
Abstract
Background:
Thyroid hormone (TH) homeostasis depends on the coordination of several key events to maintain proper local TH signaling, including iodide uptake, hormone synthesis, metabolism, and elimination. Three selenoprotein isoenzymes, deiodinases 1–3 (DIO1–3), are essential components of TH metabolism, and their activities have been identified as relevant endpoints regarding the screening of compounds influencing the TH system. Given the importance of DIO2 as the key enzyme for local activation of the prohormone T4 to the active T3 in various tissues, and limited data on selective biochemical DIO2 inhibition, there is a clear need to identify potent and selective DIO2-inhibiting compounds.
Methods:
Human-recombinant DIO2 enzyme pools were prepared from HEK293 cells overexpressing DIO2 and used as a robust enzyme source for the development, optimization, and semiautomated miniaturization of a nonradioactive DIO2 high-throughput screening (HTS) enzyme assay for the identification of DIO2-selective small molecule inhibitors. LT4 was used as substrate, and enzymatic release of iodide was colorimetrically quantified by the iodide-catalyzed Sandell–Kolthoff reaction. Eight comprehensive small molecule libraries were screened, covering ∼1/5 of the synthetic chemicals currently registered, natural products, as well as FDA-approved drugs. A total of 59,928 compounds were first screened at a single 10 µM concentration, followed by a validation screen to confirm the primary hits. Subsequently, DIO isoenzyme selectivity and cytotoxicity were evaluated.
Results:
Utilizing this highly reproducible and robust HTS test system with a determined median Z′-factor of 0.70 identified 356 primary inhibitory hits. Concentration-response experiments verified 17 potent inhibitors, further characterized regarding their DIO isoenzyme selectivity and cytotoxicity. Six potent DIO2-selective inhibitors, including two FDA-approved drugs and various novel pan-DIO inhibitors, for example, the fungicide fluazinam, were identified.
Conclusions:
Specific DIO2 inhibitors, such as the FDA-approved drugs racecadotril and ibrutinib and the tyrosine kinase inhibitor rociletinib, might serve as a future toolbox for reversible pharmacological interference with the local provision of DIO2-generated T3 from T4 during development, tissue regeneration, and various DIO2-dependent metabolic processes. Furthermore, they can serve as reference compounds for the development and validation of regulatory in vitro tests. Identified FDA-approved drugs warrant a closer look at potential disturbances of local TH availability.
Keywords
Introduction
Thyroid hormones (THs) are essential regulators of many physiological processes like embryonic development, postnatal maturation, and adult body homeostasis. They affect neurodevelopment, growth, tissue differentiation and regeneration, energy metabolism, and thereby body temperature.1–4 T4 and T3 reach target cells of TH action via specific transmembrane transporters, such as monocarboxylate transporter 8 (MCT8). 5 Three selenoprotein deiodinases (DIOs) are important determinants of local and systemic TH concentrations.6–8 Spatiotemporal cell type-specific expression patterns of the individual DIO isoenzymes are dynamically but precisely regulated over the course of the organisms’ development.6,9 Thereby they affect T3 availability at the intracellular T3 receptor ligand binding site and susceptibility of certain cell types and tissues toward T3 signaling. The DIO2 isoenzyme (iodothyronine 5′-deiodinase type 2), activating the prohormone T4 to T3, exerts a key function in the local T3 provision on the single-cell level.7,8 The short-lived DIO2, expressed at very low levels and rapidly regulated by its substrate T4, plays an important role in sensitizing cells to systemic TH concentrations 10 and enabling (patho-)physiological responses and adaptations to endogenous or exogenous challenges.
Studies on the phenotype of genetically modified mouse models and DIO variants in humans have demonstrated the eminent importance of DIO expression for the correct function and negative TH feedback regulation of the hypothalamus–pituitary–thyroid (HPT) axis as well as local TH action, especially during developmental processes of the sensory system.11,12 Targeted chemical modulation of DIO2 activity could provide novel treatment paradigms for pathologies with direct or indirect link to the thyroid hormone system (THS), altered TH status, dysregulations of the TH-dependent energy metabolism (e.g., fatty liver), emotional disorders, or even certain types of cancer.
Strategies and methods to identify unintended DIO inhibition are foreseen as an essential part of in vitro testing batteries for regulatory use. They might advance the identification of potential THS-disruptive compounds in the international efforts to support hazard identification for pharmaceuticals, industry chemicals, biocides, or pesticides.13,14 Given the importance of DIO2 for the local THS and the limited data available on the cellular or physiological impact of transient DIO2 inhibition, there is a clear need to identify potent and selective DIO2 inhibitors. 13
The classical determination setup of DIO enzyme activities in vitro follows common schemes using 125I-radioisotope-labeled TH, radiometric, chromatographic, and/or antibody-based methods, hardly applicable for unbiased high-throughput screening (HTS) methods.15,16 Thus, a nonradioactive method was developed based on an adapted modification of the classical Sandell–Kolthoff (SK) reaction, which is a versatile microdetermination method for iodide.17–19 Using murine tissue and human recombinantly expressed DIO enzyme sources, this method was further optimized and adjusted for testing effects of low molecular weight compounds and potential endocrine disruptors on the functionality of all three DIO isoenzymes on a 96-well microtiter assay format.
In the present work, an appropriate miniaturization of the SK-reaction-based DIO2 assay method was developed to suit a semiautomated HTS format using 384-well microtiter plates in a screening lab environment (Fig. 1). Albeit this new set of data was generated by a similar technical approach as successfully employed for DIO1, 20 the assay for this HTS had to be adapted to the specific mechanistic, kinetic, and functional DIO2 target properties. Hereby, our new data build on and complement our recently published HTS for identification of DIO1-selective inhibitors. Their (pre-)clinical application mainly aims toward interference with the systemic provision of circulating T3, preferentially generated by the large DIO1-expressing tissues: thyroid, liver, and kidneys. In contrast, DIO2 inhibitors are expected to primarily generate effects on the local, cellular availability of TH.

Schematic illustration of the high throughput screening (HTS) workflow for the identification, prioritization, and characterization of potent and selective small molecule DIO2 inhibitors. DIO2, deiodinase 2.
