
Abstract
Idiopathic pulmonary fibrosis (IPF) is characterized by persistent fibroblast activation and progressive extracellular matrix remodeling, leading to irreversible lung architectural distortion. Although high-throughput omics approaches have advanced understanding of IPF pathogenesis, most studies have relied on single-omics analyses, limiting cross-layer functional interpretation. Here, we applied an integrated multi-omics strategy to characterize coordinated molecular alterations in IPF fibroblasts. Primary lung fibroblasts derived from patients with IPF and control subjects were analyzed using integrated transcriptomic, proteomic, and metabolomic profiling. Publicly available RNA-sequencing data deposited in the Gene Expression Omnibus (GEO; GSE301181) were used for transcriptomic analysis, while proteomic and metabolomic analyses were newly performed in a donor-matched subset of 10 IPF patients and 10 control subjects. Differentially expressed genes (DEGs), proteins (DEPs), and metabolites were identified using standardized statistical criteria, and cross-layer integration was conducted to identify molecules showing concordant regulation. Transcriptomic analysis identified 1,689 DEGs in IPF fibroblasts, whereas proteomic profiling initially quantified 6,236 proteins; after restricting analyses to peptide-supported proteins (≥2 unique peptides), 56 high-confidence DEPs were retained. Integration of transcriptomic and proteomic datasets identified 10 peptide-supported molecules exhibiting concordant regulation at both the mRNA and protein levels. Metabolomic profiling demonstrated significant reductions in metabolites involved in redox balance, lipid metabolism, glycolysis, nitrogen metabolism, and amine metabolism, indicating broad metabolic reprogramming associated with persistent fibroblast activation. Together, these findings suggest that IPF fibroblast activation is accompanied by coordinated transcriptional, translational, and metabolic reprogramming and highlight the value of integrated multi-omics analysis for interpreting regulatory features of fibrotic fibroblasts.
Introduction
The pathogenesis of idiopathic pulmonary fibrosis (IPF) involves a self-perpetuating cycle of dysregulated fibroblast activity and progressive structural remodeling of the lung parenchyma, driven by persistent myofibroblast activation and excessive extracellular matrix (ECM) deposition (Mei et al., 2021; Sgalla et al., 2018). Although antifibrotic agents such as nintedanib and pirfenidone are currently utilized in clinical practice, their ability to slow disease progression remains limited, and they fail to halt or reverse the underlying fibrotic process, highlighting the incomplete therapeutic coverage of current treatment strategies (Denis et al., 2025; Man et al., 2024). These clinical limitations underscore the need to move beyond isolated signaling pathways and instead adopt a systems-level framework that captures the coordinated interplay between transcriptional regulation, protein abundance, and downstream metabolic reprogramming that sustains fibrotic persistence (Dasgupta, 2025; Zheng et al., 2022).
Accumulating experimental and clinical evidence has shifted the conceptual framework of IPF away from a purely inflammation-centered model toward one in which aberrant fibroblast activation and persistence constitute the primary driver of disease progression (Wynn, 2011). In physiological tissue repair, fibroblast activation is tightly regulated and resolves once injury subsides. In contrast, the fibrotic lung environment promotes the survival of activated fibroblasts and myofibroblasts that evade apoptotic clearance, thereby sustaining ECM production and matrix remodeling (King et al., 2011; Selman et al., 2001). These maladaptive cellular states are thought to arise from coordinated disturbances in intracellular signaling networks coupled with rewiring of cellular metabolic programs.
Recent advances in single-cell profiling have further revealed that fibroblasts within fibrotic lungs are not a uniform population but instead comprise multiple functionally distinct subsets with differential capacities for matrix deposition, tissue remodeling, and interaction with neighboring cell types (Peyser et al., 2019; Xie et al., 2018). This profound fibroblast activation and heterogeneity are now recognized as central to the pathogenesis of various fibrotic diseases, necessitating a more granular understanding of their functional states (Zhang et al., 2025). Consequently, this degree of cellular heterogeneity highlights an inherent limitation of analytical strategies that interrogate disease biology at only a single molecular layer.
