
Abstract
The mechanisms of Alzheimer’s disease (AD) development are complex, and the detailed roles of neuroinflammation in AD still need to be elucidated. With various genetic and environmental risk factors, the accumulation of harmful amyloid plaques containing amyloid-β (Aβ) peptides is one of the main hallmarks of AD. Recent findings show that the innate immune protein interferon-induced transmembrane protein 3 (IFITM3) binds to γ-secretase and modulates Aβ production, providing a direct link between neuroinflammation, amyloidogenesis, and the pathogenesis of AD. In this review, we explore IFITM3-mediated modulation of γ-secretase complex activity and its pivotal role in AD pathology during neuroinflammation. Furthermore, we also provide an overview of the recent growing evidence connecting the roles of infection, the immune system, and AD pathogenesis.
Keywords
Alzheimer’s disease (AD) is the most common form of dementia and features neurodegenerative symptoms, including cognitive impairment, memory loss, and language problems (Knopman et al., 2021). The development of AD is characterized by two main pathological hallmarks, amyloid plaques and neurofibrillary tangles (NFTs) (St. George-Hyslop, 2000; Perl, 2010). NFTs are formed by hyperphosphorylated tau protein, which accumulates intracellularly in neurons, whereas amyloid plaques are extracellular depositions of amyloid-β (Aβ) formed by the amyloidogenic pathway (Billings et al., 2005; Simic et al., 2016). During the amyloidogenic pathway, β-secretase first cleaves amyloid precursor protein (APP) into sAPPβ and APP-CTF (C99), and γ-secretase subsequently cleaves C99 into Aβ and the APP intracellular domain (AICD) within cellular membranes (Fig. 1) (Vassar et al., 1999; Wolfe et al., 1999). Aβ42 is the major component of amyloid plaques and is more prone to aggregation and more toxic than Aβ40 (Iwatsubo et al., 1994). Recent studies on neuroinflammation driven by microglial activation, along with the identification of innate immunity-related genes in AD, suggest that the innate immune response plays a critical role in AD development (Kinney et al., 2018).
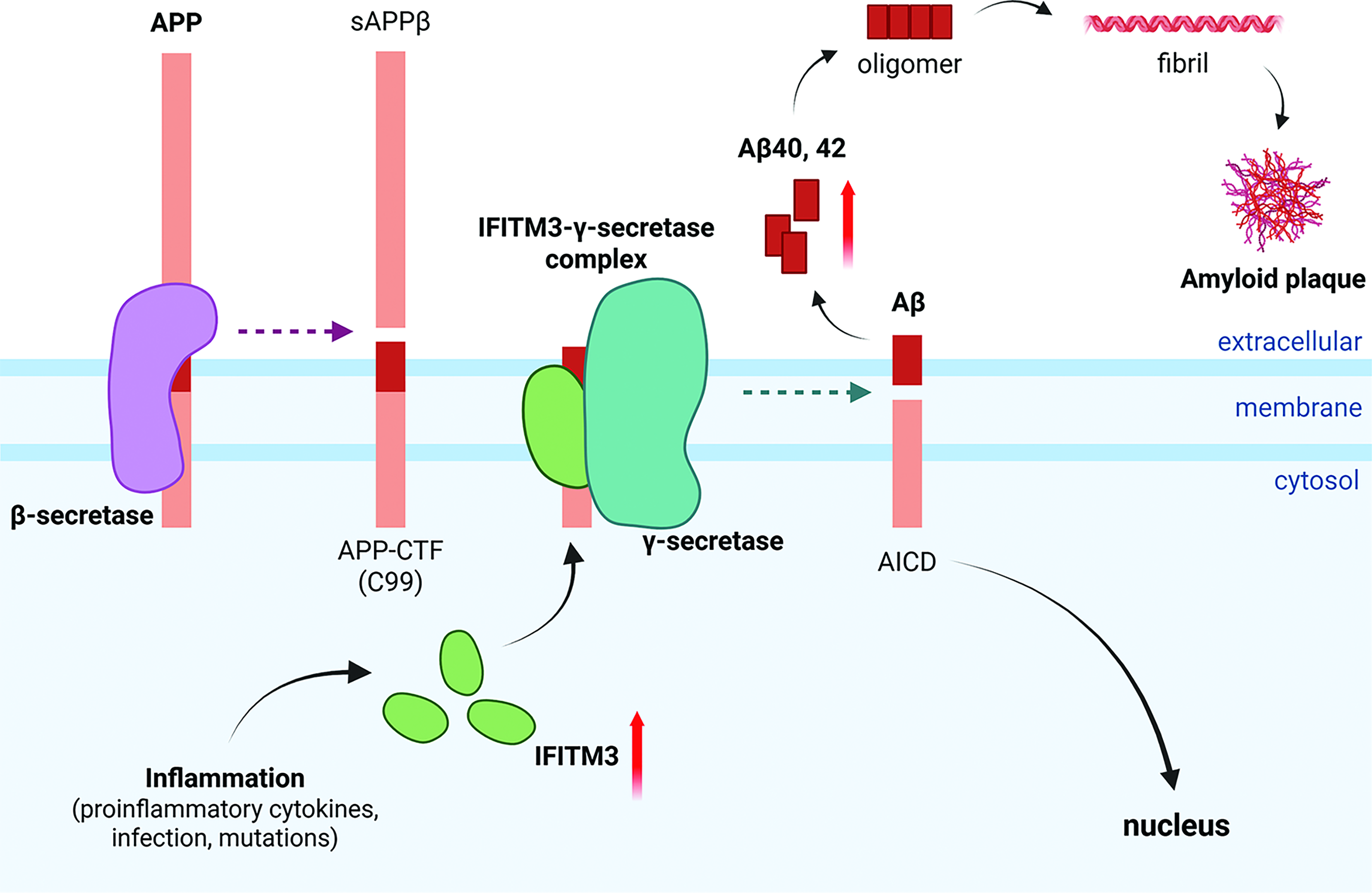
