
Abstract
This study aims to investigate the expression of the SLC25 subfamily in sepsis-associated acute kidney injury (SA-AKI) and the role of SLC25A30 in regulating PINK1/PARKIN-mediated mitophagy. Transcriptome sequencing of renal tissues from lipopolysaccharide (LPS)-induced SA-AKI rats at multiple time points revealed time-dependent differential expression of SLC25 genes. At 12 h post-LPS injection (renal injury peak), 11 differentially expressed genes were identified. Intersection with Gene Expression Omnibus datasets and Gene Ontology enrichment highlighted 11 codifferentially expressed genes enriched in mitochondrial transmembrane transport. Notably, SLC25A30 was significantly negatively correlated with KIM-1 (r = –0.96) and LCN2 (r = –0.98). SLC25A30 was significantly downregulated in SA-AKI rat renal tissues and LPS-induced HK-2 cells, accompanied by upregulated PINK1/PARKIN, excessive mitophagy (elevated LC3B-II, decreased p62), and increased renal injury markers. SLC25A30 overexpression inhibited PINK1/PARKIN, reversed excessive mitophagy, reduced KIM-1 and LCN2 levels, alleviated mitochondrial dysfunction, enhanced cell viability, and exerted cytoprotective effects. PINK1 knockdown attenuated the regulatory effect of SLC25A30 on excessive mitophagy, indicating a dependence on the PINK1/PARKIN pathway. In conclusion, downregulated SLC25A30 is closely associated with excessive mitophagy in SA-AKI. SLC25A30 overexpression inhibits excessive mitophagy via downregulating the PINK1/PARKIN pathway, improves mitochondrial function, and alleviates HK-2 cell injury, suggesting that SLC25A30 may be a novel molecular target for SA-AKI-targeted therapy.
Introduction
The definition of acute kidney injury (AKI) is the rapid decline or even loss of kidney function, which is a common heterogeneous disease. AKI has various etiologies, with serious infections and sepsis being the most common causes. Sepsis represents a life-threatening organ dysfunction arising from a dysregulated host response to infection (Poston and Koyner, 2019; Wang et al., 2018b). Approximately 70% of septic patients develop AKI. Sepsis-associated AKI (SA-AKI) has a very high incidence rate and mortality and represents a super general class of AKI encountered in the intensive care unit (Ostermann et al., 2025; Legrand et al., 2024). Increasing evidence indicates that the pathogenesis of SA-AKI is multifactorial nature and complex, involving inflammation, microcirculatory dysfunction, mitochondrial dysfunction, and metabolic reprogramming (Peerapornratana et al., 2019; Bellomo et al., 2017; Lv et al., 2024). Although current studies have made some progress, the pathophysiological mechanism underlying SA-AKI has not yet been completely validated. Further in-depth investigations are warranted to explore the pathogenic mechanism of the trait and identify candidate therapeutic strategies.
Mitophagy is a crucial mitochondrial quality control (QC) mechanism that identifies and removes senescent or damaged mitochondria. Current studies indicate that the major mitophagy pathways include the PINK1/PARKIN signaling pathway and several other receptor-mediated pathways. The beneficial effect of mitophagy mediated by these distinct pathways in AKI has been confirmed; specifically, PINK1/PARKIN-dependent mitophagy fulfills an essential function in mitochondrial QC and renal function during cisplatin toxicity or sepsis-induced AKI (Wang et al., 2018a, 2021). Furthermore, BNIP3-mediated mitophagy confers protection against ischemia-reperfusion-induced AKI (Tang et al., 2019). Mitophagy also contributes to the pathophysiological mechanism of SA-AKI (Li et al., 2024b; Sun et al., 2019). Targeted activation of mitophagy to preserve and restore mitochondrial function may provide an effective therapeutic strategy for AKI.
Solute carrier (SLC) transporters constitute a vast superfamily of transporters, encompassing over 400 proteins organized into 52 subfamilies. SLC transporters mediate the translocation of diverse substrates across cellular membranes and are vital to numerous physiological processes (Zhang et al., 2019; Nigam, 2015). With the widespread clinical application of drugs targeting SLC transporters (Lin et al., 2015), burgeoning research is now directed toward their involvement in certain diseases. The mitochondrial carrier protein family (SLC25) comprises 53 members and is the largest SLC transporter subfamily in humans. These proteins can transport various compounds across the mitochondrial inner membrane and modulate mitochondrial physiological processes, including small molecule transport, mitochondrial dynamics, and apoptosis. Consequently, the dysfunction of SLC25 transporters shows an association with the progression of a growing number of diseases (Kunji et al., 2020; Ruprecht and Kunji, 2020; Gorgoglione et al., 2019). However, few studies have investigated the role of SLC25 in SA-AKI. Given that the kidney is exceptionally rich in mitochondria and that mitochondrial dysfunction plays a critical role in the pathogenesis of SA-AKI, we propose that SLC25 may help alleviate mitochondrial dysfunction and promote recovery from SA-AKI, potentially through the regulation of mitophagy.
In this study, we aimed to investigate the mechanisms by which SLC25A30 contributes to SA-AKI, with the goal of identifying potential therapeutic targets. Using bioinformatics analysis, reverse transcription quantitative polymerase chain reaction (RT-qPCR), Western blotting, and other approaches, we verified the regulatory role of SLC25A30 in mitophagy using both in vivo and in vitro models. Our findings may provide insights into novel therapeutic strategies for SA-AKI.
Materials and Methods
Ethics statement
This study was conducted in strict accordance with the guidelines of the Ethics Committee of the First Affiliated Hospital of Harbin Medical University. All animal experimental protocols were approved by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University (Approval No.: YS296) and complied with the national and institutional guidelines for the care and use of laboratory animals. All efforts were made to minimize animal suffering and the number of animals used.
Data acquisition of transcriptome sequencing
Animal models and transcriptome analysis: Kidney tissues were harvested from rats with lipopolysaccharide (LPS)-induced septic AKI at 3, 6, 12, 24, and 48 h after intraperitoneal injection in our previous study. RNA sequencing (RNA-seq) was performed to obtain genome-wide transcriptomic profiles. In addition, a transcriptome dataset of renal tissues from rats subjected to cecal ligation and puncture (CLP) was obtained from the Gene Expression Omnibus (GEO) database (accession number: GSE256430; https://www.ncbi.nlm.nih.gov/geo/). The robust multiarray averaging method was then used to preprocess the raw data. Probes were annotated with the latest official annotations file. All bioinformatics analyses were processed and analyzed by RStudio and GraphPad Prism (8.0.1).
Establishment of septic AKI models
Male Wistar rats weighing 180–220 g (aged 8–9 weeks) were sourced from the Experimental Animal Center of Harbin Medical University, Heilongjiang Province, China. All rats were individually maintained in cages under controlled temperature conditions (21–25°C) to access standard food and water for 1 week. The SA-AKI model was induced by intraperitoneal injection of LPS. Rats were randomized to control and LPS groups (n = 4/group, repeat three times, 24 rats in total). Control and LPS group rats received intraperitoneal injections of sterile 0.9% saline (600 μL) and LPS (10 mg/kg), respectively. Twelve hours after LPS injection, the rats received i.p. anesthesia with 3% pentobarbital sodium (CAS: 57-33-0, Sigma-Aldrich; USA, 0.2 mL/100 g body weight). Blood and renal tissue were collected for further analysis.