Consistent with this limitation, many molecular candidates implicated in IPF pathogenesis have been identified predominantly through single-level omics analyses, most commonly transcriptomics. While such studies have yielded valuable insights into disease-associated gene expression patterns, they offer only a partial representation of the regulatory landscape governing fibrotic activation (Liu et al., 2016; Subramanian et al., 2020; Vogel and Marcotte, 2012). Transcriptional changes are frequently decoupled from downstream protein abundance and metabolic activity due to post-transcriptional control, protein turnover, and context-dependent metabolic rewiring, limiting the functional interpretability of transcriptome-centric observations (Liu et al., 2016; Vogel and Marcotte, 2012).
In contrast, integrative multi-omics strategies that combine transcriptomic, proteomic, and metabolomic information have been increasingly adopted in oncology and metabolic disease research to strengthen mechanistic inference through cross-layer validation (Heo et al., 2021; Reel et al., 2021). However, comparable integrative frameworks have been applied only sparingly in IPF, and a coordinated tripartite analysis encompassing gene expression, protein dynamics, and metabolic outputs has yet to be systematically established in the context of pulmonary fibrogenesis.
Accordingly, the objective of the present study was to explore fibrosis-associated regulatory features using an integrated multi-omics approach in primary lung fibroblasts from patients with IPF. By cross-referencing differentially expressed genes (DEGs), proteins (DEPs), and metabolites (DEMs) across molecular layers, we sought to identify regulatory nodes that exhibit consistent perturbation at transcriptional, translational, and metabolic levels, thereby providing an exploratory framework for interpreting molecular processes associated with IPF fibroblast activation.
Materials and Methods
Study subjects
Lung fibroblasts from patients with IPF were obtained from the biobank of Soonchunhyang University Hospital (Bucheon, Korea), following approval from the Institutional Review Board (IRB numbers: SCHCA-IRB-2018-10-034 and 201910-BR-058). Informed written consent was obtained from all participants. Subjects underwent comprehensive clinical evaluation, including medical history review, chest X-ray, high-resolution computed tomography, and pulmonary function testing. Patients with evidence of collagen vascular diseases were excluded based on clinical and laboratory assessments. The diagnosis of IPF was established according to the 2011 and 2018 ATS/ERS/JRS/ALAT guidelines (Raghu et al., 2011; Raghu et al., 2018).
Fibroblast culture
Primary lung fibroblasts were isolated from surgical biopsy specimens obtained from a donor-matched subset of 10 patients with histologically confirmed usual interstitial pneumonia and from histologically normal lung tissue of 10 control subjects who underwent resection for stage I or II lung cancer. Fibroblast culture protocols followed previously published methods (Lee et al., 2017). Briefly, tissue samples were minced and cultured in 150 cm2 flasks containing Dulbecco’s Modified Eagle Medium (Lonza Walkersville, MD, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Rockford, IL, USA), 2 mmol/L glutamine, and 1% penicillin–streptomycin–amphotericin B (Lonza, Basel, Switzerland). Cultures were maintained at 37°C in a humidified 5% CO2 incubator and serially passaged to obtain a morphologically homogeneous population of adherent fibroblasts.
Study design and sample sources
Transcriptomic analysis was based on previously generated RNA sequencing data derived from primary lung fibroblasts of 33 patients with IPF and 10 control subjects, which have been deposited in the NCBI Gene Expression Omnibus under accession number GSE301181. In the present study, proteomic and metabolomic analyses were newly performed using primary lung fibroblasts obtained from a donor-matched cohort consisting of 10 control subjects and 10 patients with IPF. The proteomic samples correspond to this same donor-matched cohort, and the proteomic data analyzed in this study are publicly available in figshare (https://doi.org/10.6084/m9.Figs.hare.24539938.v2). These paired samples were used for integrated analysis across proteomic and metabolomic layers.
Total RNA sequencing and data processing
Publicly available RNA sequencing data (GSE301181) were reanalyzed in this study. Differential expression analysis was performed using the Biomedical Analysis Workspace (hyper.schedulerju.com), a clinical bioinformatics analysis platform that implements RNA-seq normalization, differential expression testing, and functional enrichment workflows. Expression data were normalized using edgeR with the trimmed mean of M-values method and represented as log2 counts per million. Differentially expressed genes (DEGs) were identified based on an absolute log2 fold change ≥ 1 and an FDR-adjusted p-value <0.05.