APP processing by γ-secretase complexes with IFITM3, mediated by inflammation in AD. In the amyloidogenic pathway, APP is cleaved into sAPPβ and APP-CTF (C99) by β-secretase. The γ-secretase complexes with IFITM3 cleaves C99 into Aβ and AICD. Increased IFITM3 expression under inflammatory conditions can lead to increased Aβ production. The cleaved AICD translocates to the cell nucleus. The accumulation of Aβ peptides (Aβ40 and Aβ42) leads to the oligomerization and fibrilization of Aβ, and the formation of amyloid plaques, which are one of the major pathological hallmarks of AD. AD, Alzheimer’s disease; APP, amyloid precursor protein.
AD can be divided into early-onset AD (EOAD) and late-onset AD (LOAD) (Selkoe, 2001). EOAD is caused by genetic mutations in Aβ-generating genes, such as APP, PSEN1, and PSEN2, which increase the Aβ burden (Holtzman et al., 2011). In contrast, LOAD is caused by the mixture of genetic and environmental factors (Lane et al., 2018). Genetic variants in the innate immune system, such as APOE, TREM2, and TYROBP, are reported to promote neuroinflammation and impair Aβ clearance in LOAD (Huang and Xu, 2019; Griciuc and Tanzi, 2021). The TREM2 R47H mutation or the APOE ε4 allele exacerbates inflammation and microglial dysfunction in the brain (Nguyen et al., 2020; Cheng-Hathaway et al., 2018). Therefore, the dysfunctional microglia with defective phagocytic function accelerate amyloid plaque accumulation, which triggers a prolonged inflammatory response, synaptic loss, and accelerated AD progression (Hickman et al., 2008; Selkoe and Hardy, 2016). However, even with those genetic and cellular mechanisms, the precise pathologies of AD related to innate immunity are still not well understood.
Recently, the identification of interferon-induced transmembrane protein 3 (IFITM3), an innate immunity protein, as a γ-secretase modulatory protein (GSMP) modulating the γ-secretase in Aβ processing provides novel insights into the innate immune system underlying AD via Aβ production (Hur et al., 2020; Hur, 2021; Hur, 2022). Here, we discuss the γ-secretase modulation mechanism by IFITM3 and the direct connection between neuroinflammation and AD.