Evaluation of renal function
Serum creatinine (SCr) and blood urea nitrogen (BUN) were analyzed by an automatic biochemical analyzer (Vitros V5600, Johnson, USA). A 1.5-fold increase in SCr compared with the control group demonstrated a successful establishment of the SA-AKI model.
Hematoxylin and eosin staining
Renal tissues from rats were fixed with 4% paraformaldehyde for 24 h, embedded in paraffin, and sectioned at 4-μm thickness per sample. Hematoxylin–eosin (H&E) staining was performed on kidney sections following standardized procedures to observe renal pathological injury.
Immunofluorescence staining
Paraffin-embedded kidney sections were fixed in paraformaldehyde, blocked, and sequentially incubated with primary and secondary antibodies. After incubation, the sections were mounted, dried, and protected from light, and immunofluorescence images were captured using a fluorescence microscope. The results were analyzed using ImageJ software.
Cell culture and transfection
HK-2 cells were sourced from BeNa Biology (Henan Province, China). Cells were cultured in DMEM supplemented with 10% (Fetal Bovine Serum [FBS]; Procell) and 1% dual antibiotics (Seven), under 37°C/5% CO2/95% air conditions. When the cell density in the culture flask reached 70–80%, the cells were separated and passaged for subsequent experimental treatments, and the cell passage counts did not exceed 20 passages. To evaluate the SA-AKI cell model and induce mitophagy in HK-2 cells, the cells were treated with 20 μg/mL LPS (Cat.: L8880, Solarbio).
HK-2 cells in the logarithmic growth phase were seeded into 6-well plates and cultured until reaching ∼80% confluence prior to transfection. For siRNA transfection, 200 μL of serum-free medium was mixed with 8 μL of GP-transfect-Mate transfection reagent in a sterile 1.5 mL microcentrifuge tube, gently pipetted, and incubated at room temperature for 5 min. In a separate tube, 200 μL of serum-free medium was combined with 15 μL of RNA oligonucleotide (RNA oligo), gently pipetted, and incubated at room temperature for 5 min. The transfection reagent mixture was then added dropwise to the RNA oligo mixture, gently homogenized by pipetting, and incubated at room temperature for an additional 15 min. For plasmid transfection, 200 μL of serum-free medium was mixed with 6 μL of GP-transfect-Mate transfection reagent in a sterile 1.5 mL microcentrifuge tube, gently pipetted, and incubated at room temperature for 5 min. In a separate tube, 200 μL of serum-free medium was combined with 2 μg of pcDNA3.1-SLC25A30 overexpression plasmid or empty vector control plasmid, gently pipetted, and incubated at room temperature for 5 min. The transfection reagent mixture was subsequently added dropwise to the plasmid mixture, gently homogenized by pipetting, and incubated at room temperature for 15 min. Following the preparation of the transfection complexes, the original culture medium in each well of the 6-well plates was aspirated and replaced with 1.6 mL of fresh culture medium. Subsequently, 400 μL of the prepared transfection complex was added dropwise to each well, resulting in a final volume of 2 mL per well. The plate was gently swirled to ensure even distribution of the transfection complexes and then incubated at 37°C in a humidified atmosphere containing 5% CO2. Subsequently, HK-2 cells were treated with 20 μg/mL LPS for 12 h, before obtaining total RNA and protein for next-step experiments.
Cell viability assay (CCK-8 assay)
HK-2 cells in the logarithmic growth phase were seeded into 96-well plates at a final volume of 100 μL per well and cultured in a cell incubator at 37°C with 5% CO2 for 24 h to allow cell adherence. After complete attachment, the cells were treated accordingly based on experimental grouping. Following incubation, 10 μL of CCK-8 (Cell Counting Kit-8) reagent was added to each well. The plate was gently mixed by shaking and incubated in the dark at 37°C with 5% CO2 for 2 h. After incubation, the absorbance of each well was measured at a wavelength of 450 nm using a microplate reader.
Western blot analysis
Renal tissues and HK-2 cells were lysed in RIPA buffer on ice for 0.5 h. The extracted protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (12% gel electrophoresis) and transferred onto PVDF membranes, which were blocked in 5% skim milk at room temperature for 1 h, followed by incubation with primary antibodies overnight at 4°C. The following primary antibodies were used: SLC25A30 (bs-21228R; 1:500; Bioss, China), PINK1 (WL04963; 1:1000; Wanlei, China), PARKIN (WL02512; 1:1000; Wanlei, China), β-actin (WL01372: 1:1000; Wanlei, China), p62 (CY5546; 1:1000; Abways, China), KIM-1 (CY8586; 1:1000; Abways, China), LCN2 (CY6730; 1:1000; Abways, China) and LC3B (T559925; 1:1000; Abmart, China). After three washes with PBST (10 min per wash), the PVDF membranes were incubated with secondary antibodies (goat antirabbit IgG [RS030229; 1:10,000; Immunoway, USA] and goat antimouse IgG [S0002; 1:10,000; Affinity, USA]) at RT and cleaned again three times. Protein bands were visualized by treating the PVDF membranes with chemiluminescent reagents and using a chemiluminometer. Quantitative analysis was performed using the ImageJ software (version 1.45s; National Institutes of Health, Bethesda, MA, USA).