Proteomics analysis
Proteomic profiling was performed using donor-matched IPF and control lung fibroblast samples. Proteins were extracted in a urea-based lysis buffer containing protease inhibitors, and protein concentrations were determined using a BCA assay. Samples were digested using the filter-aided sample preparation method with trypsin (1:50, w/w). The resulting peptides were desalted using C18 columns and subjected to LC–MS/mass spectrometry (MS) analysis.
Peptide analysis was performed using an Orbitrap Exploris 480 mass spectrometer coupled to an Ultimate 3000 UPLC system. Peptides were separated using trap and analytical columns with a linear gradient and analyzed in data-dependent acquisition mode. Raw MS/MS data were processed using Proteome Discoverer™ (v3.1) with the UniProt Homo sapiens database and the SEQUEST HT algorithm, applying a false discovery rate (FDR) of <1% at the peptide-spectrum match/peptide level. Protein quantification was based on precursor ion intensities with total peptide amount normalization, and fold change was calculated based on protein abundance ratios. DEPs were identified using the t-test (Background Based) implemented in Proteome Discoverer™ (v3.1), with FDR-adjusted p value <0.05 and |log2 fold change| ≥ 1 as the significance criteria. To improve protein-level confidence for downstream interpretation, differential expression testing and integrative analyses were restricted to proteins supported by at least two unique peptides.
The raw proteomic data supporting the findings of this study have been deposited in the Figshare public repository and are available at https://doi.org/10.6084/m9.figshare.24539938.v2.
Metabolomics analysis
Metabolomic profiling was performed using the same donor-matched IPF and control fibroblast samples analyzed in the proteomic study. Analyses were conducted by Human Metabolome Technologies, Inc. (HMT, Tsuruoka, Japan) using capillary electrophoresis time-of-flight MS in both cationic and anionic modes as previously described (Soga et al., 2003). Briefly, cultured cells were washed, followed by methanol treatment. Milli-Q water containing internal standards (10 µM) was then added to the extract. After centrifugation (2,300 g, 4°C, 5 min), the supernatant was filtered through a 5-kDa cut-off membrane. The resulting extracts were dried under vacuum, reconstituted in Milli-Q water, and analyzed in both cationic and anionic modes. Peaks were extracted using MasterHands software and annotated against the HMT standard library based on migration time (±0.5 min) and m/z (±10 ppm). Peak areas were converted to relative peak areas using internal-standard normalization as defined by the vendor, and the peak detection limit was determined based on signal-to-noise ratio (S/N = 3). Normalized metabolite data were subjected to multivariate analyses, including principal component analysis and hierarchical cluster analysis. PCA and hierarchical clustering analysis were performed using the vendor’s statistical analysis software. Between-group differences in individual metabolite levels were evaluated using Welch’s t-test. Statistical significance of metabolite contributions to principal components was assessed as previously described (Yamamoto et al., 2014), with p values <0.05 considered significant. The metabolomics data generated in this study have been deposited in Figshare and are publicly available at https://doi.org/10.6084/m9.figshare.31032778.v1.
Gene ontology enrichment analysis
Gene ontology (GO) analysis was performed using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt; https://www.webgestalt.org/) (Zhang et al., 2005) to characterize the functional distribution of the 10 transcript–protein concordant genes across the biological process, cellular component, and molecular function categories. The human genome was used as the reference background, and results were summarized at the category level to describe the overall functional composition of the concordant gene set.
Statistical analysis
All statistical and omics analyses were performed using R and the Biomedical Analysis Workspace (hyper.schedulerju.com), a clinical bioinformatics analysis platform. Differential expression results from transcriptomic, proteomic, and metabolomic datasets were integrated using the analysis outputs described above, with FDR adjustment applied where appropriate. Correlation analyses between transcriptomic and proteomic expression levels were performed using Spearman’s rank correlation in R. A two-tailed p-value < 0.05 was considered statistically significant.
Results
Patient characteristics
The clinical characteristics of the study participants are summarized in Supplementary Table S1. A total of 43 individuals were included, comprising 33 patients with IPF and 10 control subjects. Age, sex distribution, and smoking status were comparable between the two groups. As expected, pulmonary function was significantly impaired in the IPF group, with lower predicted forced vital capacity and diffusion capacity for carbon monoxide compared with controls.