γ-Secretase
γ-Secretase is important for understanding AD since it cleaves its immediate substrate, C99, to produce Aβ during the amyloidogenic processing (Fig. 1) (Haass et al., 2012). γ-Secretase is a transmembrane protein complex consisting of at least four components: presenilin (PS), nicastrin (Nct), anterior pharynx-defective 1 (Aph-1), and presenilin enhancer 2 (Pen-2), and PS is being a catalytic subunit of γ-secretase (Kimberly et al., 2003; Li et al., 2000; Edbauer et al., 2003). PS1 has an isoform, PS2, and these two PS homologs (PS1 and PS2) constitute distinct γ-secretase complexes (Steiner et al., 2002). The catalytic activity of γ-secretase is initiated by endoproteolytic cleavage of the PS holoprotein into an N-terminal fragment (NTF) and a C-terminal fragment (CTF) (Thinakaran et al., 1996). Each mature PS then exists as a heterodimer of NTF/CTF in γ-secretase (Thinakaran et al., 1996). Only a small portion (<14%) of PS1 is present in the active γ-secretase complex (Lai et al., 2003), and it has become a primary target for reducing Aβ production in AD (De Strooper et al., 2010). For substrate cleavages, γ-secretase selectively discerns the extracellular domain length of type I membrane proteins, such as APP and Notch, whereas most intramembrane proteases bind substrates at exosites before their catalytic activity (Struhl and Adachi, 2000). One of the γ-secretase complex components, Nct, conducts this substrate recognition and recruitment step by permitting substrates with sufficiently short ectodomains to access the active site of γ-secretase (Bolduc et al., 2016). For example, in Notch signaling, Notch receptors undergo a cleavage by ADAM metalloproteases at the extracellular site after binding to Notch ligands, and produces membrane-bound NotchΔE (Brou et al., 2000). γ-Secretase then recognizes and sequentially cleaves NotchΔE into Nβ and Notch intracellular domain (NICD), which translocates to the nucleus and regulates target gene expression, HES and HEY (De Strooper et al., 1999; Iso et al., 2003). During Aβ production in amyloidogenesis, the C99 substrate sequentially engages with exosites in Nct, Pen-2, and then PS1-NTF to access the active site of γ-secretase (Fukumori and Steiner, 2016), and is subsequently cleaved into Aβ and AICD. The major Aβ species are 40 and 42 amino acids long (Aβ40 and Aβ42), while APP-CTF is cleaved into various lengths of Aβ peptides (Takami et al., 2009). A higher ratio of Aβ42 over Aβ40 (Aβ42:Aβ40) inhibits synaptic activity due to the hydrophobic and aggregation-prone feature of Aβ42 (Kuperstein et al., 2010). Thereafter, Aβ peptides form oligomeric, protofibrilar, and fibrillar structures, ultimately leading to neurotoxicity (Kuperstein et al., 2010).
γ-Secretase inhibitors (GSIs) were utilized to inhibit toxic Aβ production by γ-secretase. GSIs successfully decreased Aβ levels in the cerebrospinal fluid of humans and reduced Aβ deposition in APP-induced mice (Bateman et al., 2009; Dovey et al., 2001). These findings made GSIs an attractive therapeutic target for AD treatment. However, clinical trials using GSIs such as Semagacestat (LY-450139) and Avagacestat (BMS-708163) failed due to nonselective inhibition of other γ-secretase substrate pathways, particularly Notch signaling (Doody et al., 2013; Coric et al., 2012). Sparing Notch signaling with GSIs became critical for protecting against side effects including disruption of cell differentiation and worsening cognition in AD patients (Hur, 2022).
Because of the failure of GSI clinical trials, modulation strategies have been prioritized over selective γ-secretase inhibition. γ-Secretase modulators (GSMs) are modulatory compounds that selectively reduce the generation of aggregation-prone Aβ42 by allosterically modifying the C99 cleavage site by γ-secretase (Weggen et al., 2001). They decrease Aβ42 levels while simultaneously elevating the levels of shorter, nontoxic species like Aβ37 and Aβ38 (Hakem et al., 2024). Also, this procedure occurs without interfering with Notch signaling or causing the abnormal C99 accumulation observed in GSI trials (Crump et al., 2013). Recent findings demonstrated that GSM-mediated selective modulation relies not only on allosteric modification but also on stabilization between γ-secretase and C99 substrates (Petit et al., 2022). This refined understanding of the enzymatic interaction of γ-secretase provides insights for developing promising GSM compounds for AD treatment.
