
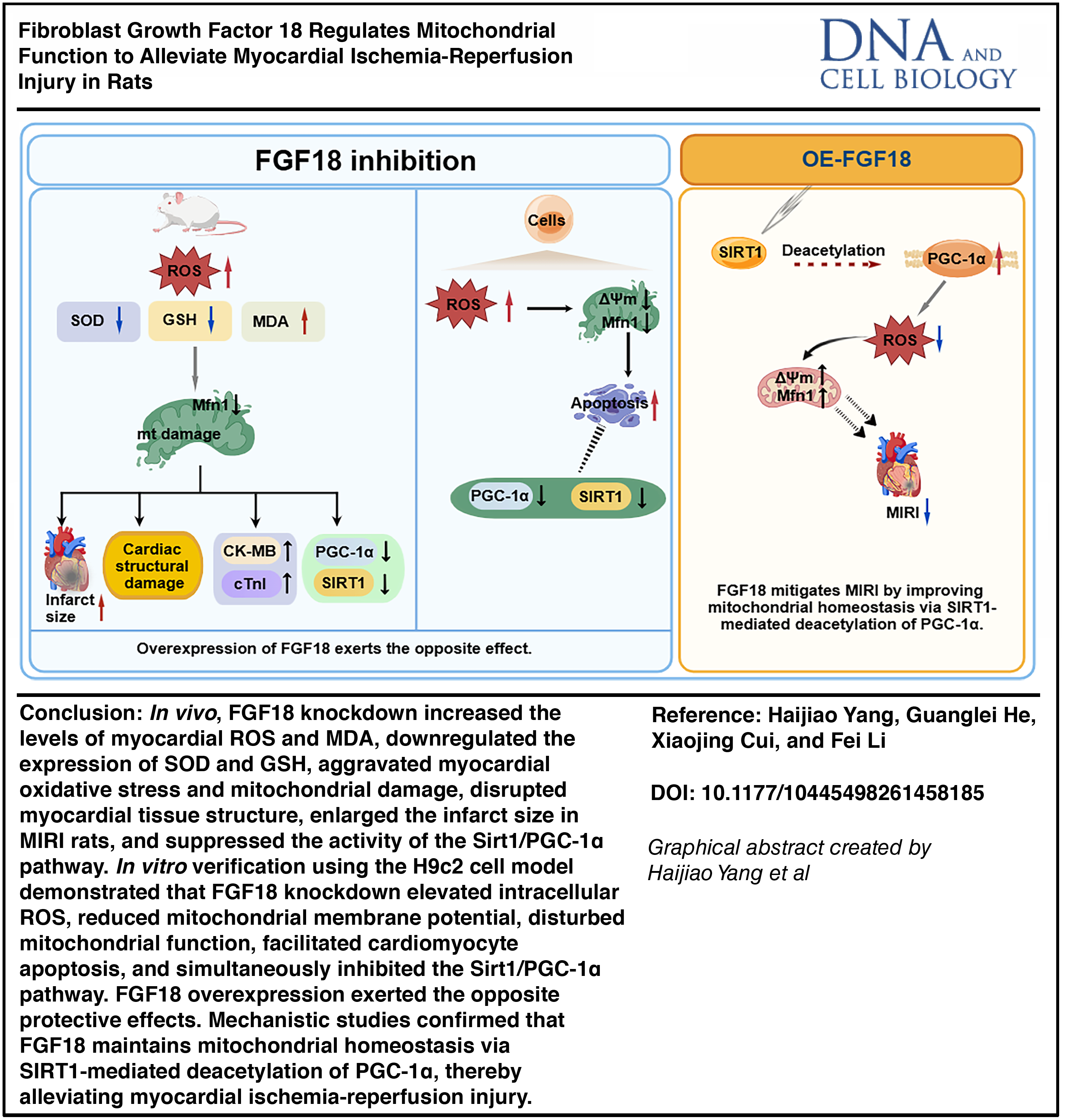
Abstract
The purpose of this work was to examine the function of fibroblast growth factor 18 (FGF18) in rat myocardial ischemia-reperfusion injury (MIRI) and elucidate its relationship to mitochondrial function through the Sirtuin 1/peroxisome proliferator-activated receptor gamma coactivator 1 (SIRT1/PGC-1α) pathway. To evaluate myocardial infarct size, pathological alterations, cardiomyocyte injury, mitochondrial state, oxidative stress, and SIRT1/PGC-1α protein expression, FGF18-knockdown and FGF18-overexpression rat MIRI models were created. H9c2 cardiomyocytes were used to create an in vitro hypoxia-reoxygenation (H/R) model, and FGF18-overexpressing H9c2 cells were given the SIRT1 inhibitor EX-527. We detected the effects of FGF18-mediated regulation of the SIRT1/PGC-1α pathway on H/R-induced alterations in H9c2 cells, including cell viability, mitochondrial reactive oxygen species (ROS) production, mitochondrial membrane potential, apoptotic rate, and the protein expression of FGF18, SIRT1, PGC-1α, and mitofusin 1 (Mfn1). Furthermore, we performed a protein immunoprecipitation (IP)-protein acetylation assay to determine whether FGF18 influences the acetylation level of PGC-1α through the regulation of SIRT1. Results showed that FGF18 overexpression upregulated SIRT1/PGC-1α/Mfn1 expression, improved mitochondrial function, reduced oxidative stress, and enhanced H9c2 survival under H/R, while FGF18 knockdown had opposite effects. Moreover, FGF18 overexpression inhibited H/R-induced PGC-1α acetylation, and SIRT1 inhibition abrogated FGF18-mediated protective effects. Collectively, FGF18 attenuates rat myocardial MIRI by alleviating oxidative stress and regulating mitochondrial homeostasis through SIRT1-mediated deacetylation of PGC-1α.