Materials and Methods
Production of recombinant human DIO enzymes
HEK293 cells, overexpressing either human-recombinant DIO2, DIO1, or DIO3, were cultured in three-layered cell culture flasks (Nunc™ TripleFlask™, Thermo Scientific); cell pellets were harvested, frozen, homogenized, and pooled as previously described.19,20 Stock enzyme solutions, activity- and protein-calibrated for a consistent and stable DIO assay readout, were prepared by sonication of enzyme pools in homogenization buffer (250 mM
Small molecule compound libraries
In total, 59,928 compounds of eight small molecule chemical libraries (Supplementary Table S1) were tested as described in detail (Supplementary Data) at the Screening Unit core facility of the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP). The whole screening library consists of a diverse set of natural products, pharmaceuticals, and synthetic compounds collected and impartially designed on the basis of the principle of maximum-common substructure. 21 Stocks of all test compounds were dissolved in dimethyl sulfoxide (DMSO).
DIO2 HTS quality parameters
DIO2 HTS data of each plate were analyzed using statistically robust estimators, Z-score, Z′-factor, repeatability, and percent activity, which served as quality and performance parameters of the HTS (see Supplementary Data).20,22
DIO2 HTS assay
DIO2 activity in the 384-well HTS was determined based on the assay principle previously described, 19 which uses the SK reaction for quantification of DIO2-catalyzed release of iodide from the substrate T4 (Supplementary Fig. S1). Compared with this original protocol, major adaptations of the reaction conditions were necessary both with respect to the miniaturization of the DIO2 384-well HTS assay format (see Supplementary Data). These were related to the specific mechanistic, kinetic, and functional DIO2 target properties and the characteristics of lower, more unstable DIO2 enzyme activity compared with the previously established DIO1 HTS format. 20 Test compounds (10 mM in 100% DMSO) were dispensed into 384-well assay plates using the acoustic Echo 650 Liquid Handler (Beckman Coulter). Per 30.03 μL total reaction volume, 20 µL of the appropriate DIO2 enzyme dilution (100 µg protein/well) were added to 30 nL of H2O, DMSO, or the test compounds using a pipetting robot (Biomek Workstation; Beckman Coulter). The DIO2 enzyme reaction was started by adding 10 µL reaction buffer (final concentrations: 0.91 µM T4, 40 mM DTT, 1 mM EDTA, 40 mM K+-phosphate buffer pH 6.8) to the wells using the pipetting robot. After incubation for 4 hours at 37°C, the enzymatic reaction was stopped by adding 50 µL of 10% acetic acid; the plates were sealed with a foil and stored overnight at 4°C. The enzymatically released iodide was quantified with the SK reaction as previously described.19,20
Compound libraries were screened in the primary screen at a single 10 µM inhibitor concentration. For quality control and hit identification, a 100% activity and a solvent control with DMSO, as well as a 0% activity control without DIO2 enzyme were applied on each test plate. To identify valid and unique primary hits, the Z-score >3.0 for each compound was included as a criterion. In addition, previously identified DIO3-inhibiting compounds (unpublished data) were eliminated to ensure prioritization on DIO2-selective hits. On each day, either 10 or 20 plates, each containing 352 test compounds and assay controls (for technical details and plate layout, see Supplementary Data), were screened simultaneously.
Concentration-dependent validation screen
A total of 356 compounds with an inhibition of more than 25% of DIO2 activity at the primary screening were selected as primary hits. Ten consecutive twofold serial dilutions (final well concentrations, 50–0.2 µM) were made across multiple plates in DMSO. Starting from these diluted master compound plates, the assay protocol was repeated as in the primary screen to assess DIO2 inhibition at varying concentrations. IC50 value determination was carried out using the four-parameter log-logistic function and the “drc” R-package for determining concentration-response curves (Supplementary Fig. S2)23,24,25. To determine effective DIO2 inhibitors, the criteria were made more stringent, requiring the activity difference to be >50% and the IC50 <25 µM.
DIO isoenzyme selectivity
Deiodination experiments for each DIO isozyme were performed under optimal reaction conditions for each enzyme (see Supplementary Data and Supplementary Table S2) in 96-well format in triplicates according to the protocol as described. 19 Here, the 17 most inhibitory compounds of the validation screen were tested in an eight-point 1:2 dilution serial, with 200 µM as the highest. 100 µM concentrations of known DIO isoenzyme inhibitors (PTU for DIO1, xanthohumol for DIO2 and DIO3) were used as positive controls in comparison with DMSO as solvent control. To compare isoenzyme-specific inhibition, IC50 values of substances for each isoenzyme were determined.
Cell toxicity assay
Cell toxic properties of compounds were tested in 96-well format using the Promega CellTox™ Green Cytotoxicity Assay 26 and HEK293 hDIO2 cells (for details, see Supplementary Data). Continuous timepoint measurements were made at 0, 4, and 24 hours after substance addition using a multiwell fluorescence plate reader (PerkinElmer, Waltham, USA).
Results
HTS quality parameters
A total of 59,928 compounds contained in eight FMP small molecule libraries (Supplementary Table S1) were screened on a total of 169 plates. For each plate, a Z′-factor was calculated using 16 technical replicates of each, the 100% DIO2 activity control, and the 0% DIO2 activity control. Altogether, this resulted in a median Z′-factor of the overall HTS of 0.70. The Z′-factor of the 18 plates of the validation HTS was 0.69—parameters that define a robust HTS assay (Supplementary Fig. S3) with a strong discrimination between both control groups. The Bland–Altman plot showed a good repeatability between two technical replicates (Supplementary Fig. S4). No systematic deviation of the replicates was seen (Supplementary Fig. S5).