Those beneficial features of GSMs led to clinical trials, such as nonsteroidal anti-inflammatory drugs (NSAIDs) for AD treatment (Sastre and Gentleman, 2010). However, most NSAIDs had problems crossing the blood–brain barrier (BBB) to be effective and did not show significant results in patients (Sastre and Gentleman, 2010; Miguel-Álvarez et al., 2015). Although many next-generation GSMs have been developed and have exhibited notable advantages in safety and efficacy during clinical trials, practical challenges remain, such as the selective modulation of APP cleavage while sparing various other γ-secretase substrates and/or effects on the human brain (Nordvall et al., 2023).
Meanwhile, γ-secretase requires at least four subunits (PS1, Nct, Aph-1, and Pen-2) (Edbauer et al., 2003), and its activity can be modulated by GSMPs, which are inherent intracellular proteins (Hur, 2022; Hur, 2021). GSMPs, such as nontranscriptional hypoxia-inducible factor-1α (Hif-1α), stress-associated endoplasmic reticulum protein 1 (SERP1), transmembrane protein 21 kDa (TMP21), and many others, have been identified to modulate the γ-secretase activity. Hif-1α increases the γ-secretase activity for Aβ and NICD production under hypoxia, which indicates that nontranscriptional Hif-1α directly binds to γ-secretase and works as a GSMP to convert inactive complexes into active forms (Villa et al., 2014; Alexander et al., 2022). SERP1 binds to the γ-secretase subcomplex to regulate the assembly of the γ-secretase holoenzyme, and increases Aβ production by preferentially recruiting APP over Notch (Jung et al., 2020). TMP21 binds with PS complexes, both PS1 and PS2, as a GSMP and regulates the catalytic activity of γ-secretase without altering the cleavage of other substrates, such as Notch (Chen et al., 2006). This finding was confirmed by increased levels of Aβ40 and Aβ42 during TMP21 suppression (Chen et al., 2006).
Recently, IFITM3 has been identified as one of the GSMPs regulating Aβ production, suggesting a direct link between inflammatory conditions and Aβ accumulation in AD (Fig. 1). A study using affinity pulldown and photolabeling approaches with GSIs and/or GSMs to isolate enzymatically active γ-secretase identified a transient binding protein, IFITM3, localized to the active γ-secretase complexes near PS1-NTF (Hur et al., 2020). IFITM3 modulates γ-secretase activity for APP cleavage when it is stimulated by proinflammatory cytokines, such as type I interferon (IFN) (IFN-α) and type II IFN (IFN-γ) in primary mouse neurons, and interleukin-1β (IL-1β) and IL-6 in primary human astrocytes. In addition, human LOAD brain samples were divided into “LOAD-high (LOAD-H)” and “LOAD-low (LOAD-L)” subgroups based on IFITM3 protein expression and γ-secretase activity (Hur et al., 2020). The LOAD-H subgroup exhibited significantly higher γ-secretase activity for Aβ40 and Aβ42 cleavage compared with healthy controls and the LOAD-L subgroup. In LOAD, genetic mutations such as TREM2 and CD33 were identified in relation to microglia-mediated Aβ clearance regulation. Thus, impaired Aβ clearance due to microglial dysfunction leads to Aβ accumulation and triggers the release of proinflammatory cytokines from microglia (Griciuc et al., 2019). Concurrently, neuroinflammation-induced IFITM3 directly interacts with the active γ-secretase complex, thereby potentiating Aβ accumulation and driving exacerbated neuroinflammation and AD progression (Hur et al., 2020). These findings indicate a novel and direct link between γ-secretase activation and amyloidogenesis under neuroinflammatory conditions in the brain (Hur, 2021).
