylation sites of CDK17 including S122, S137 and S165 are also conserved with CDK16 and CDK18. Although we could not conclude the mechanistic function of CDK18 (S98, S89, and S132) phosphorylation, several leads were found for CDK16 (S119) functionality. CDK16 (S119) is a part of the binding motif for 14-3-3 protein, certain kinases, and Cyclin Y-like 1 (CCNYL1) (Zi et al., 2015). 14-3-3 proteins are a family of conserved regulatory molecules expressed in all eukaryotic cells (Fu et al., 2000) playing an important role in mitogenic signal transduction, apoptotic cell death, and cell cycle control (Fu et al., 2000). In addition, CDK16 (S119) phosphorylation is known to regulate cell cycle through CCNYL1 and kinase binding. The mechanical aspects and the connection between CDK16 (S119) and CDK17 (S146) need to be experimentally validated in future studies.
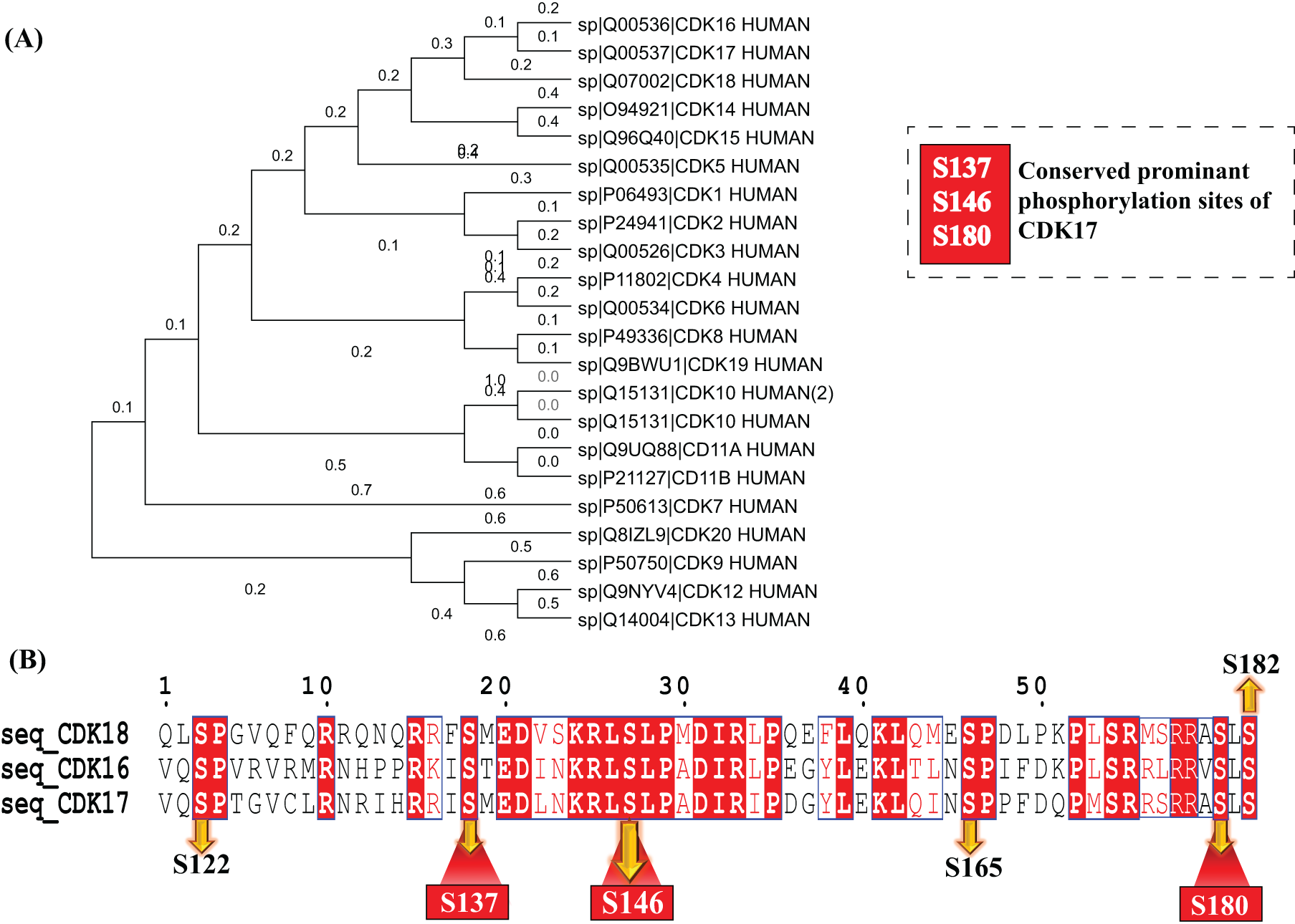
Site-specific conservation analysis of the CDK family.