HTS of comprehensive small molecule libraries to identify inhibitors of DIO2
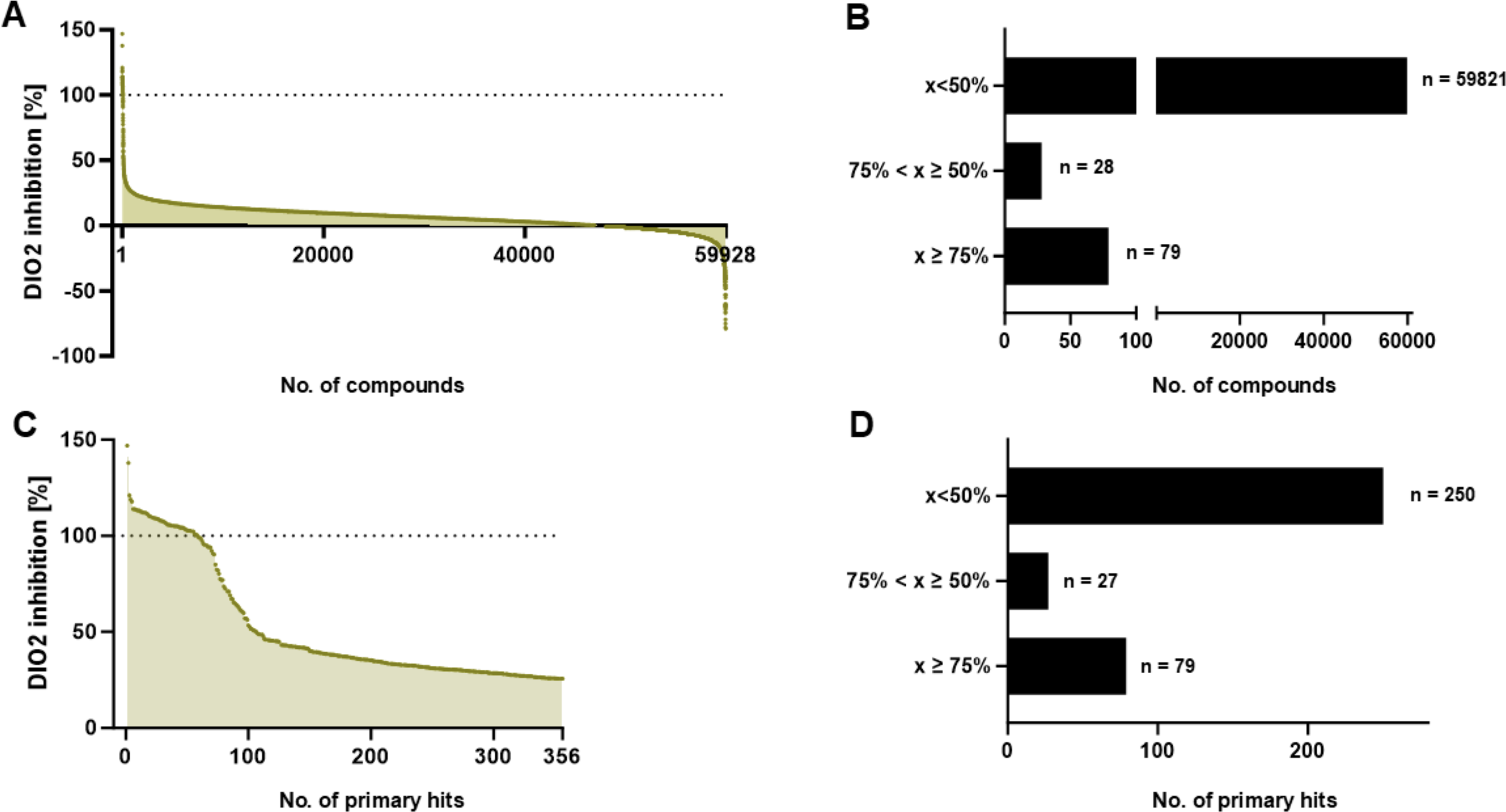
All available compounds were screened at a fixed concentration of 10 µM (Fig. 2A). At this concentration, 79 compounds showed a DIO2 inhibition of more than 75% and 107 compounds of more than 50%. For most of the screened compounds (59,821), inhibition was below 50% (Fig. 2B). A total of 356 primary DIO2 hits were selected with the Z-score set to >3 to find unique primary hits and after elimination of prior identified DIO3-inhibiting compounds to ensure to identify DIO2-selective hits. Of the 356 selected primary hits that inhibited DIO2 activity by more than 25% (Fig. 2C), 70% hits inhibited DIO2 between 25.7% and 50%, and 22% hits inhibited DIO2 by more than 75% (Fig. 2D). Among the primary hits, FDA-approved drugs, environmental chemicals, as well as novel compounds were found.
Validation HTS of primary hits
The 356 identified, most potent DIO2 inhibitory hits from the primary screen were retested on human-recombinant DIO2 homogenates for verification and to create concentration-response curves in duplicates. In total, 82 out of 356 DIO2 primary hits were verified. To identify the most effective DIO2 inhibitors, stringency of selection criteria was increased, requiring the activity difference to the control to be >50% and the IC50 <25 µM. Thus, 17 potent hits were prioritized, including a collection of drugs approved by regulatory agencies, several small molecule compounds (SMOL) reported as tools, probes, or clinical/preclinical candidates, and one fungicide listed as an active ingredient of plant protection products approved in the EU (Supplementary Table S3). Among these 17 selected compound hits, all FDA-approved drugs, the fungicide, and 13 SMOLs were chosen for further manual characterization in the 96-well format (Table 1). The initial HTS IC50 of these hits ranged between 1.3 and 15 µM. The DIO2 IC50 values determined from the concentration-response experiments could almost all be verified (Table 1) in the 96-well assay, with the exception of compounds 0 and 11 that showed markedly higher IC50 values compared with the 384-well HTS verification assay.