IFITM3
IFITM3, an innate immune protein, was first identified in a study of influenza A virus (IAV) infection (Brass et al., 2009). Upon viral infection, the innate immune response to viruses are first initiated by the sensing of viral nucleic acids (DNA or RNA genomes) via pattern recognition receptors (PRRs) (Kawai and Akira, 2006). PRRs, including the retinoic acid-inducible gene I-like receptors (RLRs), Toll-like receptors, nucleotide-binding oligomerization domain-like receptors, and cytosolic DNA sensors, induce the secretion of inflammatory factors as part of the antiviral defense mechanism (Takeuchi and Akira, 2010). These mechanisms lead to the expression of type I or II IFNs, which in turn regulate the expression of human IFITM1, IFITM2, and IFITM3 (Bailey et al., 2014; Friedman. et al., 1984). The promoter regions of the IFITM family contain interferon-stimulated response elements that respond to both type I IFN and type II IFN; however, these genes can also be expressed through IFN-independent signaling pathways (Yanez et al., 2020). Human IFITMs restrict the entry of proteins from many types of viruses, such as IAV, flaviviruses, human immunodeficiency virus-1 (HIV-1), severe acute respiratory syndrome coronavirus, and hepatitis C virus (Bailey et al., 2014; Diamond and Farzan, 2013). However, they do not restrict murine leukemia virus or the arenavirus and alphavirus assayed thus far (Bailey et al., 2014; Diamond and Farzan, 2013).
The functions of human IFITM1-3 as antiviral effectors against diverse viruses have been extensively studied (Narayana et al., 2015; Brass et al., 2009). IFITM1 is mostly localized at the plasma membrane or early endosomes to restrict viruses with early endosomal fusion, while IFITM2 and IFITM3 are localized at late endosomes or lysosomes, which restrict viruses that undergo endocytosis and membrane fusion (Narayana et al., 2015). Recent studies have also reported immune-related function of IFITM1 in immune cells from gastric cancer patients infected with Helicobacter pylori, as well as in epithelial cells of patients infected via Ephrin receptor A2 (EphA2)-mediated entry of Epstein–Barr virus (Chen et al., 2025; Yang et al., 2024). IFITM2 can also regulate the development of the mammalian neocortex in the endocytosis of crucial neural stem cells and radial glial cells (Lv et al., 2025). These findings indicate that human neurogenesis may interact with IFN signaling pathways.
In IFITM3, the rs12252-C single nucleotide polymorphism (SNP), a minor IFITM3 allele, reduced antiviral activity against IAV infection compared with wild-type IFITM3 (Denz and Yount, 2025; Everitt et al., 2012). This is because the rs12252-C allele alters a splice acceptor site, resulting in truncation of the N-terminal 21 amino acids (Everitt et al., 2012; Denz and Yount, 2025). Additionally, in the 2009 H1N1 influenza pandemic and coronavirus disease 2019 (COVID-19), the rs12252-C SNP affects disease severity as well as susceptibility, which means the N-terminal region of IFITM3 is essential for its antiviral function (Zhang et al., 2013; Ahmadi et al., 2022; Li et al., 2022).
Additionally, IFITM proteins play a role in the adaptive immune system (Yanez et al., 2020). The adaptive immune system is a highly antigen-specific defense mechanism that protects against pathogens that evade innate immunity (Bonilla and Oettgen, 2010). It also establishes long-lasting immunological memory, providing enhanced protection upon reinfection, primarily mediated by lymphocytes (T and B cells) (Bonilla and Oettgen, 2010). Initially, it remained unclear whether IFITM3 could play a role beyond the acute phase of the infection (Wakim et al., 2013). However, Wakim et al. (2013) showed that CD8+ resident memory T cells (TRM cells) in the lungs of mice maintained increased IFITM3 expression after infection with influenza. This result suggests that IFITM3 is critical for both innate and adaptive immune systems (Wakim et al., 2013).