DEGs and stepwise multi-omics integration in IPF fibroblasts
An overview of the stepwise multi-omics results generated in this study is summarized in Figure 1. Transcriptomic analysis of the GSE301181 dataset identified 1,689 DEGs in IPF fibroblasts compared with controls, comprising 1,402 upregulated and 287 downregulated genes (Supplementary Table S2). Proteomic profiling was performed using fibroblasts from a paired subset of 10 IPF patients and 10 matched control subjects. A total of 6,236 proteins were initially quantified. After restricting the dataset to proteins supported by at least two unique peptides (n = 5,765), differential abundance testing was performed, yielding 56 high-confidence DEPs for downstream interpretation (Supplementary Table S3). Integration of transcriptomic and proteomic datasets was then performed using this revised high-confidence protein set. A Venn diagram illustrating the overlap between DEGs and high-confidence DEPs is shown in Figure 2A. These concordant genes included TXNIP, FBLN1, S100A4, TRPA1, FGL2, HNMT, PLPP1, HOXA5, JUP, and TMEM35A (Table 1).
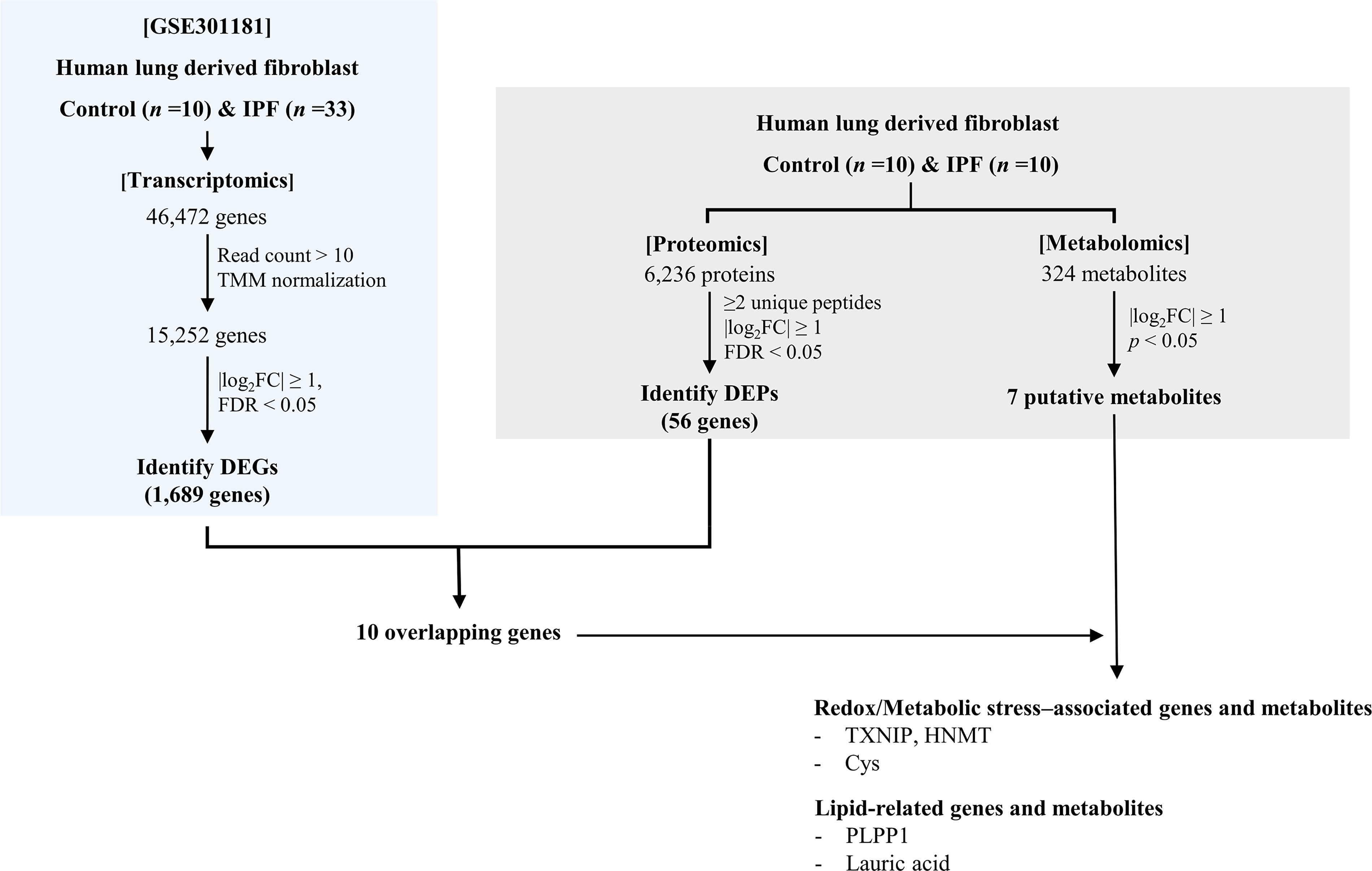
Stepwise overview of multi-omics analyses performed in idiopathic pulmonary fibrosis fibroblasts. Transcriptomic profiling identified differentially expressed genes, followed by proteomic analysis in paired samples. Integration of transcriptomic and proteomic datasets yielded concordantly regulated molecules, which were subsequently examined in relation to metabolomic alterations identified in the same fibroblast samples. DEG, differentially expressed gene; DEP: differentially expressed protein; FC, fold change; FDR, false discovery rate; IPF, idiopathic pulmonary fibrosis; TMM: trimmed mean of M-values.