Conserved co-differential regulation patterns in CDK17 phosphorylation
Furthermore, the positive (UUDD) and negative (UDDU) co-expression/co-regulation pattern of protein phosphorylation sites of other proteins with the prominent phosphorylation sites of CDK17 were compiled (Supplementary Tables S3–S8). This approach was used to comprehend the coregulation patterns (FET p value < 0.05) that may implicate signaling network modulated by CDK17. We identified 464, 837, and 308 protein phosphorylation sites that showed positive coregulation with S180, S137, and S146 of CDK17, respectively. In addition, 20, 150, and 56 protein phosphorylation sites showed negative coregulation with S180, S137, and S146 of CDK17, respectively. Interestingly, multiple proteins involved in cell cycle were found to show conserved coregulation with CDK17. Specifically, CDK17 (S180) showed positive coregulation with CDK16 (S153) and PARN (S557) (Balatsos et al., 2012) and negative coregulation with NES (S1347) and BRAF (T401) (Wang et al., 2021; Hernandez et al., 2016). In case of CDK17 (S137), positive coregulation was observed with DYNC1H1 (S4368), while negative coregulation featured WEE1 (T190) and RBL1 (S988) (Pan et al., 2021; Esposito et al., 2021; Ventura et al., 2021). Similarly, negative coregulation of CDK17 (S146) was observed with TRIM32 (S335) and CDC6 (S54) (Lazzari and Meroni, 2016; Donovan and Diffley, 1996). Collectively, these findings highlight that several coregulated phosphorylation sites associated with the prominent phosphorylation sites of CDK17 are directly linked to cell cycle control, supporting the potential functional significance of CDK17-mediated phosphorylation in cell cycle. Top 20 high-confidence protein phosphorylation sites that are positively and negatively co-regulated with the prominent phosphorylation sites of CDK17 are shown in Figure 3.

Top 20 co-differentially regulated protein phosphorylation site with the prominent phosphorylation sites of CDK17 that are positively and negatively correlated.
Furthermore, we observed that each of the prominent phosphorylation sites of CDK17 had a unique set of coregulated phosphoproteins, which prompted us to performed geneset enrichment analysis to understand significant biological functions modulated in site-specific manner. Coregulated proteins of CDK17 (S180) were enriched for unique biological functions, including regulation of cellular component size, regulation of organelle assembly, actin filament bundle organization, actin polymerization or depolymerization, regulation of DNA-templated transcription elongation, and regulation of alternative mRNA splicing via the spliceosome. Coregulated protein of CDK17 (S137) was enriched for cellular component biogenesis, cellular response to chemical stimulus, and phosphate-containing compound metabolic process. Similarly, coregulated proteins of CDK17 (S146) were enriched for cellular response to abiotic stimulus, endosomal transport, cellular response to environmental stimulus, and cellular response to osmotic stress (Supplementary Tables S9–S11). This analysis indicated the functional relevance of CDK17 in biological processes other than cell cycle, pointing to variable role of CDK17.