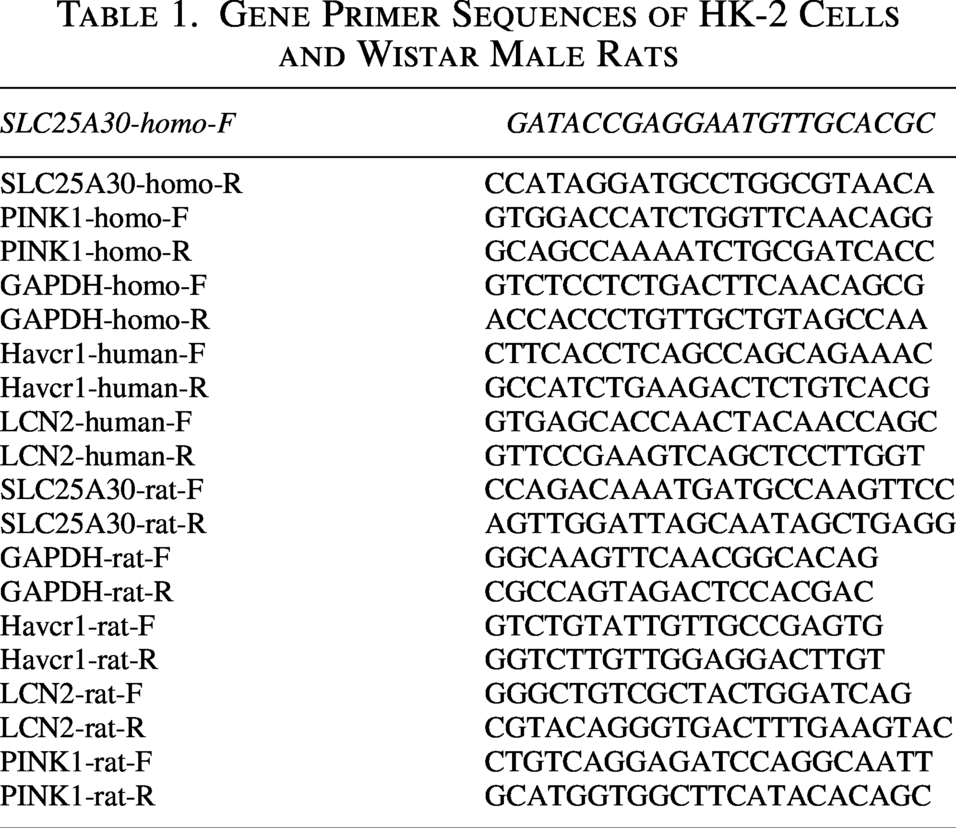
RT-qPCR detection
Total RNA was extracted from renal tissues and HK-2 cells using the SevenFast® Total RNA Extraction kit. RNA was reverse transcribed into cDNA using 5× PrimeScript RT Master Mix reagent. RT-qPCR was employed using 2× SYBR Green qPCR Master Mix II reagent under preset reaction conditions. Primer sequences are shown in Table 1. Primers were designed using Primer 5 software, with reference to the NCBI online primer design tool (Primer-BLAST, http://www.ncbi.nlm.nih.gov/tools/primer-blast) and NCBI–Basic Local Alignment Search Tool (BLAST) for primer design and sequence alignment. All primers were synthesized by Saiwen Chuangxin Biotechnology Co., Ltd (Beijing, China). The 2−△△ct method was used to quantify the normalized mRNA expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Gene Primer Sequences of HK-2 Cells and Wistar Male Rats
Mitochondrial reactive oxygen species detection
The 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) probe was diluted via serum-free medium before adding to cells and incubating in a cell culture incubator. The cells were then cleaned three times to remove DCFH-DA that did not enter the cells. Fluorescence intensity was explored using a microplate reader at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
Mitochondrial membrane potential detection
After cell culture and treatment, the old medium was discarded, and the cells were rinsed twice with sterile PBS. Subsequently, 500 μL cell culture medium was added to every sample. According to the manufacturer’s instructions, 500 μL prepared JC-1 staining working solution was added and incubated for 20 min in a cell culture incubator. The supernatant was discarded after centrifugation, and cells were cleaned twice with JC-1 staining buffer. Finally, the cells were resuspended in 500 μL JC-L staining buffer and detected using a flow cytometer.
Transmission electron microscopy
Samples were fixed for 2–4 h after collection and processed following standard procedures, including dehydration, infiltration, embedding, and staining. The ultrastructure of HK-2 cells was observed using transmission electron microscopy (TEM) (JEOL, JEM1400).
Data analysis
Data are denoted by the mean ± standard error of the mean. Comparisons between two groups were performed using Student’s t-test. Multigroup comparisons were analyzed by one-way analysis of variance followed by Tukey’s post hoc test. Statistical analysis was performed using GraphPad Prism 10.1.2 software. A p value <0.05 was considered statistically significant.
Results
Temporal differential expression of SLC25 subfamily genes in sepsis-associated AKI
First, RNA-seq was performed on renal tissues from rat models of SA-AKI at different time points (3 h/6 h/12 h/24 h/48 h), and differential gene expression analysis was conducted for the SLC25 subfamily in the transcriptome sequencing data. The results are shown in Fig. 1A–E. In the LPS 3-h group, 4 upregulated genes were identified in the SLC25 subfamily; in the LPS 6-h group, 5 upregulated and 10 downregulated genes were screened; in the LPS 12-h group, 2 upregulated and 9 downregulated genes were identified; in the LPS 24-h group, 2 upregulated and 2 downregulated genes were screened; and in the LPS 48-h group, 1 upregulated and 1 downregulated gene was identified. According to previous studies in our research group, renal injury in LPS-induced SA-AKI rats peaked at 12 h (Lv et al., 2023). Therefore, this time point was selected for subsequent analysis and validation.
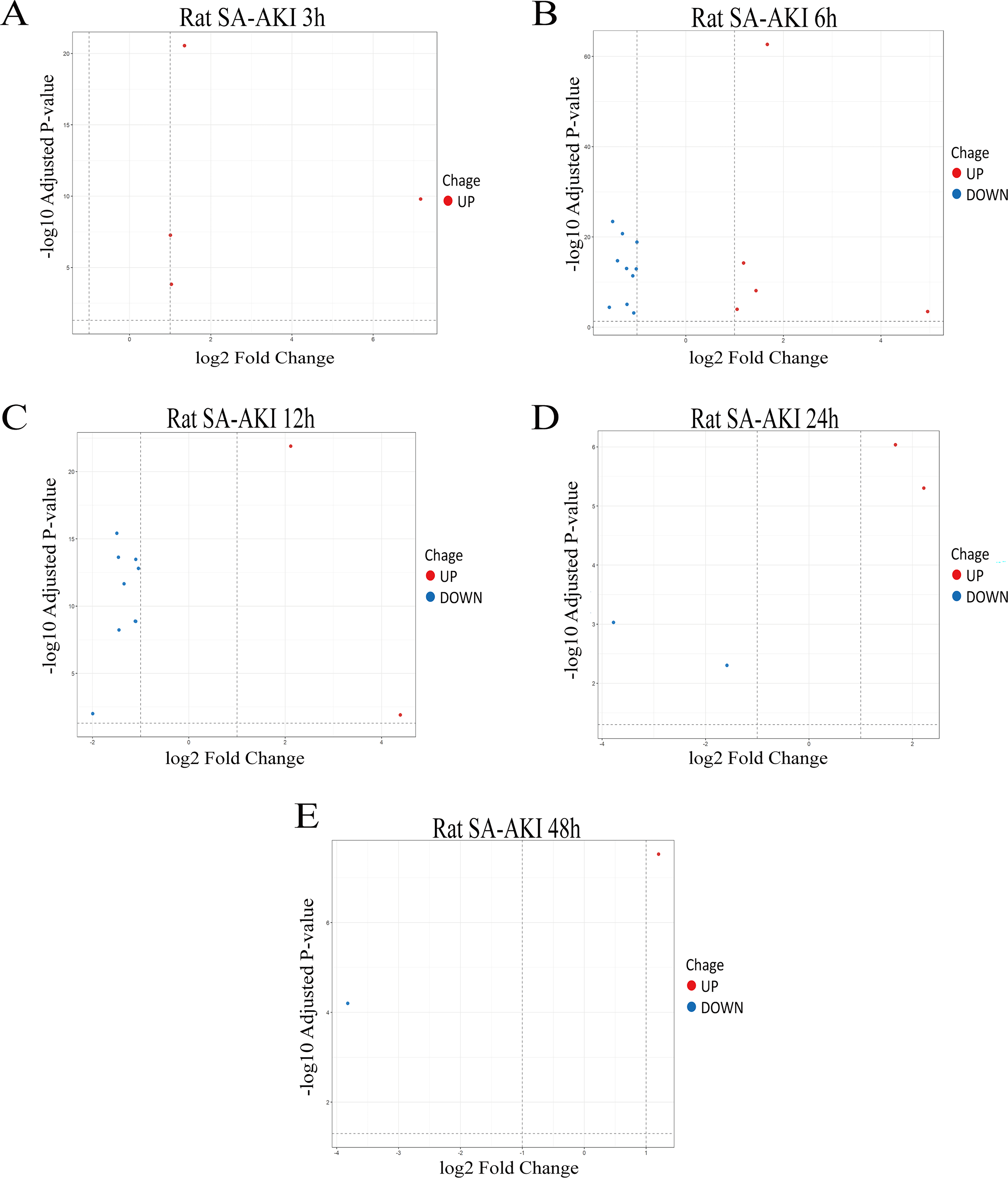
Temporal differential expression of genes in the SLC25 subfamily in SA-AKI.