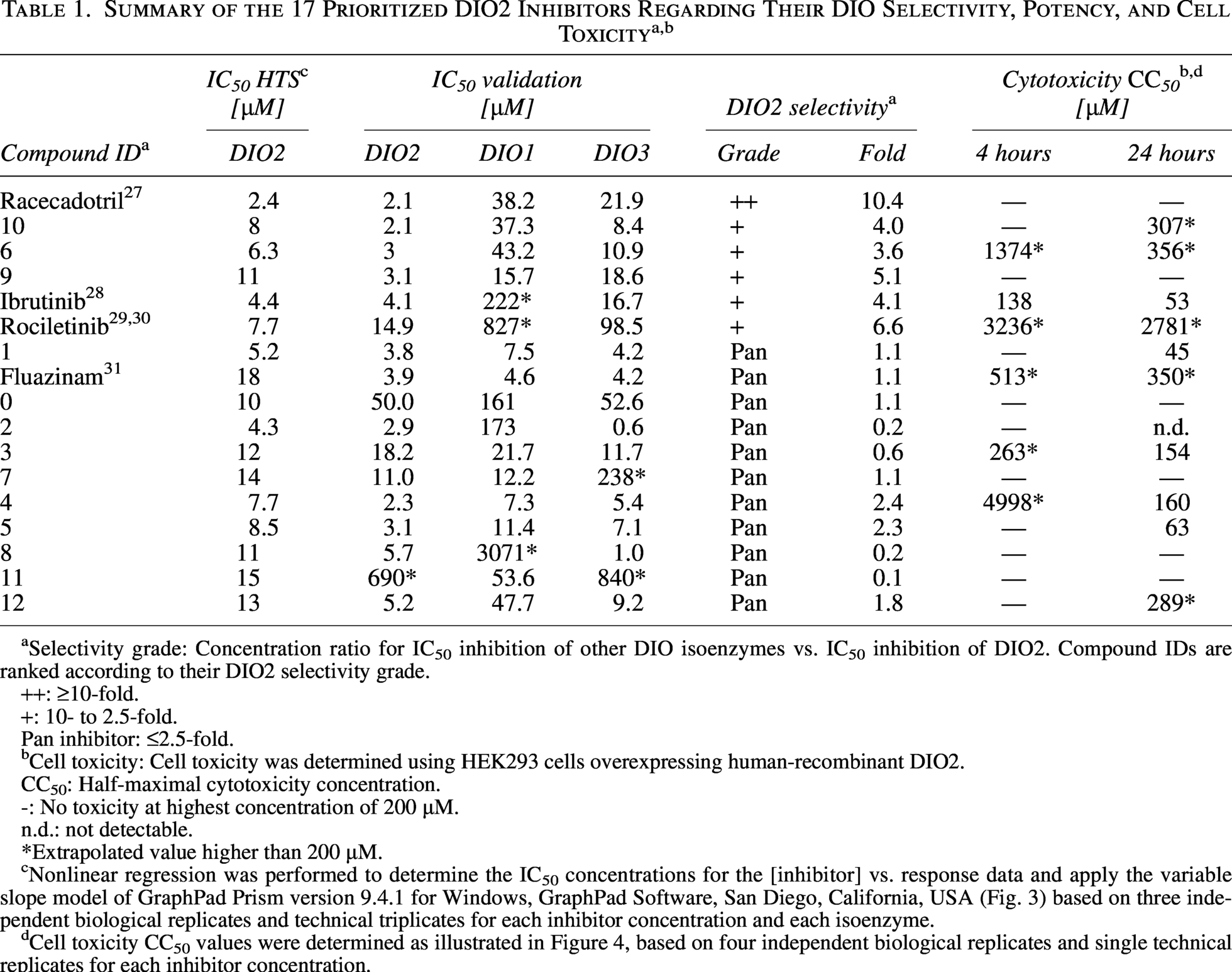
Selectivity grade: Concentration ratio for IC50 inhibition of other DIO isoenzymes vs. IC50 inhibition of DIO2. Compound IDs are ranked according to their DIO2 selectivity grade.
++: ≥10-fold.
+: 10- to 2.5-fold.
Pan inhibitor: ≤2.5-fold.
Cell toxicity: Cell toxicity was determined using HEK293 cells overexpressing human-recombinant DIO2.
CC50: Half-maximal cytotoxicity concentration.
-: No toxicity at highest concentration of 200 µM.
n.d.: not detectable.
*Extrapolated value higher than 200 µM.
Nonlinear regression was performed to determine the IC50 concentrations for the [inhibitor] vs. response data and apply the variable slope model of GraphPad Prism version 9.4.1 for Windows, GraphPad Software, San Diego, California, USA (Fig. 3) based on three independent biological replicates and technical triplicates for each inhibitor concentration and each isoenzyme.
Cell toxicity CC50 values were determined as illustrated in Figure 4, based on four independent biological replicates and single technical replicates for each inhibitor concentration.
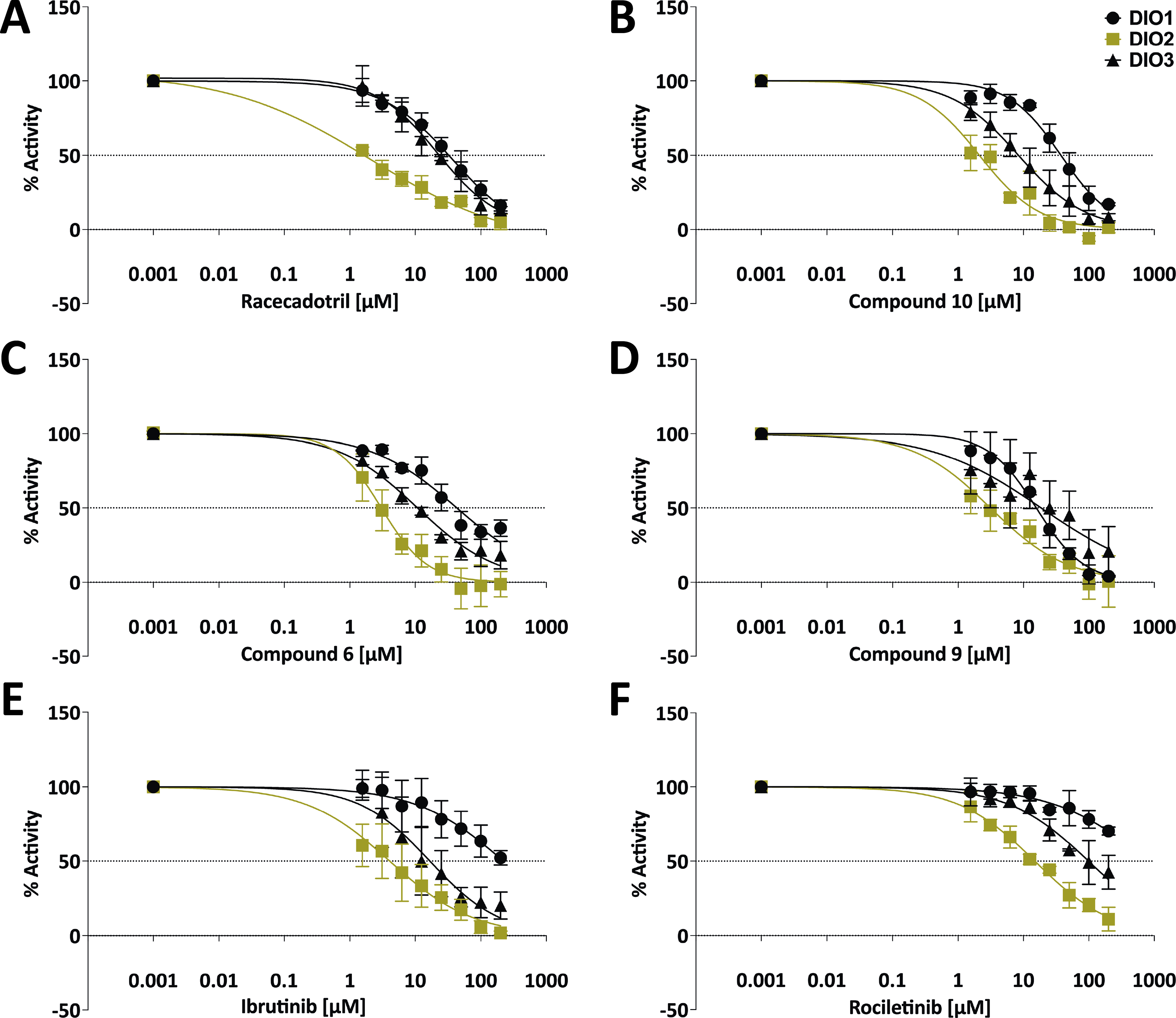
Isoenzyme selectivity of the identified DIO2-selective/preferentially selective inhibitors.