In addition to coregulation, we also analyzed the co-occurrence of CDK17 phosphorylation sites with each other. From the biological perspective, co-occurrence can show hyper/hypophosphorylation status of the proteins. From a mass spectrometry perspective, such analysis will hold ground only when we analyze phosphorylation sites on different phosphopeptides, as different sites on the same phosphopeptide are bound to co-occur. We considered these insights to analyze co-occurrence, which describes phosphorylation site propensity to undergo similar modifications (Li et al., 2017). Interestingly, two prominent phosphorylation sites, S137 and S180, commonly upregulated together with a frequency of 30 in UUDD data (Fig. 4A, Supplementary Table S12). When we looked into coregulating proteins common to these two co-occurring sites, 183 proteins were commonly co-regulated with the phosphorylation of both S137 and S180 (Fig. 4B). We performed geneset enrichment analysis with these proteins to identify their associated biological functions and pathways. Interestingly, geneset enrichment analysis showed 35 proteins specifically involved in cell cycle (Fig. 4C), further confirming the synergy in coregulation patterns directed toward cell cycle. Also, the experimental conditions in which such co-occurrence was observed showed that S137 and S180 were collectively upregulated in various cancer cell lines including GBM, neuroblastoma, cancers of lung, skin, ovary, pancreas, and breast. Therefore, we analyzed the abundance of these phosphorylation events in TCGA phosphoproteomics data through cProSite database (Wang et al., 2023). The database compared protein abundance and phosphorylation levels between cancerous and normal tissues. Although protein abundance of CDK17 remained relatively consistent across different tumor types, the prominent phosphorylation sites S137 and S180 displayed a similar regulation pattern across various cancer types (Fig. 4D). Especially due to evidenced disruption of cell cycle regulation in multiple neoplasms, this observation suggests these sites may exhibit comparable expression trends during cancer progression and potentially share related functional roles.

Co-occurrence of CDK17 phosphorylation sites.
Upstream kinases and downstream substrates of CDK17 showing co-regulation pattern
Phosphorylation is guided by kinases and phosphatases, which modulate the hyper/hypophosphorylation status of a protein. Since CDK17 itself is a kinase, it phosphorylates several downstream substrates to elicit specific signaling pathways. Among the co-regulated proteins, several upstream kinases of CDK17 were found to be positively or negatively coregulated (Fig. 5). CDK17 (S180) phosphorylation was positively coregulated with eight upstream kinases and 40 downstream substrates and negatively co-regulated with three downstream substrates. Similarly, CDK17 (S137) phosphorylation positively co-regulated with 13 upstream kinases and 64 downstream substrates, while negatively co-regulating with seven upstream kinases and 38 downstream substrates. In the case of CDK17 (S146), its phosphorylation positively co-regulated with four upstream kinases and 41 downstream substrates and negatively co-regulated with 10 downstream substrates (Supplementary Tables S13 and S14). Although coregulation events do not directly indicate potential inhibitory or inverse relationships in phosphorylation status, they suggest coordinated regulation of kinases/substrates functioning together within shared signaling pathways or biological processes. Some of the upstream kinases and substrates indicated in this study have been experimentally validated. However, most of the other interactions are semiexperimental or predictive in nature, creating scope for further experimental validation.

Upstream kinases and downstream substrates of CDK17.
Conserved co-regulation of CDK17 with its binary interactors
Our analysis also compiled the protein–protein interactors associated with the prominent phosphorylation sites of CDK17 (Fig. 6A, Supplementary Table S15). Specifically, CDK17 (S180) phosphorylation demonstrated positive co-regulation with CDK16 (S153), as well as with LARP7 (S258, S298, and T257). Similarly, the CDK17 (S137) phosphorylation exhibited positive co-regulation with multiple proteins, including LARP7 (S258), GAPDH (S210), CDK16 (S110), HDGFL3 (S121), EPHA2 (S775), and PRKACA (T196). Furthermore, the CDK17 (S146) phosphorylation showed positive co-regulation with eight distinct proteins. Among these, CDKN1B (S10) site is particularly noteworthy, as it has been previously reported to play a critical role in cell cycle regulation (Lin et al., 2019). Although protein–protein interaction is primarily established through direct interactions, validating some of the phosphorylation level interactions would be beneficial in the future for further elucidating the specific CDK17 interactions relevant to its role in cell cycle.