Identification of the key gene SLC25A30 by intersection analysis of differentially expressed genes in the LPS 12-h group and the GEO public dataset
Intersection analysis was performed between the differentially expressed genes in the LPS 12-h group and the GEO public dataset (GSE256430, mouse model of CLP-induced SA-AKI). A total of 11 common genes that were differentially expressed in both SA-AKI models were identified (Fig. 2A,B). Subsequently, Gene Ontology enrichment analysis of these differentially expressed genes was conducted using the online tool Metascape. The results showed that the enriched terms included mitochondrial transmembrane transport and mitochondrial transport (Fig. 2C). Pearson correlation analysis was then performed between the expression levels of SLC25 subfamily differentially expressed genes from the sequencing data and classical renal injury markers (LCN2, KIM-1). The results revealed that SLC25A30 was significantly negatively correlated with both KIM-1 (r = −0.96) and LCN2 (r = −0.98), suggesting that the expression changes of SLC25A30 are associated with the severity of renal injury (Fig. 2D,E).

Identification of key gene SLC25A30 via intersection of differentially expressed genes in the 12 h group with Gene Expression Omnibus (GEO) public dataset.
To further explore the molecular mechanism of SLC25A30, a protein–protein interaction (PPI) network was constructed. The results indicated that SLC25A30 may interact with mitochondrial carrier homolog 2 (MTCH2) (Fig. 2F). Previous studies have confirmed (Guna et al., 2022) that MTCH2 is a mitochondrial outer membrane protein insertase that induces mitochondrial depolarization, a prerequisite for initiating the PINK1/PARKIN signaling pathway-mediated mitophagy (Durcan and Fon, 2015). At present, SLC25A30 is specifically and highly expressed in the kidney and proximal renal tubules, but its mechanism in the PINK1/PARKIN pathway and mitophagy remains unclear (Lewis et al., 2021). Based on these findings, we hypothesized that SLC25A30 may play a critical role in the pathological injury of SA-AKI by regulating the PINK1/PARKIN pathway and mitophagy. Further verification will be performed using in vitro and in vivo SA-AKI models in subsequent studies.
Establishment of an SA-AKI rat model and validation of SLC25A30 expression changes
SPF-grade male Wistar rats (8 weeks old, weighing 180–220 g, n = 4 per group) were used in this study. The SA-AKI model was established by intraperitoneal injection of LPS (10 mg/kg, dissolved in sterile normal saline), while rats in the control group received an equal volume of sterile normal saline. Blood samples and bilateral kidney tissues were collected 12 h after LPS administration. Biochemical assay results showed that SCr and BUN levels in the LPS 12-h group were >1.5-fold higher than baseline levels compared with the control group, indicating successful establishment of the SA-AKI rat model (Fig. 3A). H&E staining was performed to further observe morphological and structural changes in rat renal tissues. Pathological images of the LPS 12-h group revealed renal tubular epithelial cell edema, tubular lumen stenosis, and resorption vacuoles in renal tubules (Fig. 3C). RT-qPCR results demonstrated that mRNA expression of SLC25A30 was significantly decreased, while mRNA levels of KIM-1 and LCN2 were markedly increased in rat renal tissues 12 h after LPS injection, which was consistent with the transcriptome sequencing results (Fig. 3B,D). Immunofluorescence staining further confirmed that SLC25A30 expression was downregulated in the LPS 12-h group compared with the control group (Fig. 3E). In summary, the SA-AKI rat model was successfully established by intraperitoneal LPS injection, and both gene and protein expression of SLC25A30 were significantly decreased in renal tissues.

Establishment of SA-AKI in vivo model and validation of SLC25A30 expression changes.
Downregulation of SLC25A30 in rat renal tissue is accompanied by upregulation of PINK1/PARKIN and activation of mitophagy during SA-AKI
Total protein was extracted from renal tissue homogenates and subjected to Western blot (WB) analysis to examine the expression changes of SLC25A30, PINK1/PARKIN, and mitophagy-related proteins. The results showed that compared with the control group, the protein level of SLC25A30 was significantly decreased in rat renal tissues after 12 h of LPS stimulation (Fig. 4A,B). Meanwhile, the protein levels of PINK1 and PARKIN were upregulated (Fig. 4A,C). In addition, mitophagy-related proteins were significantly altered, mainly including downregulation of the autophagy substrate p62 and upregulation of LC3B-II (Fig. 4A,C), suggesting the activation of mitophagy. These results indicate that during SA-AKI, the protein expression of SLC25A30 is downregulated in rat renal tissues, accompanied by upregulation of PINK1/PARKIN and activation of mitophagy.

Downregulation of SLC25A30 is accompanied by upregulation of PINK1/PARKIN and activation of mitophagy in SA-AKI.
Establishment of an SA-AKI cell model and validation of expression changes of SLC25A30, PINK1/PARKIN, and mitophagy-related proteins
To establish an SA-AKI cell model, human renal tubular epithelial cells (HK-2 cells) were stimulated with 20 μg/mL LPS for 12 h in this study. Subsequently, the expression levels of intracellular-related molecules were systematically detected by a combination of RT-qPCR and WB. Meanwhile, cell viability and injury-related indicators were measured to verify the efficiency of model establishment and explore the potential molecular mechanism.
The results showed that after stimulation with 20 μg/mL LPS for 12 h, both mRNA and protein expression of SLC25A30 were significantly downregulated in HK-2 cells, with statistically significant differences (Fig. 5A–C). Meanwhile, the protein expression of PINK1/PARKIN was markedly increased, accompanied by excessive activation of mitophagy (Fig. 5B,D). WB analysis further confirmed that the protein expression of KIM-1 and LCN2 was significantly elevated in HK-2 cells, and cell viability was remarkably decreased (Fig. 5E,F). These results indicate that the LPS-induced HK-2 cell injury model can stably simulate the cellular pathological state of SA-AKI. The downregulation of SLC25A30, upregulation of PINK1/PARKIN, excessive activation of mitophagy, and high expression of renal injury markers (KIM-1 and LCN2) are consistent with the in vivo experimental results. Based on this model, we will further investigate the effects of SLC25A30 PARKIN/PARKIN expression, mitophagy activity, and mitochondrial function in HK-2 cells under LPS stimulation to clarify the regulatory role and mechanism of SLC25A30 in SA-AKI.