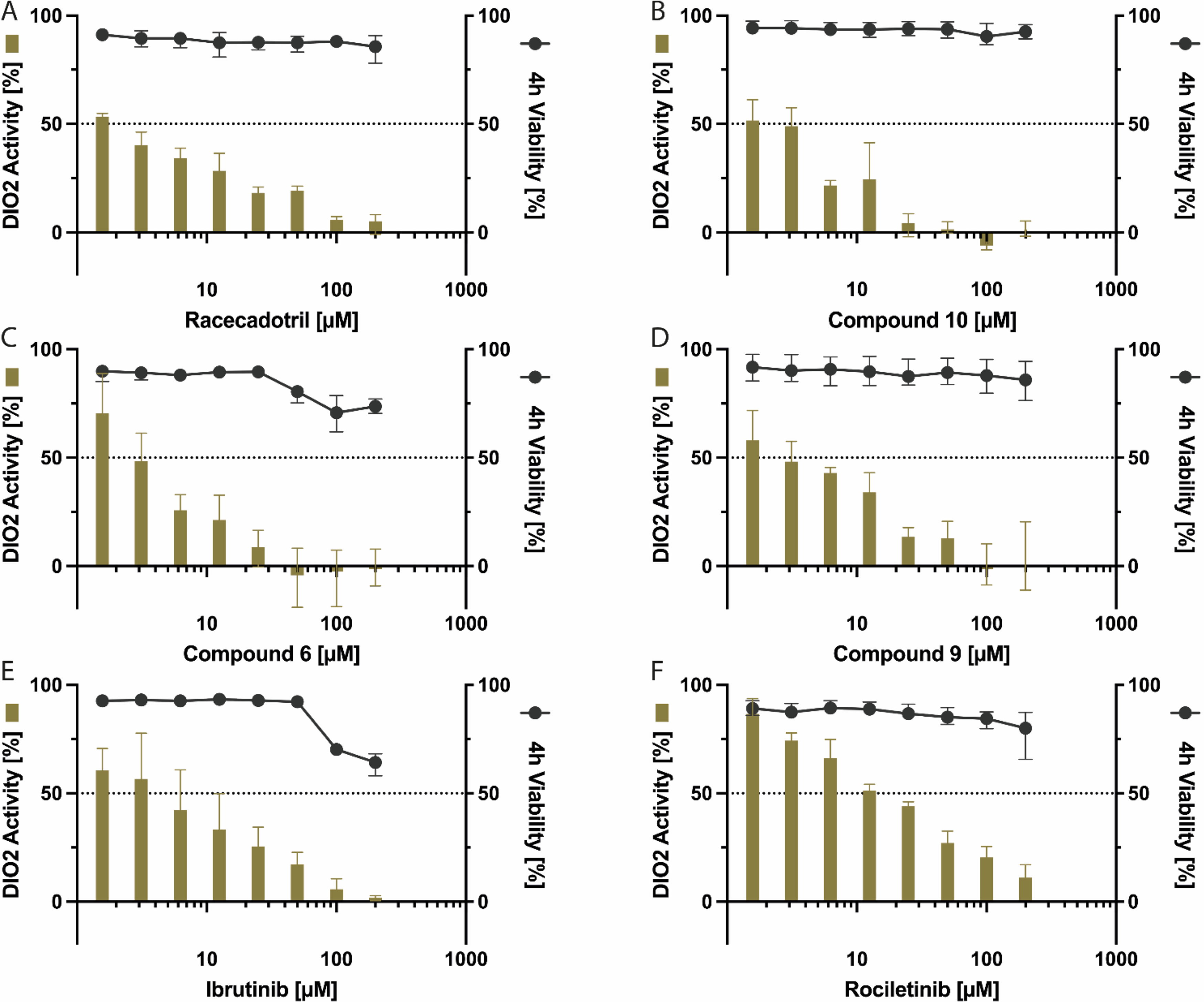
Plotted DIO2 activity (green bars) in percent against the 4 hour cell viability (black dots) of the six identified DIO2 inhibitors (panels A to F). None of these compounds showed cell toxicity in the range of their DIO2 IC50. Data represent means of four independent biological replicates, based on single technical replicates on each plate for each inhibitor concentration.