Keywords
Introduction
Reperfusion therapy is the first-line treatment for ischemic heart disease (Solola Nussbaum et al., 2022); however, restoring blood flow to myocardial regions with insufficient perfusion induces secondary cellular dysfunction and subsequent structural damage, a pathological condition defined as myocardial ischemia-reperfusion injury (MIRI) (Li et al., 2019). Myocardial ischemia triggers mitochondrial damage: the mitochondrial respiratory chain is impaired within the initial minutes of ischemia, causing an absolute elevation in reactive oxygen species (ROS), while diminished antioxidant enzyme activity leads to a relative ROS increase, forming a vicious cycle. Reperfusion further disrupts mitochondrial structure and function, resulting in reduced adenosine triphosphate (ATP) synthesis and energy depletion, which impairs normal myocardial physiological function and exacerbates necrotic areas. Moreover, ischemia-reperfusion elicits a burst of ROS production, driving oxidative stress and lipid peroxidation; excessive oxidative stress is implicated in multiple pathological processes of the myocardium, including inflammation, apoptosis, and metabolic dysfunction (Chen et al., 2020). Sirtuin 1 (SIRT1) is a pivotal regulator of cellular stress responses, with established roles in modulating oxidative stress and metabolic homeostasis. In the context of MIRI, SIRT1 exerts cardioprotective effects by preserving mitochondrial homeostasis (Ding et al., 2024). Emerging evidence further links SIRT1 activation to the suppression of cardiomyocyte apoptosis and the alleviation of endoplasmic reticulum stress, suggesting a coordinated mechanism to restore cellular integrity during reperfusion (Li et al., 2022).
Fibroblast growth factor 18 (FGF18) is a pleiotropic factor expressed in various tissues, where it plays critical roles in organ development, tissue repair, and key physiological processes such as inflammation and angiogenesis (Hu et al., 2024). Most prior studies on FGF18 have focused on skeletal, pulmonary, and neural development, with few investigating its role in cardiovascular diseases. Recent evidence, however, demonstrates that FGF18 confers cardioprotection in mouse models of pathological myocardial hypertrophy. It alleviates cardiac dysfunction and hypertrophy by attenuating oxidative stress, inhibiting cardiomyocyte apoptosis, and ameliorating fibrosis—effects primarily attributed to its preservation of redox homeostasis (Chen et al., 2023). Moreover, emerging research has explored the role of FGF18 in ischemia-reperfusion (I/R) injury, demonstrating that its expression is upregulated in renal tubules of ischemic mice and that FGF18 overexpression effectively mitigates acute kidney injury-induced renal dysfunction (Wang et al., 2024). In the liver, FGF18 markedly attenuates hepatic I/R injury, alleviates oxidative stress and inflammation, and suppresses hepatocyte apoptosis (Tong et al., 2023). However, whether FGF18 activates the SIRT1 pathway to preserve mitochondrial homeostasis and alleviate oxidative stress in MIRI remains unclear. Therefore, this study investigated the cardioprotective effects and underlying mechanisms of FGF18 in this context.
Materials and Methods
Animal feeding and grouping
Fifty-five healthy male Sprague-Dawley (SD) rats (6–8 weeks old, 250 ± 20 g) were obtained from Jinan Pengyue Laboratory Animal Breeding Co., Ltd. (License No.: SCXK(LU)20230045). All rats were housed in a specific pathogen-free facility under a 12-h light/dark cycle, at 20–26°C and 50–60% relative humidity. Following a 7-day acclimatization period, all experimental procedures were initiated. All protocols were approved by the Animal Ethics Committee of Binzhou Medical University (Approval No.: 2024-L094) and conducted in strict accordance with the national Regulations for the Administration of Laboratory Animals.
Adeno-associated virus (AAV) transfection
Rats were restrained in a fixator with their tails exposed. The tail veins were dilated by swabbing the tail from root to tip with 75% alcohol-soaked cotton balls. A needle was then inserted into the caudal vein at the middle-lower segment, and 100 μL of AAV9 vectors were slowly and consistently injected: FGF18-overexpressing AAV9 (OE-FGF18, 3 × 1013 vg/mL), FGF18-knockdown AAV9 (sh-FGF18, 2.6 × 1013 vg/mL), or blank control AAV9. The sh-FGF18 sequence was 5′-GCAAGGAGTGTGTGTTCATCG-3′, and OE-FGF18 encoded the full-length FGF18 cDNA sequence (NM_019199.2). sh-NC and OE-NC served as blank control vectors. Three weeks post-injection, rat hearts were harvested, and FGF18 protein expression was detected by Western blot.
Construction of a rat MIRI model
Healthy adult male rats were anesthetized via intraperitoneal injection of ketamine (100 mg/kg). The rats were then intubated, mechanically ventilated, and placed in a supine position on a thermostatically controlled heating pad with electrocardiographic monitoring. Following thoracotomy and adequate exposure of the heart, the left anterior descending coronary artery was ligated with a 7-0 silk suture. Ischemia was confirmed by significant ST-segment elevation on the electrocardiogram (ECG) and maintained for 30 min, after which the ligature was released to allow 2 h of reperfusion (Dou et al., 2019; Xu et al., 2020). Upon completion of the protocol, rats were euthanized by cervical dislocation under CO2 anesthesia. Heart tissue and blood samples were collected immediately. A portion of the cardiac tissue was snap-frozen in liquid nitrogen, while the remainder was fixed in 4% paraformaldehyde. Fixed tissues were dehydrated through a graded ethanol series, embedded in paraffin, and cut into 3-μm serial sections. The sections were then deparaffinized in xylene and rehydrated for subsequent pathological staining.