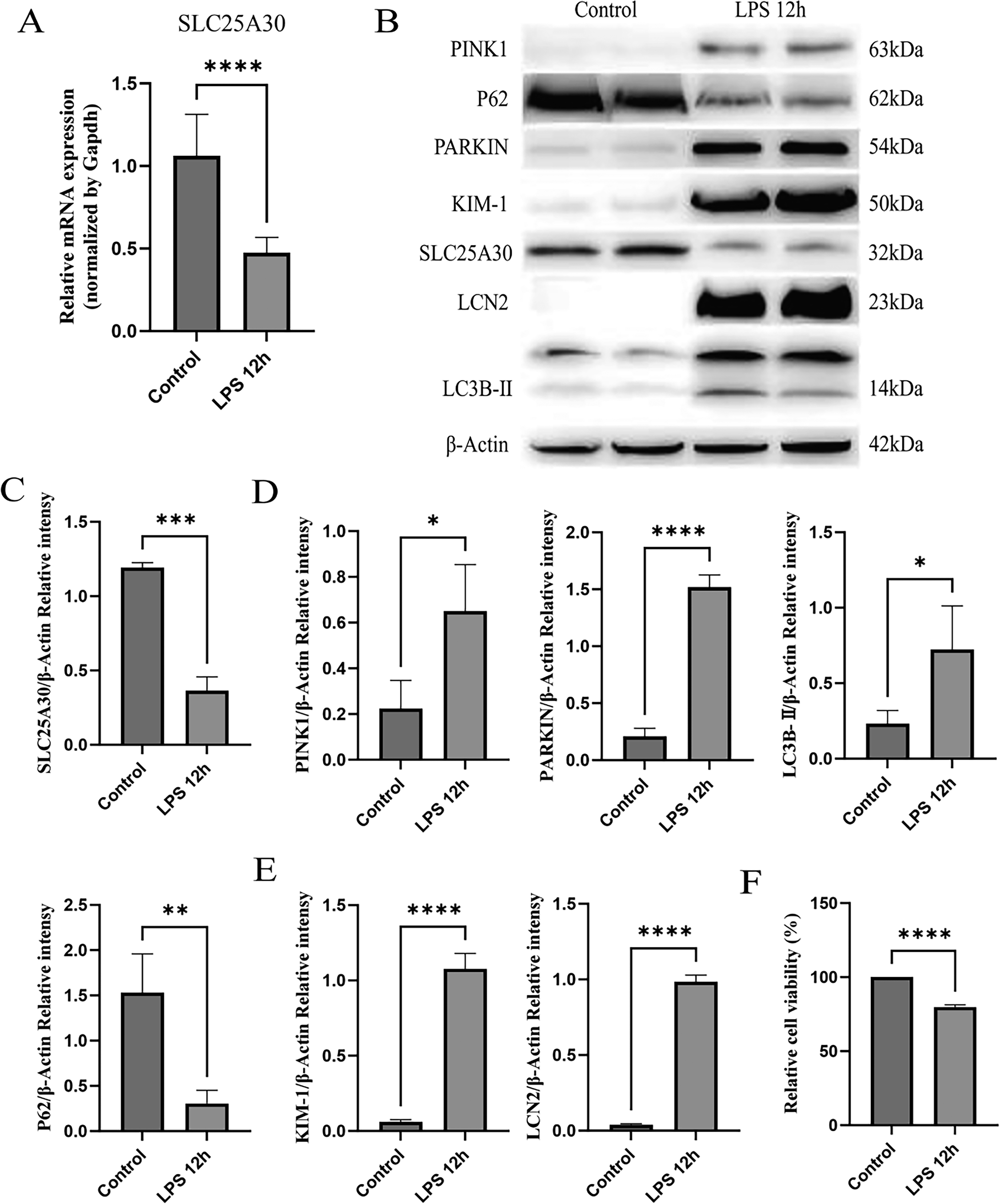
Establishment of SA-AKI cell model and validation of expression changes of SLC25A30, PINK1/PARKIN, and mitophagy-related proteins.
Overexpression of SLC25A30 inhibits excessive mitophagy activation by downregulating PINK1/PARKIN expression, ameliorates LPS-induced mitochondrial dysfunction, and alleviates HK-2 cell injury
Both in vivo and in vitro experimental results demonstrated that LPS induced downregulation of SLC25A30 at both mRNA and protein levels in rat renal tissues and HK-2 cells, accompanied by upregulation of PINK1/PARKIN expression, excessive activation of mitophagy, and aggravated renal and cellular injury. Therefore, in this study, SLC25A30 overexpression was achieved by plasmid transfection to investigate the effects of upregulated SLC25A30 expression on the PINK1/PARKIN pathway and excessive mitophagy activation (Fig. 6A,B). The results showed that upregulation of SLC25A30 reversed LPS-induced upregulation of PINK1/PARKIN and excessive mitophagy activation, reduced the protein levels of renal injury markers KIM-1 and LCN2, and alleviated HK-2 cell injury (Fig. 6A–F). To verify whether this cytoprotective effect was dependent on the PINK1/PARKIN pathway, HK-2 cells were transfected with si-PINK1 for 48 h to successfully knock down PINK1 mRNA expression. The results showed that knockdown of PINK1 led to decreased protein expression of PARKIN and LC3B-II, as well as increased protein expression of p62 (Fig. 7A–C). These results indicated that SLC25A30 overexpression inhibits excessive mitophagy activation by downregulating the PINK1/PARKIN pathway, thereby alleviating LPS-induced HK-2 cell injury.
