
Abstract
Background:
Constitutively active mutations (CAMs) in the thyrotropin receptor (TSHR) are the major cause of nonautoimmune hyperthyroidism. TSHR is a key regulator of thyroid hormone synthesis, which is essential for skeletal formation, bone turnover, and craniofacial development. In addition to its role in thyroid hormone regulation, thyrotropin and its receptor have been proposed to independently influence bone formation and resorption. However, the specific effects of constitutively active TSHR mutations on cranial and skeletal development have not been investigated in mouse models.
Methods:
Cranial morphometry, micro-computed tomography, and three-point bending tests were performed in established TSHR knock-in mouse models carrying patient-derived TSHR D633H or M453T CAMs. Homozygous TSHR D633H mice exhibit mild, transient hyperthyroidism at 2 months of age, more pronounced in females, whereas homozygous TSHR M453T mice develop a more severe, iodine-dependent hyperthyroid phenotype.
Results:
Both TSHR CAM lines showed altered craniofacial morphology, particularly in nasal bone dimensions, resulting in a shorter snout compared with wild-type (WT) controls. The incidence of malocclusion was significantly increased in both homozygous and heterozygous mice, regardless of sex. TSHR D633H mice displayed no significant changes in femoral or tibial bone structure or biomechanical properties. In contrast, TSHR M453T mice exhibited hyperthyroidism-dependent alterations in trabecular bone mineral density (BMD) and architecture, while cortical bone was unaffected. Body and tail lengths were unchanged in TSHR D633H mice. M453T homozygous mice had reduced tail length at weaning in an iodine-dependent manner, which normalized with age.
Conclusions:
This in vivo study demonstrates that TSHR CAM-induced hyperthyroidism alters craniofacial morphology and increases the incidence of malocclusion in mice. Structural changes and altered BMD in M453T mice were dependent on the severity of hyperthyroidism. These findings highlight the role of TSHR signaling and thyroid status in craniofacial and skeletal development, warranting further mechanistic investigation.
Introduction
Hyperthyroidism, defined as the excess production of thyroid hormones (THs), affects approximately 3% of individuals over a lifetime, 1 and is associated with increased risks of atrial fibrillation, fractures, and mortality, particularly in older individuals.2,3 Hyperthyroidism is most commonly caused by activation of the thyrotropin receptor (TSHR), either via stimulating autoantibodies (Graves’ disease) or constitutively active mutations (CAMs).4,5 TSHR CAMs can lead to sporadic or familial forms of nonautoimmune hyperthyroidism (NAH), resulting in chronically elevated TH levels. 5
TSHR is the main regulator of thyroid growth and hormone synthesis. Although TSHR is primarily expressed in thyroid follicular cells, it is also found in extrathyroidal tissues, including osteoclasts, osteoblasts, and chondrocytes,6–9 supporting potential direct roles for TSH (thyrotropin) and TSHR signaling in bone metabolism and skeletal development, independent of THs.
Bone develops through two main processes: intramembranous and endochondral ossification. Intramembranous ossification occurs primarily in flat bones, such as those of the skull, while endochondral ossification forms long bones. The craniofacial skeleton develops from both processes. 10 In adulthood, bone remodeling maintains mineral homeostasis and skeletal integrity by replacing old or damaged bone. 11
The TH axis plays a critical role in skeletal development and remodeling. Altered thyroid status results in growth abnormalities, bone loss, and increased fracture risk.2,12 In an in vivo study, TH-supplemented, euthyroid TSHR knockout mice exhibited high bone turnover, suggesting that TSH and its receptor may independently regulate bone formation and resorption. 13
In humans, rare gain-of-function TSHR mutations have been associated with skeletal abnormalities, including frontal bossing, premature closure of fontanelles, craniosynostosis, advanced bone age, and shortened fifth metacarpals and middle phalanges.8,10 Clinical and subclinical hyperthyroidism are also linked to reduced bone mineralization and craniofacial manifestations, often due to craniosynostosis.14,15 A recent case report described the first known germline TSHR D633H in an individual with hydrocephalus and Chiari malformation associated with craniosynostosis. 16
Despite these clinical observations, to our knowledge, the effects of TSHR CAMs on skeletal development and maintenance have not been investigated in mouse models, and data on bone morphometry in human cases are limited. Here, we hypothesize that TSHR CAMs can have a direct impact on skeletal and craniofacial development.
To address this knowledge gap, our previous work with knock-in (KI) mouse models demonstrated age-, sex-, genotype-, and iodine intake-dependent NAH, which can lead to papillary thyroid carcinoma.17,18 The TSHR D633H homozygous (HOM) mice developed mild, transient hyperthyroidism at 2 months of age, with a more pronounced phenotype in females. 17 In contrast, TSHR M453T HOM mice developed a more severe hyperthyroid state that was dependent on dietary iodine levels. Female HOM mice on a sufficient-iodine diet exhibited more severe hyperthyroidism than those on a high-iodine diet. 18
To investigate the role of constitutively active TSHR mutations in skeletal development, we analyzed our previously generated TSHR KI mouse models with patient-derived TSHR D633H and M453T CAMs. We performed micro-computed tomography (µCT), a three-point bending test, and a cranial morphometry assessment using predefined anatomical landmarks.