Animal grouping
To establish FGF18-knockdown and FGF18-overexpressing rat models, rats were randomized into five groups (n = 5): Sham, OE-NC, OE-FGF18, sh-NC, and sh-FGF18. Rats in the Sham group received 100 μL of normal saline, while the other four groups were injected with 100 μL of the corresponding AAV9 vector.
To investigate the effects of FGF18 on MIRI, rats were randomized into six groups (n = 5): Sham, MIRI, OE-NC + MIRI, OE-FGF18 + MIRI, sh-NC + MIRI, and sh-FGF18 + MIRI. Rats in the Sham group underwent coronary artery threading without ligation. The MIRI group was subjected to MIRI modeling as described above. The remaining four groups underwent MIRI modeling at 3 weeks post-AAV9 injection.
TTC staining
The rat chest cavity was opened to fully expose the heart, and a perfusion needle was inserted into the left ventricle for rapid phosphate buffered saline (PBS) perfusion. The heart was then excised and frozen at −80°C for 10 min. Subsequently, it was cut into five uniform longitudinal thin sections below the ligation site with a surgical blade. Sections were incubated in 2,3,5-Triphenyltetrazolium Chloride (TTC) staining solution at 37°C for 30 min, washed with PBS, and fixed in 4% paraformaldehyde for 24 h. The stained sections were photographed, and the area of infarction was quantitatively analyzed using the ImageJ software.
ELISA detection
At 120 min of reperfusion, 1 mL of abdominal aortic blood was collected from each rat using a syringe and transferred to centrifuge tubes. Blood samples were allowed to stand at room temperature for 2 h and then centrifuged at 1,000 × g for 15 min at 4°C. The supernatant was collected and aliquoted into microcentrifuge tubes for subsequent Enzyme-Linked Immunosorbent Assay (ELISA) assays according to the manufacturer’s instructions.
Hematoxylin and eosin (H&E)staining
After routine deparaffinization and rehydration, tissue sections were stained with hematoxylin for 5 min, followed by differentiation in acid ethanol. Sections were then rinsed with distilled water, stained with eosin, dehydrated through graded ethanol, and cleared in xylene. Finally, the sections were mounted with neutral balsam and photographed under a light microscope at 200× magnification.
Dihydroethidium staining
After euthanasia, rat cardiac tissues were rapidly excised, precooled on a −20°C freezing stage for 10 min, embedded in optimal cutting temperature compound, and frozen in metal molds. Tissues were sectioned into 5-μm-thick slices using a refrigerated cryostat and immediately mounted on precooled glass slides. Sections were incubated with dihydroethidium (DHE), followed by 4′,6-diamidino-2-phenylindole (DAPI) for 15 min at room temperature. After air-drying, the sections were mounted with anti-fade mounting medium and imaged under a confocal laser scanning microscope.
Observation of mitochondrial morphology
Fresh myocardial tissues were cut into 1 mm³ cubes and fixed in glutaraldehyde fixative at 4°C overnight in the dark, then post-fixed with 1% osmium tetroxide for 2 h. Tissues were subsequently dehydrated, embedded, polymerized, and sectioned using standard procedures, followed by staining. Images were acquired under a transmission electron microscope.
Cell culture
H9c2 cells (CL-0089) were obtained from Procell Company and cultured in dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified incubator with 5% CO2. Cell identity was verified by STR profiling. The culture medium was changed every 2 to 3 days, and cells were passaged upon reaching confluence. Cells from passages 3 to 5 were used for subsequent experiments.
Lentiviral transfection
sh-FGF18, OE-FGF18, and corresponding negative control (sh-NC and OE-NC) lentiviral vectors were purchased from DELIVECTORY Bioscience. The sh-FGF18 target sequence was 5′-GCAAGGAGTGTGTGTTCATCG −3′, and OE-FGF18 encoded the full-length FGF18 cDNA sequence (NM_019199.2). sh-NC and OE-NC were blank lentiviral vectors (LV-SFFV-EGFP and pLV2-CMV-MCS-EGFP-IRES-Puro, respectively). For transfection, the original medium was aspirated, and virus diluent containing 4 μg/mL polybrene was added at a multiplicity of infection of 10. After 16 h of incubation, the culture medium was refreshed. Transfection efficiency was confirmed by detecting FGF18 expression via Western blot analysis.
Cell grouping
To establish FGF18-knockdown and FGF18-overexpressing H9c2 cell lines, cells were randomized into five groups: Control, OE-NC, OE-FGF18, sh-NC, and sh-FGF18. Cells in the Control group were cultured under normal conditions, while the remaining four groups were transfected with the corresponding lentiviral vectors.
To investigate the effects of FGF18 on hypoxia/reoxygenation (H/R)-induced injury in H9c2 cells, cells were randomized into six groups: Control, H/R, OE-NC + H/R, OE-FGF18 + H/R, sh-NC + H/R, and sh-FGF18 + H/R. Cells in the Control group were cultured under standard conditions. Cells in the H/R group were exposed to hypoxic conditions (0.1% O2, 5% CO2, 94% N2) for 6 h, followed by reoxygenation in a humidified incubator with 5% CO2 at 37°C for 12 h (Cui et al., 2024); the remaining four groups were exposed to H/R after lentiviral transduction.
To explore the mechanism underlying the effects of FGF18 on H/R-induced H9c2 cells, cells were randomized into five groups: Control, H/R, OE-FGF18 + H/R, OE-FGF18 + H/R + EX-527, and H/R + EX-527. Cells in the control, H/R, and OE-FGF18 + H/R groups were treated as described above. Cells in the OE-FGF18 + H/R + EX-527 group were transfected with lentiviral vectors, then subjected to H/R modeling and incubated with 10 μM EX-527 during H/R (Jiang et al., 2021); cells in the H/R + EX-527 group were treated with 10 μM EX-527 during H/R (Jiang et al., 2021).