Integrated analysis of transcriptomic and proteomic alterations in IPF fibroblasts.
Transcript–Protein Concordance Analysis of 10 Molecules in IPF
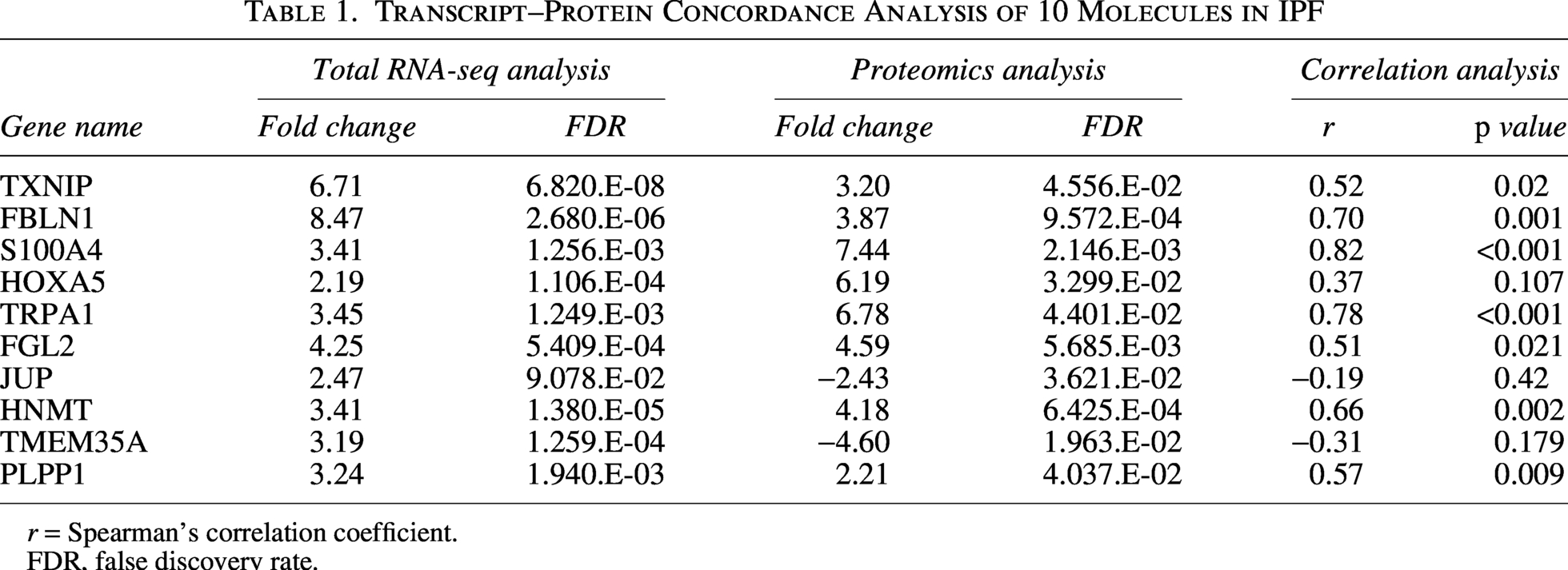
r = Spearman’s correlation coefficient.