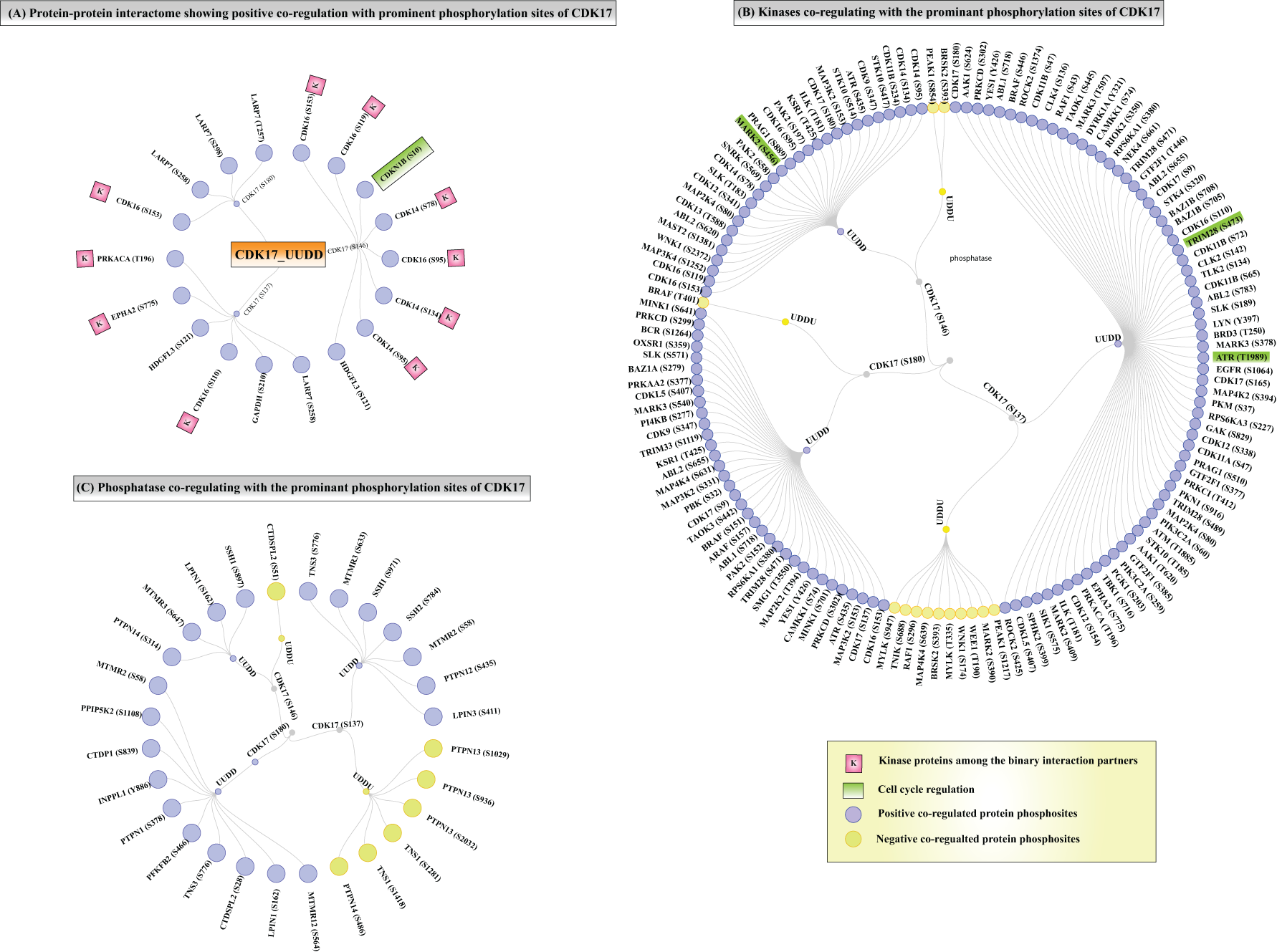
Binary interactors, subsidiary kinases, and substrates co-regulated with CDK17.