CCK-8 assay
H9c2 cells (5 × 103 cells/well) were seeded into a 96-well plate. After adhesion, cells were subjected to the corresponding treatments. 24 h after treatment, cell viability was assessed using a CCK-8 assay: 100 μL of fresh medium containing 10% CCK-8 solution was added to each well and incubated for the recommended time. Absorbance was then measured. After a further 2 h incubation in a humidified CO2 incubator, absorbance was measured at 450 nm using a microplate reader. The cell survival rate (%) was calculated as follows.
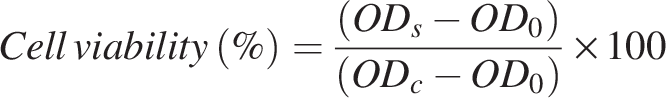
Flow cytometry
To assess mitochondrial membrane potential, H9c2 cells were seeded into six-well plates according to the experimental groups. JC-1 staining solution (1 mL) was added to each well, and cells were incubated for 20 min at 37°C. Cells were then trypsinized, washed twice, and resuspended in 1 mL of PBS. Mitochondrial membrane potential was measured using a flow cytometer.
For apoptosis detection, cells were stained with 5 μL of Annexin V-FITC and 5 μL of PI for 30 min at room temperature in the dark, then analyzed by flow cytometry.
For mitochondrial ROS detection, cells were incubated with 500 nM MitoSOX Red for 20 min at 37°C in the dark, washed, and then analyzed by flow cytometry.
Western blot
Total protein was extracted from cells and tissues using radioimmunoprecipitation assay (RIPA) lysis buffer, and protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit. Samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. After blocking with 5% nonfat milk for 2 h, membranes were incubated overnight at 4°C with primary antibodies against SIRT1 (1:1,000), PGC-1α (1:5,000), Mfn1 (1:2,000), FGF18 (1:2,000), and β-actin (1:20,000). The following day, membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (H + L) (1:1,000) or HRP-conjugated goat anti-mouse IgG (H + L) (1:1,000) for 1 h at room temperature. Protein bands were visualized using enhanced chemiluminescence (ECL) luminescent reagent and imaged with a gel imaging system. Protein band gray values were quantified using ImageJ software.
IP protein acetylation assay
IP was performed using a Protein A + G magnetic bead kit (P2179S, Beyotime). After cell lysis, the supernatant was collected and divided into the Input and IP groups. The Input group was denatured at 95°C for 10 min and stored at −80°C for subsequent use. The IP group was incubated with the primary antibody against the target protein overnight at 4°C with rotation. On the next day, magnetic beads were added to the protein–antibody mixture and incubated for 1 h at 4°C with rotation. After washing with pre-cooled phosphate buffered saline with tween-20 (PBST), 5 × loading buffer was added, followed by boiling for 5 min for subsequent Western blot analysis. Primary antibodies used for Western blot were as follows: PGC-1α (1:5,000), β-actin (1:20,000), and acetylated lysine antibody (1:800).
Statistical analysis
Statistical analyses and graphing were performed using SPSS 25.0 and GraphPad Prism 8.0. All data are presented as mean ± SD. Intergroup differences were assessed by one-way analysis of variance. p < 0.05 was considered statistically significant.
Results
FGF18 attenuates myocardial injury in rats with MIRI
Three weeks after AAV9 injection, myocardial tissues were harvested. FGF18 protein expression was significantly increased in the OE-FGF18 group and decreased in the sh-FGF18 group (Fig. 1A, B). To investigate the effect of MIRI on FGF18 expression, a MIRI model was established. Western blot analysis demonstrated that MIRI significantly downregulated FGF18 expression. In contrast, FGF18 expression was markedly upregulated in the OE-FGF18 + MIRI group, whereas it was further reduced in the sh-FGF18 + MIRI group (Fig. 1C, D). TTC staining revealed that FGF18 overexpression significantly reduced myocardial infarct size, while FGF18 knockdown exacerbated it. Myocardial infarct area was significantly smaller in the OE-FGF18 group than in the OE-NC group, whereas it was markedly larger in the sh-FGF18 group than in the sh-NC group (Fig. 1E, F). Histopathological analysis (Fig. 1G) revealed myocardial injury in MIRI groups. Compared with the normal architecture in sham controls, MIRI rats displayed disorganized myocardial fibers, loss of fascicular structure, interstitial erythrocyte extravasation, and uneven staining. These pathological changes were markedly ameliorated by FGF18 overexpression but exacerbated by FGF18 knockdown. Consistent with histological findings, serum levels of cardiac injury biomarkers CK-MB and cTnI were significantly elevated following MIRI (Fig. 1H, I). FGF18 overexpression effectively reduced these elevations, whereas FGF18 knockdown further increased both CK-MB and cTnI levels.