Immune System in AD
Microglia and astrocytes are important innate immune system components in the brain (Ransohoff and Brown, 2012). In AD, these glial cells proliferate, cluster around Aβ and NFTs, and release inflammatory mediators such as proinflammatory cytokines, which cause neuroinflammation in the brain (Walters et al., 2016; Serrano-Pozo et al., 2011; Gao et al., 2023). Moreover, a subset of microglia with a specific transcriptional characteristic, termed disease-associated microglia, is localized in the vicinity of the Aβ plaques (Keren-Shaul et al., 2017). These cells play a protective role by sensing neurodegeneration-associated molecular patterns across various neurodegenerative diseases and participating in the clearance of harmful Aβ in AD (Keren-Shaul et al., 2017; Deczkowska et al., 2018). Failure of phagocytic Aβ clearance due to microglial dysfunction is considered to exacerbate disease pathology in AD transgenic mice (Lachish et al., 2022). Genetic risk factors in LOAD are closely linked to microglial dysfunction, resulting in severe accumulation of Aβ, while excessive inflammation aimed at clearing Aβ can backfire and provoke Aβ production instead (Heneka et al., 2025). Likewise, facilitated Aβ production in microbial infections, such as herpes simplex virus type 1, supports a link between neuronal infection and Aβ accumulation in AD and suggests that Aβ may function as a part of the innate immune response (Zhao et al., 2024). This paradoxical response suggests that Aβ can also act as an antimicrobial peptide (AMP) in the brain’s defense system (Kumar et al., 2016).
The discovery of the innate immune response triggered by the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway also supports that innate immunity is associated with AD development (Chung et al., 2024). The cGAS–STING pathway plays a major role in detecting DNA viruses, viral infection, and cancer, thereby triggering subsequent immune responses, including the secretion of type I IFN (IFN-α and IFN-β) and inflammatory cytokines (TNF, IL-1β, and IL-6) (Chen et al., 2016). Evidence showing that an abnormal cGAS–STING pathway is involved in neuronal disorders led researchers to hypothesize that the cGAS–STING response is associated with the amyloidogenic pathway in the brain and is mediated through IFITM3 regulation (Chung et al., 2024). Higher signals of cGAS and STING were detected with Aβ-clustered microglia (Chung et al., 2024). Similarly, the senescence marker genes (Cdkn1a and Cdkn2a) were increased in the humanized Aβ and tau double knock-in (AppNL-G-F/hTau) mice group compared with wild-type (Chung et al., 2024). Aβ-treated microglia showed greater cGAS activation and increased levels of inflammation markers compared with control microglia (Chung et al., 2024). Enhanced expression of IFITM3 in cGAMP-treated neurons indicated that Aβ generation modulated by IFITM3 is linked to the immune response, especially by the cGAS–STING pathway in AD (Chung et al., 2024). This finding is also supported by the decreased Aβ burden in pharmacological STING inhibition (Chung et al., 2024). A similar study also showed that Aβ25–35 can induce the co-localization of IFITM3 and STING in microglia by immunofluorescence imaging (Wu et al., 2023).
Little is known about the role of adaptive immunity in AD. Gate et al. (2020) showed that the number of CD8+ T effector memory CD45RA+ cells (TEMRA cells) is increased in peripheral blood mononuclear cells (PBMCs) from mild cognitive impairment (MCI) and AD patients, and that CD8+ TEMRA cell levels are negatively correlated with cognitive scores. Moreover, single-cell RNA sequencing (scRNA-seq) of CD8+ TEMRA cells from PBMCs showed that adaptive immunity, cytokine signaling, and T cell receptor (TCR) signaling are increased by pathway analysis, and the differential expression of IFITM3 is increased in patients with MCI or AD (Gate et al., 2020). Given that the presence of more than two T cells with the same TCR sequence indicates “clonal expansion” after recognizing cognate antigen, single-cell TCR sequencing identified that clonally expanded CD8+ TEMRA cells in the cerebrospinal fluid of AD patients (Gate et al., 2020). This study establishes novel insights into the role of adaptive immunity in AD pathologies and provides evidence linking IFITM3 to adaptive immune response in AD.
IFITM3, γ-Secretase, and AD
Hur et al. highlighted innate immunity as a crucial mechanism in AD pathogenesis and suggested that further studies exploring the immune system and neuroinflammation through IFITM3–γ-secretase complexes in AD are essential (Hur, 2021; Hur, 2022; Hur et al., 2020). Hur et al. (2020) showed that knockdown of IFITM3 reduced Aβ production in cells. The number of amyloid plaques was also significantly decreased in the cortex and hippocampus of Ifitm3–/–; 5xFAD transgenic AD mice. IFITM3 modulates γ-secretase activity for APP cleavage in primary mouse neurons and primary human astrocytes following stimulation by proinflammatory cytokines (type I IFN (IFN-α), type II IFN (IFN-γ), IL-1β, and IL-6) (Hur et al., 2020). Since then, numerous subsequent studies have further highlighted the interaction between amyloidogenesis and AD development via IFITM3.