Hit characterization to identify selective DIO2 inhibitors
Racecadotril, ibrutinib, rociletinib, fluazinam, and 13 SMOLs were analyzed regarding their DIO isoenzyme selectivity and cell toxicity (Supplementary Table S4) to identify DIO2-selective inhibitors. Isoenzyme selectivity of the selected compounds was determined up to 200 µM on DIO1, DIO2, and DIO3 homogenates in 96-well format. Only racecadotril, an FDA-approved drug, was identified as a DIO2-selective inhibitor with an IC50 value of 2.1 µM, whereas five other compounds (#10, #6, #9, ibrutinib [FDA-approved drug], and rociletinib [development terminated]) showed preferentially selective DIO2 inhibition with increasing IC50 values (2.1–14.9 µM) (Fig. 3). In addition, racecadotril showed the highest DIO2-selective inhibition without any cell toxicity (Table 1). The remaining 11 compounds were classified as pan-DIO-inhibitors. The fungicide fluazinam inhibited all three isoenzymes to a similar extent (IC50 ∼ 4 µM), a potency comparable to the pan-DIO inhibitor xanthohumol. 19
Discussion
DIO2 controls local intracellular T3 production, availability, and action during development and in adulthood. The outstanding feature of DIO2 expression and enzyme activity among the DIO isoenzymes is its specific, complex, and dynamic spatiotemporal pattern. 8 DIO2 is predominantly present in the brain, primarily in astrocytes, but also in radial glial cells, brain barriers, Cajal–Retzius cells, and the hypothalamus.32–34 DIO2 is further expressed in the pituitary, 35 skin, 36 retina,37–39 brown adipose tissue, 40 and skeletal muscle. 42 In addition, DIO2 is a member of the fetal gene program 6 with a causative role in the developing heart and pathogenesis of several heart diseases.41,43 The expression of DIO2 enzyme activity is necessary for the maintenance of important physiological functions, and anomalous expression and enzyme activity are associated with a number of pathological processes. 44 For example, overexpression or upregulation of DIO2 has been described in various types of tumors,36,45,46 as well as in idiopathic pulmonary fibrosis, 47 diabetic complications,48,49 and adipose tissue during obesity or pregnancy.50–52 The importance of tight local control of T3 availability by DIO2 raises great interest in the development of isozyme-selective DIO inhibitors for pharmacological interventions.6,53 It can be assumed that local reduction of T3 concentration by selective DIO2 inhibitors limits growth and favors cell differentiation during development and tissue remodeling.
On the one hand, this potent role in physiological and pathophysiological processes in humans makes DIO2 an attractive target for pharmacological modulation. On the other hand, unintended interference, for example, by environmental compounds or pharmaceuticals, may result in unforeseen adversities. Addressing both aspects requires a robust method for assigning the character of a DIO2 inhibitor to a chemical. We have attempted to take both aspects into account by selecting test libraries covering a broad chemical spectrum in the screening (Supplementary Table S1), thereby fully exploiting the potential of a DIO2 HTS.
HTS assays are not yet available for the vast majority of key targets of the HPT axis involved in biosynthesis, distribution, transport, metabolism, and action of TH and the regulation of the THS. Most assays are based on the determination of concentrations of either TH or enzymatically released iodide and require the use of radioactive 125I tracer molecules or expensive mass spectrometry.15,54 This drastically limits the ability to miniaturize and automate assays for THS-related endpoints. DIO2 is a hormone-metabolizing enzyme operating in vivo at nano- to picomolar T4 concentrations, that is expressed at very low levels, has a short half-life, and is considerably more labile than its two related isoenzymes, DIO1 and DIO3. HEK293 hrDIO2 overexpressing cells served as enzyme source for this nonradioactive assay, which was successfully upscaled from a manual 96-well plate to a semiautomatic 384-well plate format, allowing reproducible measurements of DIO2 enzyme activity as demonstrated by the excellent HTS quality parameters (Supplementary Figs. S3-S5). This robust assay performance was achieved despite the fact that no DIO2-selective inhibitor was available as “positive control” to date, in contrast to the previously performed DIO1 HTS, 20 in which PTU served as potent classical DIO1-selective inhibitor.
For the primary compound library screen, a fixed single concentration of 10 µM was chosen in a single run, with the intention to focus on DIO2-selective and potent compounds of interest in a (patho-)physiological concentration range relevant for the THS. The subsequent automated concentration-dependent validation screen of the primary hits, followed by a manual concentration-dependent screen for the top 17 DIO2 inhibitors, was complemented by the analysis of their DIO isoenzyme selectivity and cytotoxicity for HEK293 hDIO2 cells. Accordingly, no false-negative compounds were further examined or evaluated in detail, indicating the possibility of existence of additional DIO2-selective inhibitors.
The robust and stable performance of this automatable application of the SK reaction enabled us to sensitively measure the DIO2 activity. This, in turn, allowed for the identification of DIO2-selective inhibitors from a total of 59,928 compounds covering ∼1/5 of the synthetic chemicals currently registered for production and use.55,56 Racecadotril was identified as the most potent DIO2-selective inhibitor devoid of cytotoxicity. Several other compounds, the approved drug ibrutinib and the receptor tyrosine kinase inhibitor rociletinib, showed preferentially selective DIO2 inhibition compared with their interference with DIO1 and DIO3, while the fungicide fluazinam and several other compounds acted as pan-DIO inhibitors. As such, this is one of the most extensive chemical inhibition studies of DIO2 activity and considerably extends previous studies.
A 96-well adaptation and application of the SK reaction-based DIO assay method 19 for the screening of more than 1800 compounds from the ToxCast libraries of potential endocrine-disrupting chemicals (EDCs) at an initial single maximal concentration of 200 µM for possible interference with the three DIO enzymes revealed 158 compounds which inhibited DIO2 more than 50%, whereas only 20 compounds were DIO2 selective. 57 These US EPA-tested compound libraries were mainly focused on coverage of toxicity, regulatory, environmental exposure chemical inventories.58–60 Within these 20 DIO2-selective compounds, there is no compound overlap with our study. However, in the screen of Olker et al., 57 fluazinam was also determined to be a potent DIO pan-inhibitor (IC50 2.2 µM), which is in agreement with our observation. The other preferentially selective DIO2 inhibitors of our HTS were not contained in their compound collection.
We also identified some compounds that exceeded the 100% control activity, up to almost 180%, which might suggest an action as DIO2 “activators” (Fig. 2A). However, these compounds might also exhibit chemical properties resulting in interference with the SK reaction detecting iodide. Gallamine-triethiodide, 1,1-dimethyl-4-phenylpiperaziniumiodide, and iodoacetamide were such compounds that showed an“activation” of DIO2 of more than 120% in our HTS, with this measured activation profile attributable to their chemical composition, including iodide. Both possibilities, activation or interference, have not been tested so far in detail, but compounds containing iodine were excluded from the validation screen and the further characterization.