FDR, false discovery rate.
Functional characterization of transcript–protein concordant genes
GO analysis was performed using the 10 transcript–protein concordant genes to summarize their category-level distribution across biological process, cellular component, and molecular function domains (Fig. 2C–E). In the biological process domain, the concordant genes were primarily assigned to metabolic process categories, along with broader categories related to biological regulation and response to stimulus. In the cellular component domain, these genes were primarily annotated to extracellular space, cytosol, vesicles, and membrane-enclosed lumen. In the molecular function domain, the most frequent assignments included protein binding, ion binding, and metabolism-related functional categories such as transferase and enzyme regulator activities.
Correlation of transcript–protein concordant genes
Spearman correlation analysis between transcriptomic expression and protein abundance was performed for the 10 transcript–protein concordant molecules retained after high-confidence peptide filtering. Most candidates showed positive cross-layer correlations, with statistically significant positive correlations for TXNIP, FBLN1, S100A4, TRPA1, FGL2, HNMT, and PLPP1 (Table 1). HOXA5 showed a positive but nonsignificant correlation, whereas only JUP and TMEM35A showed negative correlations (Table 1).
Integrated metabolomic profiling
To relate metabolite alterations to the revised transcript–protein concordant set, we evaluated the seven reduced DEMs in the context of the 10 peptide-supported concordant molecules (TXNIP, FBLN1, S100A4, HOXA5, TRPA1, FGL2, JUP, HNMT, TMEM35A, and PLPP1). The DEMs (Cys, lauric acid, triethanolamine, glyceraldehyde 3-phosphate, 2,3-diphosphoglyceric acid, octopamine/dopamine, and dimethylaminoethanol) spanned redox balance, lipid metabolism, glycolysis, nitrogen metabolism, and amine metabolism (Table 2). Within the concordant set, TXNIP and HNMT aligned with a redox/metabolic stress axis (Han et al., 2021; Horton et al., 2001), PLPP1 aligned with lipid-related processes (Xu et al., 2000), and TRPA1 and FGL2 aligned with stimulus-response signaling (Liu et al., 2010; Yang et al., 2024), whereas FBLN1, S100A4, and HOXA5 represented an activated fibroblast/ECM-related signature (Liu et al., 2019; Lv et al., 2024; Xia et al., 2017). Together, these results describe concurrent cross-layer changes between the concordant transcript–protein signature and metabolite depletion in IPF fibroblasts.
Seven Metabolites Significantly Reduced in IPF Fibroblasts Compared with Controls among 324 Detected Metabolites
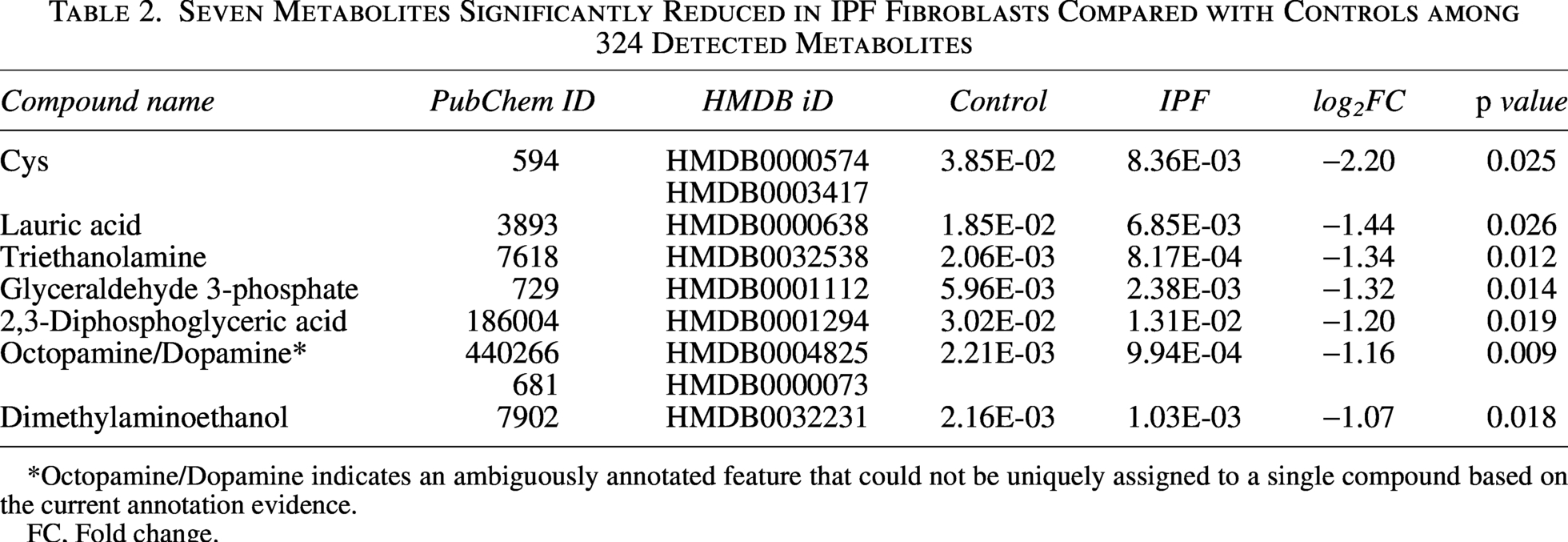
Octopamine/Dopamine indicates an ambiguously annotated feature that could not be uniquely assigned to a single compound based on the current annotation evidence.