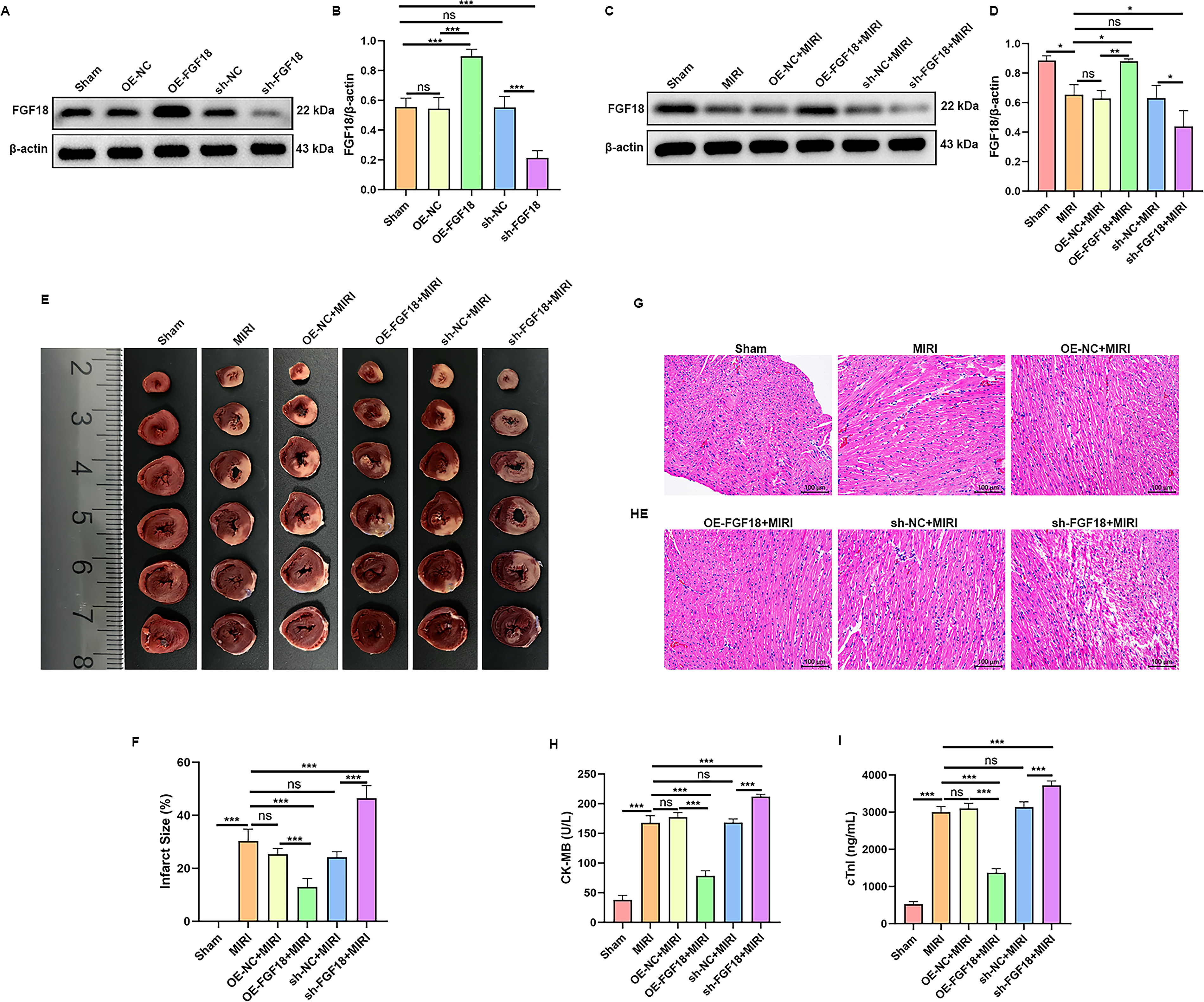
FGF18 alleviated myocardial injury in rats with myocardial ischemia-reperfusion. AAV9-FGF18 was used to establish FGF18-overexpressing rat models, while AAV9-shFGF18 was applied for FGF18-knockdown models. Three weeks later, myocardial tissues were collected for
FGF18 attenuates myocardial ischemia-reperfusion injury via modulating oxidative stress and mitochondrial homeostasis
To elucidate the underlying mechanism, oxidative stress in myocardial tissue was first assessed. DHE fluorescence staining revealed a significant increase in ROS following MIRI (Fig. 2A, B). This increase was attenuated by OE-FGF18 but exacerbated by sh-FGF18. Oxidative stress was evaluated by measuring serum levels of superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and malondialdehyde (MDA) (Fig. 2C–E). Compared with the Sham group, MIRI significantly reduced SOD and GSH-Px levels and increased MDA levels. FGF18 overexpression attenuated these changes, as evidenced by elevated SOD and GSH-Px and reduced MDA, whereas FGF18 knockdown exacerbated the oxidative stress response.

FGF18 alleviated MIRI in rats by reducing oxidative stress and regulating mitochondrial homeostasis. AAV9-FGF18 was used to establish FGF18 overexpressing rat models, while AAV9-shFGF18 was applied for FGF18 knockdown models. Three weeks post-viral injection, a rat MIRI model was established.
Transmission electron microscope (TEM) revealed the impact of MIRI and FGF18 manipulation on myocardial ultrastructure (Fig. 2F). Sham-operated rats exhibited intact intercalated discs and mitochondria with well-organized cristae. In contrast, MIRI induced marked ultrastructural damage, including disrupted intercalated discs, mitochondrial vacuolization, and loss of cristae. OE-FGF18 attenuated these injuries, preserving the integrity of both intercalated discs and mitochondria. Conversely, sh-FGF18 exacerbated the MIRI-induced structural damage.
The SIRT1/PGC-1α axis is a key regulator of mitochondrial homeostasis. Accordingly, its protein expression in response to FGF18 manipulation was assessed. Western blot analysis showed that MIRI significantly downregulated the protein levels of SIRT1, PGC-1α, and the mitochondrial fusion protein Mfn1 (Fig. 2G–J). This downregulation was effectively reversed by FGF18 overexpression. Conversely, FGF18 knockdown further suppressed the expression of these proteins.