BBB dysfunction and decreased cerebral blood flow have been shown in many AD patients (Love and Miners, 2016; van de Haar et al., 2016). These observations suggest that vascular disorders may exacerbate AD (Faraco et al., 2016). Most recently, it was reported that inhibiting IFITM3 in astrocytes suppresses neuroinflammation and neuronal damage in cerebral ischemia–reperfusion injury (Ni et al., 2025). Cerebrovascular Aβ deposition and its correlation with IFITM3 in AD were also investigated (Feng et al., 2025). Cerebrovascular endothelial cells (CVECs) maintain central nervous system homeostasis by forming the BBB via high-resistance tight junctions and regulating molecular transport (Daneman and Prat, 2015). CVECs also modify permeability at the BBB in response to inflammatory stimuli (McConnell and Mishra, 2022). Higher IFITM3 expression surrounding Aβ plaques in the CVECs was detected by immunofluorescent analysis in both APP/PS1 overexpressing (APP23/PS45 double transgenic mice) and 5xFAD transgenic AD mouse models (Feng et al., 2025). In scRNA-seq data analysis, IFITM3 was also upregulated in the CVECs of AD patients (Feng et al., 2025). When endothelial IFITM3 expression was reduced at the cortex and hippocampus, the Aβ plaques were decreased in APP23/PS45 mice (Feng et al., 2025). Moreover, improved cognitive abilities in AD mice with knockdown of IFITM3 suggested that decreasing Aβ deposition in cerebral blood vessels through endothelial IFITM3 knockdown can be a therapeutic approach in AD (Feng et al., 2025).
Additionally, a history of chronic periodontitis in patients positively correlates with a higher rate of AD development (Chen et al., 2017). The brains of AD patients also exhibited accumulation of Porphyromonas gingivalis (Pg), and one possible pathway is the disruption of the BBB induced by Pg. Therefore, researchers hypothesized that Pg infection is correlated with AD development through neuroinflammation (Dominy et al., 2019). Increased Aβ production, activated microglia and astrocytes, and cerebral IFITM3 levels were also shown in Pg-induced periodontitis mice. These mice showed impaired spatial memory and learning ability in the Morris Water Maze test (Kong et al., 2025). These studies collectively indicated that the brain immune system, infections, Aβ production mediated by IFITM3–γ-secretase complex or IFITM3, and AD pathogenesis are interconnected. These complex interactions further enhance neuroinflammation and contribute to AD progression.
Conclusion
The identification of IFITM3 as a γ-secretase modulatory protein demonstrates its role as a mediator between the toxic molecular deposition of Aβ and neuroinflammation in AD. Since LOAD patients can be categorized by IFITM3 expression levels, IFITM3 could also serve as a potential biomarker for indicating a severe inflammatory condition in AD patients, and a potential intervention targeting the interaction between IFITM3 and PS1 can be a promising therapeutic target for AD. However, the exact interaction sites between IFITM3 and PS1-NTF within the IFITM3–γ-secretase complexes for APP and other potential substrates remain unknown. The broader implication and function of IFITM3 in glial cells, vascular cells, and endothelial cells in AD, as well as its original viral restriction mechanisms in the brain, require further investigation to fully understand AD pathology. In conclusion, IFITM3 establishes a crucial link between neuroinflammation and Aβ production in AD. Further precise molecular mechanism studies on the IFITM3-mediated modulation of γ-secretase are essential for the development of novel AD therapeutics.
Authors’ Contributions
J.-Y.H. conceptualized the article; J.-S.C. wrote the initial draft of the article; and J.-S.C. and J.-Y.H. revised the article.
Footnotes
Acknowledgment
The figure was made with BioRender.com.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (RS-2025-25436818, RS-2021-NR060141) (J.-Y.H.).