FC, Fold change.
Discussion
In this study, we performed an integrated multi-omics analysis of lung fibroblasts derived from patients with IPF, combining transcriptomic, proteomic, and metabolomic data to identify fibrosis-associated regulatory features across multiple molecular layers. Cross-layer integration revealed a limited set of molecules showing concordant regulation at both the mRNA and protein levels, indicating coordinated transcriptional and translational changes associated with persistent fibroblast activation. In parallel, metabolomic profiling demonstrated consistent reductions in metabolites involved in redox balance, lipid metabolism, glycolysis, nitrogen metabolism, and amine metabolism, highlighting broad metabolic reprogramming in IPF fibroblasts. Notably, coordinated alterations in proteins and metabolites related to redox balance, lipid-related processes, and stimulus–response programs illustrate how transcriptional, proteomic, and metabolic layers converge on shared pathogenic features. Together, these findings suggest that fibroblast pathogenicity in IPF reflects multi-layer molecular reorganization rather than isolated alterations at a single omics level.
The relatively small overlap between transcriptomic and proteomic alterations highlights the limitations of single-omics approaches in capturing functionally relevant disease mechanisms. Because mRNA abundance does not consistently predict protein levels due to post-transcriptional regulation, differences in protein stability, and context-dependent translational control, candidates exhibiting concordant regulation across multiple molecular layers are more likely to reflect biologically meaningful regulatory processes (Liu et al., 2016; Vogel and Marcotte, 2012). Although several studies have applied multi-omics approaches to IPF lung tissues and patient cohorts (Pattaroni et al., 2024; Ruan et al., 2023), and others have combined multi-omics with single-cell transcriptomic analyses to infer cell-type–specific features (Jiang et al., 2025), systematic integration of transcriptomic, proteomic, and metabolomic data within the same, well-defined fibroblast model remains limited in IPF. In this context, the present study provides an integrated analytical framework that enables direct, cross-layer validation of molecular alterations associated with fibroblast activation, extending beyond descriptive single-omics observations.
Beyond transcriptomic and proteomic concordance, metabolomic profiling provides a functional context for interpreting fibroblast activation in IPF. The depletion of metabolites spanning redox balance, lipid metabolism, glycolytic intermediates, nitrogen metabolism, and amine metabolism is consistent with broad metabolic remodeling accompanying persistent fibroblast activation, rather than an isolated alteration in a single pathway. In this framework, the concordant transcript–protein set offers an interpretable scaffold linking metabolic changes to established programs of fibroblast activation and extracellular remodeling.