FGF18 ameliorates H/R-injury in cardiomyocytes by improving mitochondrial homeostasis
Lentivirus-mediated manipulation successfully altered FGF18 expression in H9c2 cells, as confirmed by Western blot (Fig. 3A, B). To assess functional outcomes, cells were subjected to H/R. FGF18 overexpression significantly enhanced cell viability (Fig. 3C) and attenuated H/R-induced apoptosis (Fig. 3D, E). Conversely, FGF18 knockdown reduced viability and promoted apoptosis. Oxidative stress and mitochondrial function were next assessed. ELISA revealed that, compared with H/R alone, FGF18 overexpression significantly reduced MDA and increased SOD and GSH-Px levels, whereas FGF18 knockdown exacerbated these oxidative stress markers (Fig. 3F–H). Consistently, mitochondrial ROS production surged following H/R, an effect that was attenuated by FGF18 overexpression and further augmented by FGF18 knockdown. (Fig. 3I, J). Western blot analysis showed that H/R downregulated the expression of FGF18, SIRT1, PGC-1α, and Mfn1 (Fig. 3K–O). Notably, FGF18 overexpression effectively reversed these H/R-induced suppressions, whereas FGF18 knockdown further diminished their expression. Collectively, these results demonstrate that FGF18 attenuates H/R injury by mitigating oxidative stress and restoring mitochondrial homeostasis.
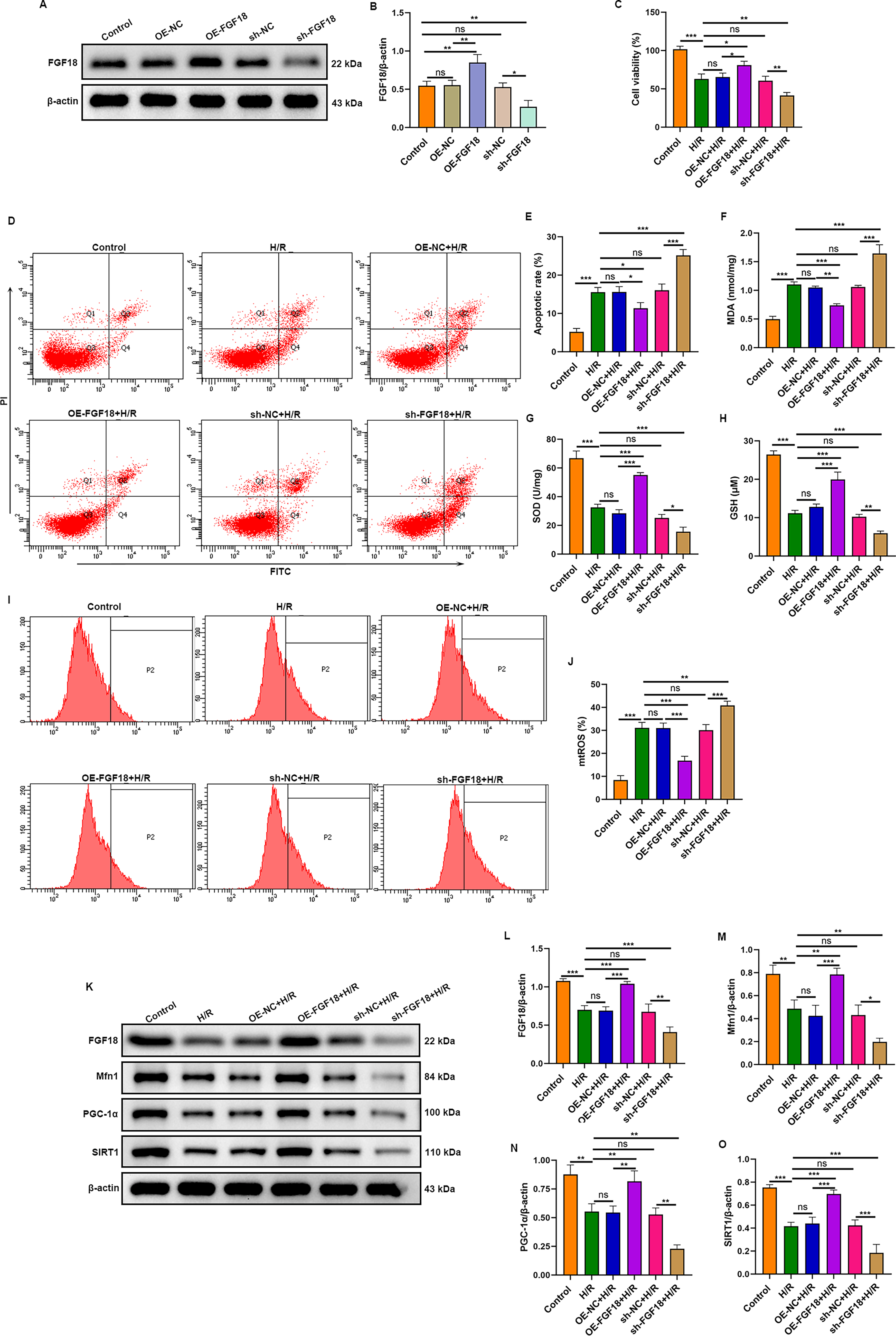
FGF18 attenuates H/R-induced apoptosis in H9c2 cells by improving mitochondrial homeostasis. H9c2 cells were transfected with lentiviruses for FGF18 knockdown or overexpression.
FGF18 improves mitochondrial homeostasis via SIRT1-mediated PGC-1α deacetylation to alleviate H/R-injury in H9c2 cells
To delineate the mechanistic role of SIRT1, its specific inhibitor EX-527 was employed. The CCK-8 assay demonstrated that H/R impaired cell viability. This impairment was mitigated by FGF18 overexpression but exacerbated by SIRT1 inhibition, which abolished the protective effect of FGF18 (Fig. 4A). Consistent with this, H/R induced mitochondrial dysfunction, characterized by elevated ROS and decreased mitochondrial membrane potential (increased JC-1 green fluorescence). FGF18 overexpression attenuated these dysfunctions. SIRT1 inhibition not only intensified mitochondrial damage beyond H/R levels but also abolished the protective effects of FGF18 (Fig. 4B, C). Consequently, this aggravated mitochondrial dysfunction led to a significant increase in cardiomyocyte apoptosis (Fig. 4D).