In the present study, racecadotril was identified as the most selective and potent DIO2 inhibitor, and ibrutinib, rociletinib, as well as compounds # 6, 9, and 10 as preferential DIO2 inhibitors (Table 1, Fig. 1, Supplementary Table S3). These compounds are structurally different and cannot yet be clustered according to their chemical building blocks or obvious functional group features apart from the observation that most of them have complex polycyclic (aromatic) ring structures substituted with space-demanding (polar) groups. Their voluminous chemical structures might effectively mimic essential features and functional groups of the iodothyronine substrate(s) of DIO2. Furthermore, their space-demanding functional groups might strongly interfere with the essential selenocysteine and the other hydrophobic and charged amino acids constituting the active site of DIO2, 61 partially explaining their high inhibitory potency in the low-µM concentration range. Key features of the reaction mechanisms and essential active site residues of the thioredoxin fold, supporting the selenocysteine-dependent deiodination of iodothyronines, have been identified,10,62–65 but exact X-ray structural data have only been reported for the mouse Dio3 cytoplasmic catalytic domain with cysteine replacing the active site selenocysteine.66,67 The recently published mouse Dio2 structure, 61 which exhibits a high overall similarity to the mouse Dio3 structure, 66 but owns several Dio2-selective features, possibly related to its distinct reaction mechanism, may be helpful for further investigation of mechanistic aspects of DIO-selective inhibitors.
The approved drug racecadotril has one of the lowest IC50 values out of all tested compounds, with a DIO2 IC50 value 10-fold lower than for DIO3 and 18-fold lower than for DIO1 (Table 1). Rociletinib is less potent with a DIO2 IC50 value of 14.9 µM. However, this pharmacologically active compound also showed high DIO2 selectivity with 6.6- and 55.5-fold lower IC50 compared with DIO3 and DIO1. Ibrutinib, an approved cancer drug, inhibits each DIO isoform to different extents. The lowest IC50 value is observed for DIO2 (4.1 µM), followed by DIO3, and DIO1 seems to be inhibited only partially at the maximum concentration of 200 µM. However, cell viability is reduced at higher concentrations after 4 hours (Fig. 4) and 24 hours, but not yet at the DIO2 IC50 concentration. The SMOLs # 9, 10, and 6 likewise inhibited DIO2 selectively in the range between 2.1 and 3.1 µM with no or negligible cell toxicity. The DIO-IC50 value of the investigated compounds is lower than their half-maximal cytotoxicity concentration (CC50) value; thus, the DIO2 inhibition is not essentially due to the influence of cell toxicity (Supplementary Table S4). This is also reflected in the IC50 values for each isoenzyme of approximately 4 µM. In the cell toxicity assay, time- and concentration-dependent cell toxicity can be observed for the SMOLs # 6 and 9, but not at the respective DIO IC50 concentration (Fig. 4; Supplementary Table S4).
Racecadotril is a specific inhibitor of enkephalinase used for the treatment of diarrhea. 68 Ibrutinib and rociletinib are tyrosine kinase inhibitors.28–30 Use of some tyrosine kinase inhibitors has been associated with hypothyroidism,69–75 and some of them act as noncompetitive inhibitors of the TH transmembrane transporter MCT8. 76 Ibrutinib is used to treat chronic lymphocytic leukemia/lymphoma, and while rociletinib was originally developed for lung cancer, its clinical development has been terminated. 29 To date, there are only two case reports describing an increased need for TH in 80- and 86-year-old thyroidectomized women during treatment with ibrutinib.72,73 After discontinuation of ibrutinib, the levothyroxine dose, required to maintain a regular TSH concentration, could be decreased. It was concluded that ibrutinib-induced hypothyroidism was reversible. Accordingly, the authors of both case reports hypothesized that the patients’ hypothyroidism was caused by the induction of DIO3, which converts T4 to rT3, and the inhibition of DIO2, which converts T4 to T3. In our isoenzyme comparison of ibrutinib, we detected a strong DIO2 inhibition of human recombinantly expressed DIO2. Simulation of any counter-regulation in the complex in vivo THS will require more complex in vitro or in vivo experimental models that co-express both DIO2 and DIO3 enzymes. Currently, there is no detailed information available, whether these three FDA-approved drugs, identified as DIO2-selective inhibitors, also interfere with other components of the THS, such as TH distributor proteins, transmembrane transporters, or T3 receptors.
Recently, the cephalosporin antibiotic cefuroxime was identified as a novel selective Dio2 inhibitor based on computational Dio2 homology modeling using the crystal structure of the cytoplasmic catalytic domain of mouse Dio3 as a template.46,66 Effective inhibition of Dio2 in cellular in vitro models and in mouse models was demonstrated, with notable effects on cellular and serum TH concentrations and HPT feedback regulation. Sagliocchi et al. described the related compounds cefazolin, cefaclor, and ceftriaxone as ineffective with respect to Dio2, 46 which was consistent with our primary HTS results showing negligible DIO2 inhibition of 5%, 5%, and 16%. Only ceftazidime, which was also described as a Dio2 inhibitor, was not recognized as a DIO2 inhibitor in our primary HTS screening. These differing results could be due to different compound concentrations used, given that our primary screen was performed only once with 10 µM and that divergent DIO2 enzyme assay conditions and readouts were used (hrDIO2 as the enzyme source and different protein concentrations, incubation volumes, time, and readouts) (see Supplementary Table S2). Sagliocchi et al. tested cefuroxime and its analogues using 0.1 mg of homogenized sonicated mouse brown adipose tissue/protein as enzyme source, homogenized and sonicated in 0.25 M sucrose, 1 mM EDTA, 0.1 M NaPO4 and 10 mM DTT, 1 hour preincubation of protein with 30 µM cefuroxime, followed by 6 hours incubation in phosphate-EDTA buffer (pH not stated) in the presence of 20 nM T4-13C6 as a substrate. T3-13C6 product formation served as a readout of determination of Dio2 enzyme activity analyzed by liquid chromatography-tandem mass spectrometry.