Within the concordant set, several candidates provide mechanistic anchors for interpreting the metabolite depletion as part of an activated fibroblast state. TXNIP has been linked to fibroblast-to-myofibroblast differentiation and redox-associated signaling in fibrotic contexts, which is compatible with reduced thiol/redox-related metabolites such as cysteine (Han et al., 2021). HNMT, although classically studied in histamine metabolism, may reflect broader rewiring of amine-related metabolic regulation accompanying activated cellular states (Horton et al., 2001), consistent with alterations in amine-related metabolites. PLPP1 regulates lipid phosphate metabolism and lysophospholipid signaling (Xu et al., 2000), providing a plausible context for the observed depletion of lipid-related metabolites such as lauric acid within a broader lipid-handling shift. TRPA1 and FGL2 highlight stimulus–responsive pathways that may influence fibroblast behavior within a profibrotic microenvironment (Liu et al., 2010; Yang et al., 2024). In parallel, FBLN1, S100A4, and HOXA5 have established links to ECM remodeling and profibrotic fibroblast phenotypes (Liu et al., 2019; Lv et al., 2024; Xia et al., 2017), supporting the interpretation that metabolic remodeling co-emerges with stable extracellular and activation programs. Taken together, these observations favor a process-level interpretation in which metabolic stress adaptation, stimulus-response signaling, and extracellular remodeling programs co-occur in activated IPF fibroblasts. This framework is consistent with the well-recognized decoupling between mRNA abundance and protein levels across omics layers, which limits strict one-to-one mapping between molecular readouts (Liu et al., 2016; Vogel and Marcotte, 2012).
To provide a clinico-molecular context, we performed exploratory Spearman correlation analyses within the IPF cohort (n = 10). These analyses suggested inverse associations of lauric acid with FVC% predicted and DLco% predicted and positive associations of HNMT, PLPP1, and HOXA5 transcript levels with FVC% predicted (Supplementary Table S4). Given the limited sample size, these findings should be interpreted as hypothesis-generating and require validation in larger independent cohorts.
Several limitations of this study should be acknowledged. Functional validation experiments were not performed to establish causal roles for individual candidates identified through cross-layer integration. In addition, mechanistic interpretation of the metabolomic alterations remains limited because targeted functional assays to directly quantify redox capacity, lipid handling, or pathway-specific flux were not performed. The limited sample size of the proteomic and metabolomic analyses may also reduce statistical power and affect the stability of the detected signals, and the lack of validation in independent cohorts or in vivo models may limit the generalizability of the findings. Therefore, the multi-omics findings presented here should be considered exploratory and hypothesis-generating. Future studies incorporating targeted functional assays, larger sample sizes, and external validation will be required to substantiate the mechanistic relevance of the multi-omics candidates identified here.
Overall, this study suggests that persistent activation of IPF fibroblasts is accompanied by coordinated reprogramming at the transcriptomic, proteomic, and metabolomic levels. The integrated multi-omics analysis provides supportive evidence that adaptive metabolic regulation may represent an important component of fibrotic pathobiology, although these findings should be interpreted cautiously given the limited sample size and lack of external validation.
Conclusion
This study suggests that IPF fibroblasts undergo coordinated transcriptional, translational, and metabolic reprogramming. Integrated multi-omics analysis identified a peptide-supported set of concordantly regulated transcript–protein features and accompanying depletion of metabolites spanning redox balance, lipid metabolism, glycolysis, nitrogen metabolism, and amine metabolism. Given the limited sample size of the proteomic and metabolomic cohorts, these findings should be regarded as exploratory and hypothesis-generating, and they provide a framework for future mechanistic and translational studies targeting fibroblast persistence in IPF.
Authors’ Contributions
Conceptualization: S.W.P. and J.-U.L.; Data curation: J.-U.L., M.K.K., and S.-L.P.; Investigation: J.-U.L. and M.K.K.; Validation: E.J.S., J.-U.L., and M.K.K.; Visualization: J.-U.L. and M.K.K.; Methodology: J.-U.L.; Resources: E.J.S., S.-L.P., and W.S.P.; Formal Analysis: E.J.S. and S.-L.P.; Software: J.-U.L.; Writing—original draft: M.K.K., E.J.S., and J.-U.L.; Writing—review and editing: All authors; Supervision: J.-U.L. and S.W.P; Project administration: J.-U.L.; Funding acquisition: S.W.P. and J.-U.L. All authors have read and approved the final version of the article.
Footnotes
Acknowledgments
The authors thank the participants of the study as well as all the study staff for their contributions to the study.
Disclosure Statement
The authors have no potential conflicts of interest to disclose.
Funding Statement
This research was supported by the National Research Foundation of Korea (NRF) through the Basic Science Research Program funded by the Ministry of Education (RS-2025-25419099) and a grant funded by the Korea government (MSIT) (RS-2023–00274884), as well as the Soonchunhyang University Research Fund.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.