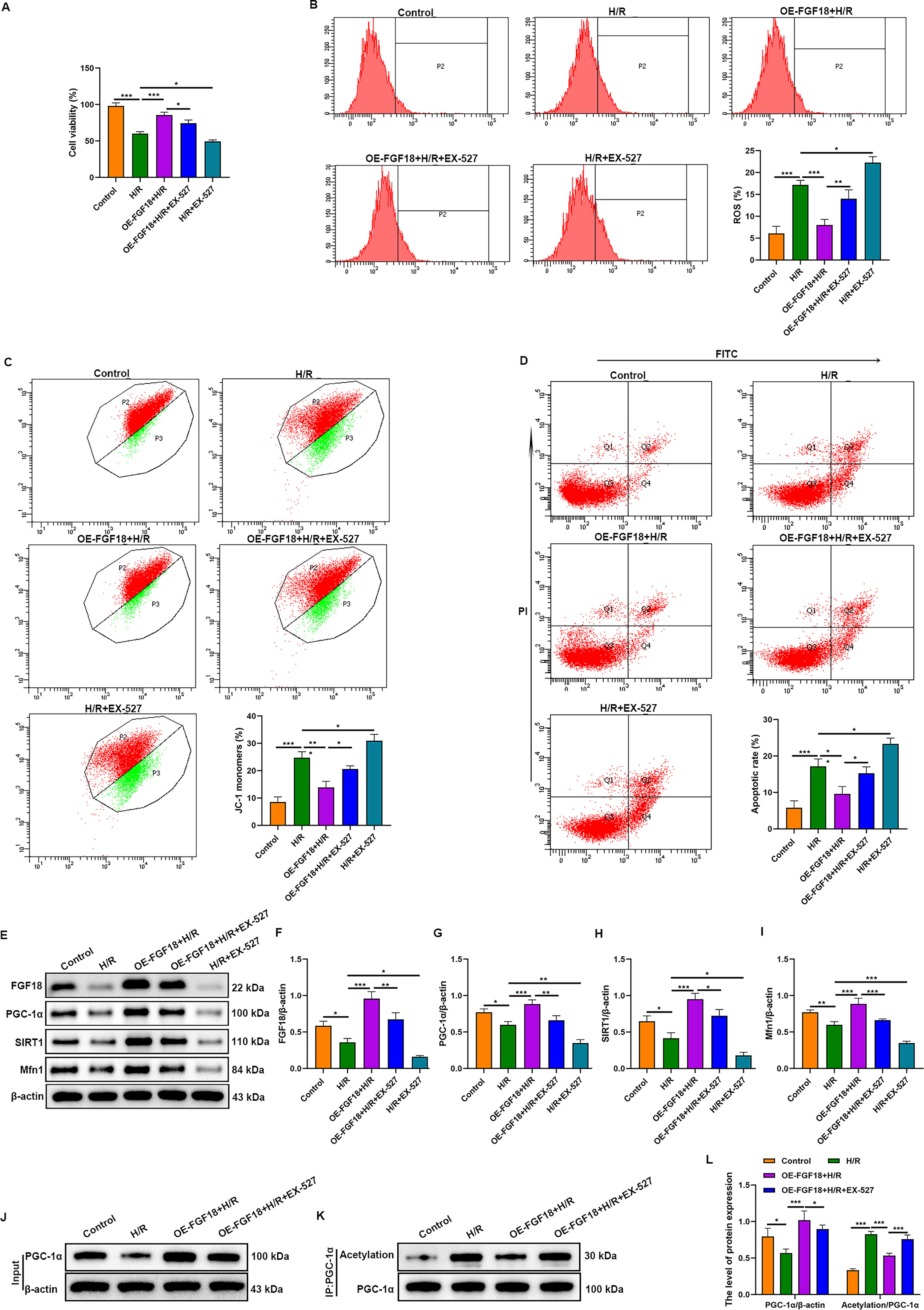
FGF18 regulates SIRT1-mediated PGC-1α deacetylation, maintains mitochondrial homeostasis, and inhibits hypoxia/reoxygenation-induced cardiomyocyte apoptosis. H9c2 cells were transfected with lentiviruses for FGF18 overexpression. Subsequently, the cells were cultured under hypoxic conditions (0.1% O2, 95% N2, 5% CO2) for 6 h, followed by incubation at 37°C with 5% CO2 for 12 h to establish the H/R model. Cells in the OE–FGF18 + H/R + EX-527 group and H/R + EX-527 group were incubated with 10 μM EX-527 during the H/R process.
Western blot analysis confirmed that H/R downregulated the expression of FGF18, SIRT1, PGC-1α, and Mfn1 in H9c2 cells (Fig. 4E–I). This suppression was counteracted by FGF18 overexpression. Notably, SIRT1 inhibition not only reduced the expression of these proteins but also abolished the upregulatory effect of FGF18 overexpression.
SIRT1, a key mammalian NAD+-dependent deacetylase, mediates PGC-1α deacetylation to enhance mitochondrial function (Pang et al., 2024). To investigate whether FGF18 acts via SIRT1-mediated PGC-1α deacetylation in MIRI, we performed an IP protein acetylation assay. The results showed that FGF18 overexpression inhibited H/R-induced PGC-1α acetylation, whereas SIRT1 inhibition reversed this effect (Fig. 4J–L), indicating that FGF18 ameliorates MIRI by activating SIRT1 to enhance PGC-1α deacetylation.