Subsidiary kinases and phosphatases showing conserved co-regulation with CDK17 phosphorylation
Multiple other kinases and phosphatases (that do not directly mediate CDK17 phosphorylation) were also found to be co-regulated with CDK17 differential phosphorylation (Fig. 6B). Specifically, CDK17 (S180) phosphorylation exhibited positive co-regulation with 35 kinases and negative co-regulation with BRAF (T401). CDK17 (S137) phosphorylation demonstrated positive co-regulation with 67 kinases and negative co-regulation with 10 kinases. The CDK17 (S146) phosphorylation displayed positive co-regulation with 28 kinases, while it was negatively co-regulated with PEAK1 (S854) and BRSK2 (S393) (Supplementary Table S16). Notably, TRIM28 phosphorylation (S473) has been previously implicated in cell cycle regulation (Krischuns et al., 2018; Chang et al., 2008). In addition, ATR phosphorylation at T1989 is known as an inhibitory modifier in cell cycle progression (Blackford and Jackson, 2017; Goodarzi et al., 2003), while MARK2 has been reported to mediate cell cycle arrest (Natarajan et al., 2022; Ponnusamy et al., 2022). These findings provide valuable insights into the potential regulatory role of CDK17 phosphorylation in controlling cell-cycle-associated signaling networks. Among phosphatases that showed conserved co-regulation with CDK17 phosphorylation (Fig. 6C), CDK17 (S180) phosphorylation exhibited positive co-regulation with 10 phosphatases; CDK17 (S137) phosphorylation showed negative co-regulation with 3 phosphatases and positive co-regulation with 7 phosphatases. Similarly, CDK17 (S146) phosphorylation was negatively co-regulated with CTDSPL2 (S51) and positively co-regulated with four phosphatases. These findings suggest a potential involvement of phosphatase-mediated regulatory mechanisms in modulating CDK17 phosphorylation.
Discussion
CDKs are key regulators of critical cellular functions like the cell cycle, transcription, and metabolism, with their activity controlled by cyclins, upstream kinases, and phosphatases (Lukasik et al., 2021). Within this family, PCTKs form a conserved but less understood subgroup, characterized by a core catalytic domain and flanking regions that aid in cyclin binding (Dixon-Clarke et al., 2017). Despite its inclusion in the CDK family, CDK17 remains largely understudied. The molecular mechanisms, mutation-based studies, and disease association of CDK17 are not well reported, with its crystallographic structure yet to be derived. As CDK17 shares evolutionary conservation with other members of the PCTAIRE family, site-specific conservation is observed among the members, with S146, S137, and S180 being the prominent and conserved sites. However, co-regulation of possible regulatory network guiding CDK17 phospho/dephosphorylation is currently underexplored. Given this knowledge gap, we undertook a phosphoproteomics-centric approach to explore the phosphoregulatory network of CDK17. Traditional biochemical assays are limited in capturing the global co-regulation dynamics of phosphorylation sites; thus, we utilized a statistical framework to construct co-regulation patterns using large-scale phosphoproteome dataset integration and harmonization to uncover novel insights into CDK17 functionality. We identified S180, S137, and S146 as prominent phosphorylation sites in CDK17. Interestingly, S180 and S137, though located on different phosphopeptides, frequently co-occur across various biological experiments, and their expression patterns were found to be similar across multiple cancers. This suggests potential co-regulatory functions and involvement in common cellular pathways. Also, sequence conservation of these prominent phosphorylation sites [CDK17 (S137) with CDK18 (S89) and CDK16 (S110)] is in the β-helix region, while CDK17 (S180) [conserved with CDK16 (S153) and CDK18 (S132)] implicates the scope for the homology modeling and functional analysis.