In our recent comparable SK-reaction-based HTS for the human DIO1 enzyme, a series of DIO1-selective potent inhibitors were identified using the same set of small molecule libraries. 20 Surprisingly, no overlap was observed between the 15 top-prioritized DIO1-selective compounds with IC50 < 1 µM, which were more potent than the bonafide DIO1-selective inhibitor PTU and the top 17 DIO2 inhibitors characterized in this work. The characterized set of the most potent DIO1 inhibitors also included two drugs approved for medical use, that is, edaravone (FMP405651) and pranlukast (FMP405931), which have not yet been reported to interfere with the THS or its other components in a clinically relevant way. These findings indicate that the first stages of the chosen unbiased HTS for the two closely related human 5′-DIOs initially allowed to identify two distinct isoenzyme-selective sets of potent inhibitors. However, their further characterization may reveal some overlaps in selectivity, including the identification of pan-inhibitors that also effectively inhibit the third isozyme, 5-DIO (DIO3). Even when the features and application perspectives for selective DIO1 and/or DIO2/DIO3 inhibitors may overlap in part, their potential biochemical, preclinical, and regulatory applications will diverge. Obviously, the target DIO2 has quite different biological and also high clinical relevance compared with the target DIO1. While DIO1 preferentially contributes to the production of circulating T3 and the degradation of TH and its metabolites, especially rT3, its tissue-specific activity also represents a relevant drug target in the context of (systemic) T3 provision, especially under hyperthyroid conditions.
The increasing volume and diversity of globally produced and marketed chemicals55,56 has prompted the establishment of various types of screening assays, using panels of in silico, in vitro, and in vivo testing of novel chemicals and drugs for hazard identification by regulatory authorities77,78 to efficiently obtain comprehensive information for a large number of compounds that could affect the THS. Especially, validated test methods of THS endpoints are lacking to address a potential exposure impact on the developing brain. 13 The successful development, adaptation, and application of the SK-reaction-based HTS platform for DIO2, one of the key targets of the THS potentially affected by EDCs or drugs, illustrates the feasibility to validate in vitro-based approaches for comprehensive testing of large numbers of chemicals for regulatory purposes using automatable resource-conserving technologies. This approach can remarkably reduce and prioritize the number of compounds to be selected for further testing in more complex in vitro models (e.g., organoids) and eventually in suitable experimental animal models. In this way, the regulatory acceptance of the 3R principle (replacement, reduction, refinement) can be met, while meaningful scientific information on the target specificity and potency of those potentially relevant adverse compounds is received. The extensive information obtained from HTS data also allows to develop and interrogate impactful quantitative/qualitative structure–activity relationship (QSAR) models for the tested targets in silico, thus providing valuable complementary information to that obtained from molecular and mechanistic in vitro and in vivo animal experimental studies.79,80
Conclusions
In the present study, we investigated a diverse, unbiased set of small molecule compounds that also included novel compounds as well as various building blocks and substructures of potential drugs. Accordingly, it might be possible to develop DIO2-selective new or repurposed pharmaceuticals using these inhibitory compounds as drug leads, which could offer a shortcut by characterizing the hazard potential of existing substances already on the market or in clinical use compared with new drug candidates. However, the “toolbox” of specific modulators for potential pharmacological interventions is still limited to a small number of compounds, partially restricted regarding accessibility due to commercial interests. In contrast to the concept of intentional pharmacological intervention, disruption of DIO activities by environmental chemicals or unrecognized drug side effects could have drastic effects on human health and, in particular, on pre- and perinatal TH-dependent (neuro-)development.81–83 These new DIO2 inhibitory compounds can be employed as target-selective reference compounds for regulatory in vitro test methods in systematic screening of chemicals for nutritional, pharmaceutical, toxicological, and environmental purposes. 13 Prospective insights are now possible with the availability of these selective DIO2 inhibitors into mechanistic and molecular aspects of catalytic details of isoenzyme-specific 5′-deiodination and potential target-selective drug leads.
With the help of new selective DIO2-inhibiting compounds, it would not only be possible to provide pharmacological interventions but also to gain mechanistic insights by means of targeted modulation of discrete target cell-specific metabolic pathways, for example, in time- and concentration-dependent manner. Specific DIO2 inhibitors might serve as useful tools to reversibly interfere with those changes both at the experimental level or as potential clinically relevant game-changers to modulate local T3 availability in vivo. Such an option might effectively complement insights solely relying on and obtained from experimental studies using global or conditional irreversibly genetically modified mouse models in attempts to unveil underlying molecular mechanisms of local TH metabolism and action. The local control of TH availability via DIO2-selective inhibition may be of etiological and therapeutic value in various diseases.
Authors’ Contributions
C.F.: Methodology, investigation, validation, formal analysis, data curation, visualization, and writing—original draft. N.W.: Investigation and data curation. Y.Z.: Investigation and data curation. R.S.: Investigation, data curation, visualization, and review. A.F.: Investigation, validation, and data curation. E.K.W.: Methodology, data curation, writing—review and editing, and visualization. M.N.: Data curation, formal analysis, visualization, project planning, and writing—review and editing. J.P.v.K.: Writing—review and editing, conceptualization, project planning and coordination, and data analysis and review. C.S.: Investigation and data curation. S.K.: Investigation and data curation. K.R.: Methodology, formal analysis, data curation, visualization, writing—review and editing, conceptualization, and project planning. J.K.: Data analysis, writing, review, revision and editing, project administration, supervision, funding acquisition, conceptualization, and project planning and coordination.
Footnotes
Acknowledgments
We would like to thank Prof. Dr. M. Ristow, Institute Director, for allowing us to share the necessary spatial, logistical, and administrative resources of the institute. We thank Dr. Edgar Specker, head of the Core Facility Compound Management of the FMP Screening Unit, for his valuable support and critical discussions and AnalytiCon Discovery—a division of BRAIN Biotech AG, Potsdam, Germany, for the provision of the CBB7 library consisting of natural products and natural product-derived small molecules.
Author Disclosure Statement
The authors C.F., N.W., Y.Z., R.S., A.F., E.K.W., M.N., J.P.v.K., C.S., S.K., K.R., and J.K. declare no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the article; or in the decision to publish the results.
Funding Information
This research was part of the project ATHENA (Assays for the identification of Thyroid Hormone axis-disrupting chemicals) funded by the EU Horizon 2020 program, grant number 825161, which is gratefully acknowledged (J.K.). Further funding for this work was received by the Deutsche Forschungsgemeinschaft within the framework of CRC/TR 296 Locotact P16 to J.K. and E.K.W. The DIO2 HTS assay has been awarded the 2021 prize of the state of Berlin for promotion and research on replacement and supplementation methods for animal experiments in research and teaching (C.F., K.R., and J.K.), providing additional funding support for this project.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.