Our findings demonstrate that FGF18 preserves mitochondrial function and inhibits cardiomyocyte apoptosis via SIRT1-mediated deacetylation of PGC-1α, thereby attenuating H/R injury.
Discussion
FGF18 regulates diverse processes including development, tissue repair, and disease progression (Figueroa et al., 2019). While its protective role against ischemia-reperfusion injury has been established in hepatic and renal models (Wang et al., 2024; You et al., 2025), its function in the heart remains unexplored. To address this, we employed gain- and loss-of-function approaches in a rat model of MIRI. FGF18 overexpression markedly reduced infarct size, suppressed the release of cardiac injury biomarkers (cTnI and CK-MB), and improved histopathological outcomes, indicating a cardioprotective effect.
Cardiac function is highly energy-dependent, a demand primarily met by cardiomyocyte mitochondria. Ischemia-reperfusion injury severely disrupts mitochondrial structure and function, a process mediated largely by oxidative stress. The resulting overproduction of ROS impairs the electron transport chain, damages mitochondrial components, and ultimately triggers cell death (Bhatti, et al., 2017; van der Pol, et al., 2019). In our MIRI model, TEM revealed characteristic mitochondrial damage—including vacuolation, cristae loss, and disrupted intercalated discs—alongside decreased Mfn1 expression. FGF18 overexpression counteracted these changes, preserving mitochondrial and tissue ultrastructure, upregulating Mfn1, and reducing oxidative stress markers (ROS, MDA). Conversely, FGF18 knockdown exacerbated both mitochondrial injury and oxidative damage.
SIRT1, a key regulator of cellular homeostasis, modulates diverse processes including oxidative stress, metabolism, and apoptosis through interactions with partners such as PGC-1α (Huang et al., 2022). In the heart, SIRT1 overexpression confers robust protection against oxidative injury by enhancing antioxidant defenses (e.g., manganese superoxide dismutase) via transcription factors like FoxO1 (Zhang et al., 2020). Its cardioprotective role extends to mitigating inflammatory cardiotoxicity by suppressing inflammasome activation (Sun et al., 2020) and inhibiting apoptosis through both p53-dependent (Xu et al., 2019) and PGC-1α-mediated pathways (Tang et al., 2019). Collectively, these studies establish SIRT1 as a central guardian of cardiomyocyte viability under stress. PGC-1α is a master regulator of mitochondrial biogenesis and oxidative metabolism in high-energy-demand tissues, including the heart (Halling and Pilegaard, 2020). It functions downstream of SIRT1 within the classical SIRT1/PGC-1α signaling axis, which is central to mitochondrial homeostasis. SIRT1-mediated deacetylation activates PGC-1α, thereby driving mitochondrial biogenesis and enhancing functional capacity (Peng et al., 2023). This study confirmed that SIRT1 and PGC-1α protein levels were reduced in MIRI, and that FGF18 overexpression reversed, whereas FGF18 knockdown further suppressed their expression; however, the mRNA levels of SIRT1 and PGC-1α were not examined. Therefore, whether FGF18 regulates SIRT1 and PGC-1α at the transcriptional level or via posttranscriptional/protein stability mechanisms remains unclear. Future studies will employ qRT-PCR to assess SIRT1 transcript expression, thereby clarifying the regulatory hierarchy and molecular mechanism by which FGF18 modulates SIRT1.
To delineate the underlying mechanism, an in vitro H/R model was established in H9c2 cells. H/R injury decreased cell viability and mitochondrial membrane potential, while increasing mitochondrial ROS and apoptosis, concomitant with the downregulation of FGF18, SIRT1, PGC-1α, and Mfn1. FGF18 overexpression rescued these phenotypic and molecular alterations. Critically, the SIRT1 inhibitor EX-527 abolished the protective effects of FGF18. Taken together, our data establish that FGF18 alleviates myocardial ischemia-reperfusion injury by activating the SIRT1/PGC-1α pathway, thereby attenuating oxidative stress and mitochondrial dysfunction.
The SIRT1/PGC-1α pathway, a central regulator of mitochondrial and metabolic homeostasis, is subject to multifaceted regulation—including phosphorylation, acetylation, and transcriptional control—that modulates its activity. Key upstream inputs include the adenosine monophosphate-activated protein kinase (AMPK) pathway, which transduces mechanical signals (e.g., via Piezo1) to enhance mitochondrial function through SIRT1/PGC-1α activation (Chen et al., 2026), and the PI3K/AKT pathway, which can upregulate SIRT1 expression in disease contexts (Liao et al., 2022). In addition, ERK1/2 signaling has been shown to influence PGC-1α acetylation status via SIRT1 (Collier and Schnellmann, 2020). This study confirms that FGF18 alleviates myocardial ischemia-reperfusion injury by regulating SIRT1-mediated PGC-1α deacetylation, providing a novel therapeutic target for the clinical treatment of MIRI.
This study has several limitations. First, clinical data validating FGF18 expression in patient tissues are lacking. Second, the specific PGC-1α deacetylation sites downstream of FGF18-activated SIRT1 remain undefined, pending mass spectrometry identification.
Authors’ Contributions
Conceptualization, writing—original draft, and writing—review and editing: H.Y.; Data curation: G.H.; Methodology: X.C.; Supervision: F.L.
Ethical Considerations
All animal experiments fully complied with the ARRIVE guidelines, the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978), and the relevant principles specified in the Standards for Laboratory Animal Care and Use. The experiments were approved by the Ethics Committee of Binzhou Medical University (Approval No.2024-L094).
Data Availability
The data from this study are available upon reasonable request to the corresponding author.
Footnotes
Acknowledgments
The authors acknowledge the funding support from
Disclosure Statement
All authors declare no conflict of interest.
Funding Information
This research was supported by
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.