Through our predictive co-regulation model, we identified a total of 19 upstream kinases and 164 downstream substrates associated with prominent phosphorylation sites of CDK17. Among these, kinases such as BRSK2 (Li et al., 2012; Zhou et al., 2012) and MKI67 (Taylor et al., 2010) are known to be involved in cell cycle regulation, reinforcing the likelihood that CDK17 participates in similar biological processes. Among downstream targets, CDK17 could potentially phosphorylate 185 phosphorylation sites on 164 substrates. For instance, TACC3 plays spatiotemporal roles throughout the cell cycle and has been identified as a candidate cancer biomarker due to its involvement in oncogenesis and therapeutic potential upon inhibition (Saatci and Sahin, 2023). PRC1 affects tumor cell proliferation and migration by modulating F-actin, a critical polymer involved in cell adhesion, migration, apoptosis, and invasive growth (Liang et al., 2019). Similarly, WEE1 is a well-known kinase crucial for DNA damage checkpoints and cell cycle control, especially in cancer (Ghelli Luserna di Rora et al., 2020). CP110, which regulates centrosome duplication and separation, has also been shown to be phosphorylated by CDKs both in vitro and in vivo, indicating potential substrate overlap and mechanistic similarity with CDK17 (Chen et al., 2002). Gene enrichment analysis also revealed multiple co-regulated protein enriched across multiple biological processes including RNA metabolism, transport, ubiquitination, and signaling, reflecting broad regulatory potential rather than cell cycle exclusivity. In addition to substrate prediction, we identified 144 kinases and 28 phosphatases that coregulate with CDK17 phosphorylation. This broad interaction network suggests a complex and tightly controlled regulatory environment. Furthermore, we catalogued eight binary interactors of CDK17, where S180, S137, and S146 interacted with two, six, and four proteins, respectively, highlighting the potential of CDK17 as a molecular scaffold for signaling pathways.
While our findings shed light on the previously unexplored phospho-signaling network of CDK17, they also underline certain limitations. Most notably, the absence of a solved crystal structure for CDK17 prevents a detailed understanding of how phosphorylation might alter its conformation or interactions. Resolving the three-dimensional structure of CDK17, particularly in its phosphorylated state, would offer critical insights into its regulation, substrate specificity, and therapeutic targeting. In addition, it is worth noting that related CDKs, such as CDK7, require phosphorylation on T170 to form a stable dimer with Cyclin H and transition cells into the S phase by activating CDK2/Cyclin E and CDK1/Cyclin B complexes (Lukasik et al., 2021). CDK7 phosphorylation on S164, although not essential for cell cycle progression, has been shown to favor its role in transcriptional regulation (Martinez et al., 1997). Furthermore, CDK7 is implicated in DNA repair processes, reinforcing the versatility of CDKs in maintaining genomic stability. Drawing parallels, CDK17 may also possess multifunctional roles that are yet to be revealed.
Conclusions
In conclusion, our study represents the first phosphorylation-centric comprehensive analysis of CDK17 regulation, providing foundational hypothesis-generating insights into its signaling landscape. The co-regulatory network of kinases, phosphatases, substrates, and interacting proteins identified here suggests potential involvement in critical biological pathways such as the cell cycle, apoptosis, and cancer, positioning CDK17 as a candidate for future therapeutic exploration with the pending experimental validation. Ultimately, unraveling the dynamics of CDK17 phosphorylation may contribute substantially to our understanding of apoptosis-related disorders, cancer biology, and targeted therapy development. This approach is extendable to multiple other understudied proteins and experimental platforms to draw meaningful attention to mechanisms that can otherwise be difficult to elucidate.
Associated data
All data are available along with the article and in its supplementary files.
Authors’ Contributions
A.B.: Conceptualization, formal analysis, investigation, methodology, visualization, writing—original draft, and writing—review and editing; F.L.: Writing—review and editing; A.P.G.: Data curation; P.B.S.: Software; R.R.: Conceptualization, formal analysis, and methodology; P.R.: Investigation, supervision, validation, and writing—review and editing.
Footnotes
Acknowledgments
The authors acknowledge Yenepoya (deemed to be) University, Mangalore, for providing infrastructure for the Center for Integrative Omics Data Science.
Disclosure Statement
The authors declare no competing interests. The research was conducted without any commercial or financial involvement that could influence the outcomes or interpretation of the data.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.