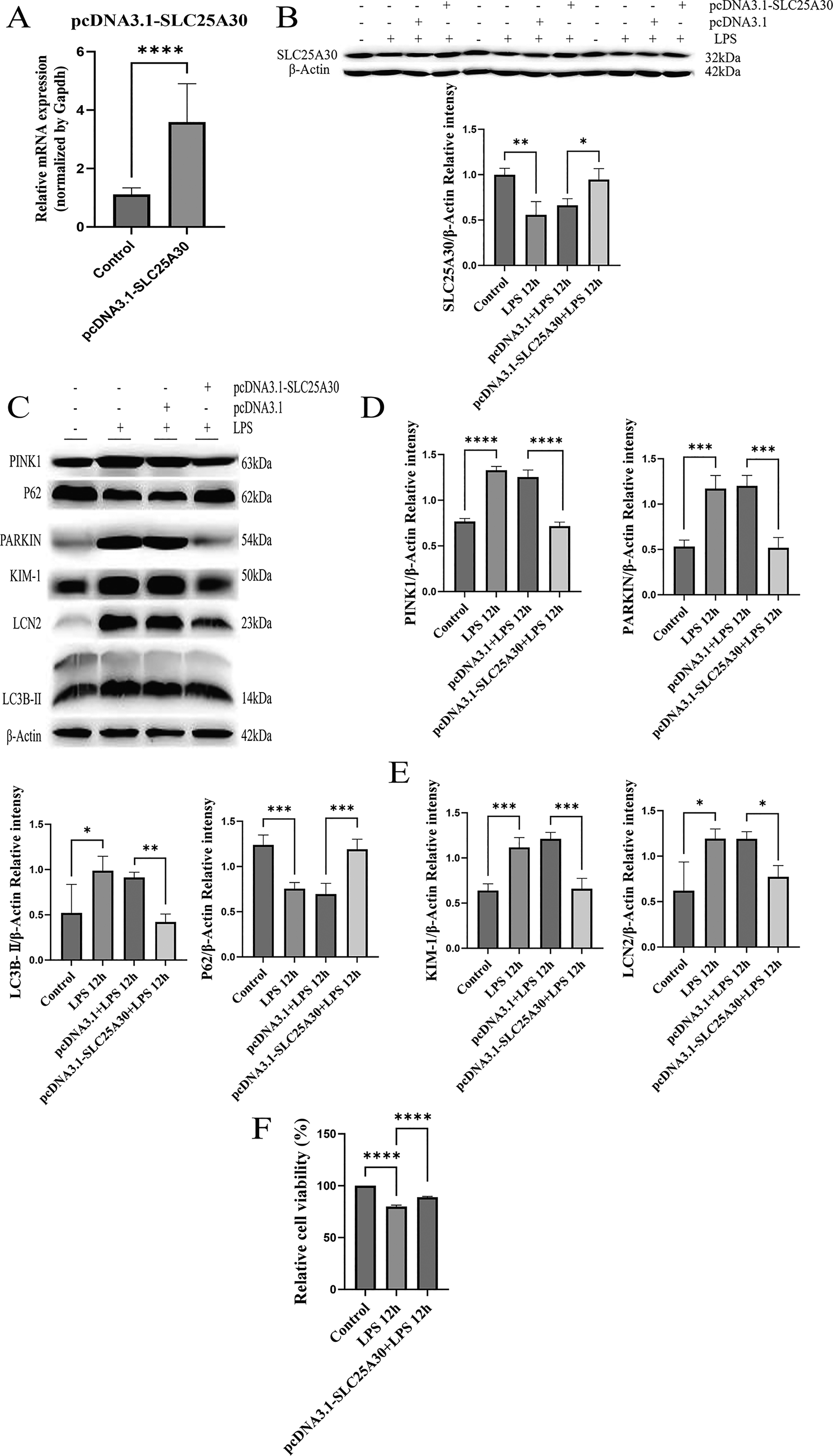
Overexpression of SLC25A30 inhibits excessive activation of mitophagy by downregulating the expression of PINK1/PARKIN, thereby reducing cell damage.
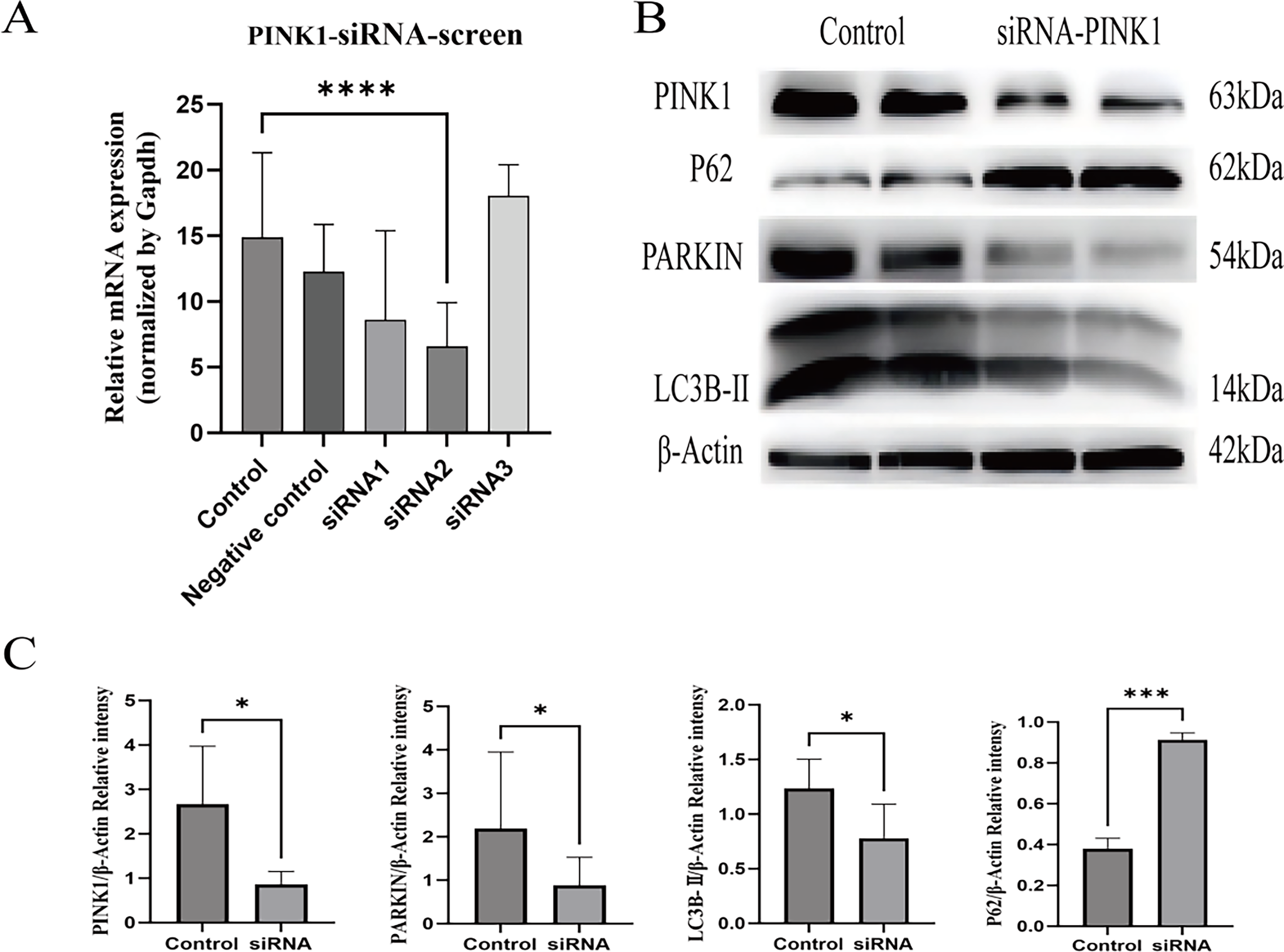
Effect of PINK1 knockdown by si-PINK1 on the expression of PINK1/PARKIN pathway and mitophagy-related proteins.
Subsequently, we further investigated the effects of SLC25A30 overexpression on mitochondrial morphology and function. TEM observations revealed that compared with the LPS group, HK-2 cells overexpressing SLC25A30 exhibited fewer autophagosomes and phagocytic vacuoles and a significantly increased number of normal mitochondria (Fig. 8A–D). Mitochondrial functional assays showed that SLC25A30 overexpression attenuated LPS-induced reduction of mitochondrial membrane potential (MMP) and decreased the level of reactive oxygen species (ROS) in HK-2 cells (Fig. 8E–H). These results demonstrated that SLC25A30 overexpression ameliorates LPS-induced mitochondrial dysfunction in HK-2 cells.

Effect of SLC25A30 overexpression on mitochondrial morphology and function in HK-2 cells.
In conclusion, this study confirmed that overexpression of SLC25A30 inhibits excessive mitophagy activation by downregulating the PINK1/PARKIN signaling pathway, improves LPS-induced mitochondrial dysfunction, alleviates injury in HK-2 cells, and thereby exerts a renoprotective effect.
Discussion
SA-AKI is a common complication among patients in the intensive care unit, characterized by poor prognosis and high mortality, and has become a major global public health concern (White et al., 2023). Current clinical management primarily includes the administration of antimicrobial agents, vasoactive support, fluid resuscitation, and renal replacement therapy (Zarbock et al., 2023a). However, specific targeted therapies that significantly improve long-term patient outcomes remain lacking (Zarbock et al., 2023b). Accumulating evidence has demonstrated that mitochondrial damage is a central mechanism underlying renal tubular epithelial cell injury during SA-AKI (Li et al., 2025), and the resulting mitochondrial dysfunction plays a critical role in the pathophysiological progression of the disease (Qiao and Cui, 2022; Jayaraman et al., 2025; Yuan et al., 2025; Miao et al., 2021; Xu et al., 2023). Although recent advances have been made in therapeutic strategies targeting mitochondrial dysfunction (Lai et al., 2025), no studies to date have reported the involvement of SLC25A30 in the regulation of mitophagy or its role in the pathological process of SA-AKI.
Research on the role of the SLC25A family in mitochondrial physiology and pathology has continued to emerge in recent years. Bresciani et al. found that the loss of SLC25A47 function could impair mitochondrial function in hepatocytes, which may act as a potential target for mitigating liver fibrosis progression (Bresciani et al., 2022). Nakanishi et al. demonstrated that SLC25A3 could regulate oxidative stress in nonalcoholic fatty liver disease (Nakanishi et al., 2023). Surpassed only by the heart in terms of oxygen consumption, the kidney is exceptionally rich in mitochondria. Mitochondrial dysfunction is highly relevant to the pathogenesis of SA-AKI (Parikh et al., 2015). Furthermore, aberrant expression of members of the SLC25 family has been identified as a potential biomarker for mitochondrial dysfunction in brain tissue (Babenko et al., 2018); alterations in their expression may ameliorate mitochondrial dysfunction by modulating mitochondria-related phenotypes, thereby influencing the initiation and progression of various diseases (Yu et al., 2025; Gao et al., 2025; Nakanishi et al., 2023). However, the association between SLC25 family members and SA-AKI has not been elucidated in previous studies. In the present study, time-series transcriptomic analysis revealed differential expression of the SLC25 subfamily at various time points during SA-AKI. Subsequent intersection analysis with GEO datasets and correlation analyses identified SLC25A30 as a key regulatory gene. This finding not only expands the functional characterization of the SLC25 subfamily in kidney diseases but also provides novel experimental evidence and a research perspective for deciphering the mitochondrial regulatory mechanisms underlying SA-AKI.
Our previous studies have confirmed that renal injury in LPS-induced SA-AKI rat models peaks at 12 h, and the differentially expressed genes at this time point exhibit the highest intersection efficiency with datasets from CLP-induced mouse models, demonstrating both pathological representativeness and species conservation. Correlation analysis revealed that SLC25A30 was significantly negatively correlated with the classical renal injury markers KIM-1 and LCN2. Notably, SLC25A30 is specifically highly expressed in renal proximal tubular epithelial cells (Lewis et al., 2021), which are the most vulnerable regions in SA-AKI, further suggesting that SLC25A30 may be involved in the pathological regulation of SA-AKI. Furthermore, PPI network analysis suggested a potential interaction between SLC25A30 and MTCH2. MTCH2 is a nuclear-encoded transporter protein localized to the mitochondrial outer membrane and has been shown to induce mitochondrial depolarization (Guna et al., 2022). Mitochondrial depolarization is a prerequisite for the initiation of PINK1/PARKIN-mediated mitophagy (Durcan and Fon, 2015) and serves as a critical trigger for pathological overactivation of mitophagy. Our experimental findings show that downregulation of SLC25A30 is accompanied by activation of the PINK1/PARKIN pathway and excessive enhancement of mitophagy. Therefore, we hypothesize that SLC25A30 may regulate PINK1/PARKIN-mediated mitophagy, thereby influencing MMP homeostasis.
The experimental results demonstrated that during SA-AKI, both the mRNA and protein expression levels of SLC25A30 were significantly downregulated in rat kidney tissues, accompanied by marked activation of the PINK1/PARKIN signaling pathway and abnormally elevated levels of mitophagy. The excessive activation of mitophagy at this time point occurred concomitantly with elevated levels of SCr and BUN, histopathological renal injury, as well as increased expression of renal injury markers. These findings suggest that alterations in SLC25A30 expression are closely associated with the severity of renal injury in SA-AKI and that excessive activation of mitophagy may participate in the pathological regulation of renal damage, thereby providing an experimental foundation for subsequent mechanistic investigations. Notably, previous studies have confirmed that mitophagy exerts renoprotective effects by clearing damaged mitochondria and maintaining mitochondrial homeostasis (Hu et al., 2025; Aggarwal et al., 2016; Tran et al., 2011). However, the present study revealed that although mitophagy was activated in SA-AKI models, it exhibited an excessive state that was paradoxically associated with aggravated renal injury. This seemingly contradictory finding suggests that mitophagy may exert a “double-edged sword” effect in SA-AKI: Moderate mitophagy facilitates the elimination of dysfunctional mitochondria and preserves cellular homeostasis, whereas excessive mitophagic activation may lead to aberrant clearance of healthy mitochondria, disrupt mitochondrial network integrity, and exacerbate energy metabolic disturbances and cellular damage. The downregulation of SLC25A30 may relieve its negative regulation of the PINK1/PARKIN pathway, resulting in uncontrolled mitophagic activation and thereby contributing to the pro-injurious effects during the pathological progression of SA-AKI. This discovery provides a novel perspective for understanding the complex regulatory mechanisms of mitophagy in SA-AKI.
To further elucidate the molecular mechanism by which SLC25A30 regulates SA-AKI, an in vitro cellular model of SA-AKI was established by stimulating HK-2 cells with LPS to mimic the pathological microenvironment of sepsis in vivo. Subsequently, SLC25A30 expression was upregulated via plasmid-mediated overexpression to validate its protective effect against LPS-induced cellular injury. In vitro experimental results demonstrated that after 12 h of LPS stimulation, HK-2 cells exhibited significantly downregulated mRNA and protein expression of SLC25A30, accompanied by activation of the PINK1/PARKIN signaling pathway, abnormally elevated levels of mitophagy, decreased cell viability, and markedly increased injury markers. This expression and regulatory pattern were entirely consistent with the findings observed in the in vivo rat model at 12 h, further confirming that downregulation of SLC25A30, activation of the PINK1/PARKIN pathway, and excessive mitophagy collectively contribute to the pathological process of SA-AKI. Consistently, Hu et al. (Hu et al., 2025) reported that mitophagy reaches its physiological peak at 8 h following LPS stimulation in HK-2 cells, whereas sustained mitochondrial damage can induce pathological overactivation of mitophagy. The selection of the 12-h time point in the present study precisely captures the critical transition phase of mitophagy from physiological adaptation to pathological overactivation. This temporal window not only provides complementary validation to previous studies from a temporal perspective but also establishes an ideal experimental model for elucidating the negative regulatory role of SLC25A30 on the pathological overactivation of mitophagy.
Furthermore, by overexpressing SLC25A30 at this specific time point, we were able to more precisely assess its regulatory effect on the pathological activation of mitophagy, thereby further refining the temporal regulatory characteristics of mitophagy in the in vitro SA-AKI model. Functional validation results demonstrated that overexpression of SLC25A30 significantly reversed LPS-induced excessive mitophagy, confirming the negative regulatory role of SLC25A30 on mitophagy. To verify whether this regulatory effect is dependent on the PINK1/PARKIN signaling pathway, pathway blockade was performed using siRNA-mediated knockdown of PINK1, which resulted in significant inhibition of mitophagic activity, confirming that SLC25A30 regulates mitophagy through the PINK1/PARKIN pathway. Consistently, Li et al. (Li et al., 2024a) also demonstrated in LPS-induced HK-2 cells that the PINK1/PARKIN signaling pathway specifically mediates mitophagy activation, and our findings are in accordance with this report. On the other hand, further TEM observation revealed that overexpression of SLC25A30 significantly increased the number of morphologically normal mitochondria and reduced the formation of autophagosomes encapsulating damaged mitochondria. Moreover, it alleviated LPS-induced MMP depolarization and decreased ROS generation. Jin et al. (Jin et al., 2025) demonstrated that knockdown of AHA1 attenuates ischemia-reperfusion-induced mitochondrial dysfunction and oxidative damage by inhibiting excessive mitophagy, elevating MMP, and reducing ROS levels. In the present study, overexpression of SLC25A30 similarly ameliorated MMP and oxidative stress status through inhibition of excessive mitophagy, which is highly consistent with the aforementioned findings. These results suggest that SLC25A30 may play a critical role in regulating the transition of mitophagy from physiological adaptation to pathological activation.
Mitophagy is a core mechanism of mitochondrial QC, primarily responsible for the selective elimination of damaged mitochondria and the maintenance of mitochondrial homeostasis, thereby ameliorating mitochondrial dysfunction. Previous studies have indicated (Aggarwal et al., 2016; Su et al., 2023) that moderate activation of mitophagy represents a protective response of the organism to early-stage injury. Mitophagy activation relies on the PINK1/PARKIN pathway and other receptor-mediated mechanisms (Su et al., 2023). Wang et al. found that the mitophagy PINK1-PARK2 pathway plays a protective role in sepsis-induced AKI. In vivo and in vitro experiments have illustrated that PINK1/PARK2 deficiency significantly reduced mitophagy and aggravated renal tubular injury and sepsis-related AKI (Wang et al., 2021). Evidence indicated that PINK1-PRKN/PARK2-dependent mitophagy is critical for renal tubular cell survival and mitochondrial QC in ischemic and nephrotoxic AKI (Tang et al., 2018). Yang et al. reported that the aldose reductase inhibitor WJ-39 could promote mitophagy through the PINK1/PARKIN signaling pathway, thereby reducing renal tubular injury in diabetic nephropathy (Yang et al., 2024). Evidence indicates that chicoric acid, a caffeic acid derivative, activated mitophagy through the Nrf2/PINK1/PARKIN pathway, alleviating renal tubular damage in mice with high-fat diet-induced chronic kidney disease (Ding et al., 2022). However, when mitochondria are subjected to sustained or severe injury, mitophagy can be pathologically overactivated, thereby triggering apoptosis and exacerbating tissue damage (Liu et al., 2023; Cao et al., 2023). This excessive activation leads to aberrant clearance of healthy mitochondria, disrupts mitochondrial network integrity, induces metabolic dysregulation, and ultimately aggravates mitochondrial dysfunction and cellular injury (Liu et al., 2023; Cao et al., 2023). In the present study, LPS stimulation of HK-2 cells for 12 h in an in vitro model demonstrated that downregulation of SLC25A30 was accompanied by PINK1/PARKIN signaling pathway-mediated excessive activation of mitophagy, resulting in decreased cell viability, increased ROS generation, and reduced MMP. Notably, Jia et al. (Jia et al., 2021) also reported in a rat model of chronic kidney disease induced by unilateral ureteral obstruction that excessive activation of PINK1/PARKIN-mediated mitophagy further aggravated renal injury. Similarly, in the LPS-induced SA-AKI rat model, we observed that excessive activation of mitophagy mediated by this pathway exacerbated renal damage, which is highly consistent with the aforementioned findings. These observations suggest that mitophagy exhibits a “double-edged sword” effect in SA-AKI: Moderate activation of mitophagy confers protective effects, whereas excessive activation may exacerbate injury through aberrant mitochondrial clearance and disruption of energy metabolic homeostasis. Downregulation of SLC25A30 may relieve its negative regulatory effect on the PINK1/PARKIN pathway, leading to uncontrolled activation of mitophagy, thereby contributing to the pro-injurious effects in the pathological progression of SA-AKI.
In conclusion, the present study demonstrated through both in vivo and in vitro experiments that SLC25A30 expression is significantly downregulated at the peak of injury in SA-AKI. Overexpression of SLC25A30 suppresses excessive mitophagy and ameliorates mitochondrial dysfunction via negative regulation of the PINK1/PARKIN signaling pathway, thereby identifying SLC25A30 as a novel therapeutic target for SA-AKI. Future studies incorporating clinical cohorts are warranted to further evaluate its translational value as a biomarker and as a basis for intervention strategies.
Authors’ Contributions
M.X.: Designed this study and conducted experiments. J.L. and Y.D.: Participated in the data collection. M.X., W.L., and Y.J.: Participated in the review of the article. M.X.: Conceptualization, methodology, validation, writing—original draft, and writing—review and editing. Y.W.: Methodology and resources. W.L.: Supervision, software, and project administration. Y.D.: Formal analysis and investigation. J.L.: Investigation and data curation. R.D.: Visualization. X.M.: Investigation. M.Y.: Investigation. Y.J.: Resources, writing—review and editing, and supervision.
Footnotes
Data Availability Statement
Disclosure Statement
The authors declare no competing or financial interests.
Funding Information
The authors declare that no funds, grants, or other support were received during the preparation of this article.
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.