Materials and Methods
Mouse models
The mouse models used in this study have been described previously.17,18 Experiments were performed on a mixed C57BL/6J and C57BL/6Ncr genetic background, with wild-type (WT) littermates as controls. Experimental groups were generated by heterozygous (HET) breeding. Analyses were performed after backcrossing the founders two to five times to a C57BL/6NCrl background. Animal technicians, blinded to genotype, were instructed to report malocclusion requiring tooth trimming. Mice with malocclusion were excluded from breeding.
Mice were housed in individually ventilated cages under controlled conditions (12-hour light/dark cycle, at 21 ± 1°C) at the Central Animal Laboratory, University of Turku, with ad libitum access to water and pelleted chow. Diets varied by model and age: TSHR D633H mice: the 6-month cohort received SDS RM-3 (Special Diet Service, 0.4 mg/kg iodine); the 2-month cohort received Teklad 2018 global 18% protein chow (Envigo). TSHR M453T mice: Received either Teklad TD.230152 (Envigo) or Teklad TD.230025 (modified from TD.120363 with potassium iodate supplementation). Iodine content in sufficient-iodine diets ranged from 0.3 to 0.4 mg/kg and in high-iodine diets was 6 mg/kg, with minor variations reflecting commercial availability. Mice were euthanized with CO2, at 2 or 6 months of age, and blood was collected via cardiac puncture. Right femora, left tibiae, and skulls were fixed in 10% formalin in phosphate-buffered saline (PBS). Left femora were stored at −20°C in PBS.
Study approvals
All experiments were conducted in accordance with protocols authorized by the National Animal Experiment Board of Finland (license number: ESAVI 35039/2019).
Cranial measurements
Skulls were cleared of soft tissue before measurements. Linear distances were taken from the dorsal aspect of the skull using predefined landmarks and a caliper with 0.02 mm precision.19,20 Measurements were as follows: (1) nasal bone rostral point to the intersection of the nasofrontal suture in the midline, (2) nasal bone rostral point to the intersection of the coronal and sagittal sutures (bregma), (3) span between the processus nasalis ossis incisivi, (4) nose tip to the caudal end of the curvature of the occipital bone, and (5) maximum skull width.
Bone analysis
µCT of left tibiae was performed using a Skyscan 1272 X-ray CT scanner (Bruker, Kontich, Belgium) following Bouxsein et al. (2010). 21 Reconstruction of cross-sectional images was performed with NRecon software version 1.7.5.6 (graphics processing unit recon engine 1.7.5), and data analysis was done by CT Analyzer version 1.20.8.0 (Bruker). Scanning parameters were resolution 5.0 mm, X-ray tube voltage 70 kV, tube current 142 µA, and a 0.5 mm aluminum filter. The object was rotated and imaged in steps of 0.40° (total of 482 images covering 192.8° rotation). Reconstruction parameters were dynamic range of attenuation coefficient 0.0001–0.10 units, smoothing level 2, beam hardening correction 70%, and ring artifact reduction 7. Cross-sectional images were post-aligned using DataViewer 1.6.0.0 (Bruker) prior to analysis.
Trabecular bone was analyzed in the femoral metaphysis, starting 50 layers (250 µm) below the lower surface of the growth plate and extending 150 layers (750 µm). Cortical bone was excluded.
Cortical bone was analyzed in the tibial diaphysis from 750 layers (3750 µm) below the growth plate and extending for 50 layers (250 µm). A binary mask generated via binary conversion of the selected area was used to limit bone mineral density (BMD) analysis to cortical bone tissue. Ceramic calibration standards (0.25 and 0.750 g/cm3) were used for BMD calibration.
Three-point bending test
Mechanical testing was performed using the Universal Testing Machine LR 300 Plus (Lloyd Instruments Ltd., UK) and Nexygen Plus software (Lloyd Instruments Ltd.). Left femora were placed with the anterior surface facing down on 6-mm-spaced supports. A compressive load (0.2 mm/s) was applied to the shaft until fracture.
Statistical analysis
Data are presented as mean ± standard deviation (SD). p-values of <0.05 were considered statistically significant. One-way analysis of variance (ANOVA) with Šídák’s multiple-comparisons test was used (GraphPad Prism 10; GraphPad Software, San Diego, CA, USA). Figures were created with BioRender.com and GraphPad Prism 10.
Results
Overview of the TSHR CAM mouse models and hyperthyroidism phenotypes
To investigate the impact of TH excess on cranial and skeletal development, we analyzed two previously characterized TSHR CAM KI mouse models17,18 carrying the D633H or M453T mutation (Fig. 1A). Thyroid function tests (TFTs) for both models have been published in detail in our previous work.17,18,22 TSHR D633H mice developed a transient hyperthyroid state at approximately 2 months of age and showed normal birth weight, body length, and tail length during early development (Supplementary Fig. S1). In contrast, TSHR M453T mice exhibit a more severe hyperthyroid state that is sex- and iodine-dependent. Specifically, HOM females fed a sufficient-iodine diet had lower body weight and shorter tail length at 3 weeks of age than WT littermates; both parameters normalized by 8 weeks. No significant growth differences were observed in M453T HOM males or in females receiving a high-iodine diet (Supplementary Fig. S1). 18
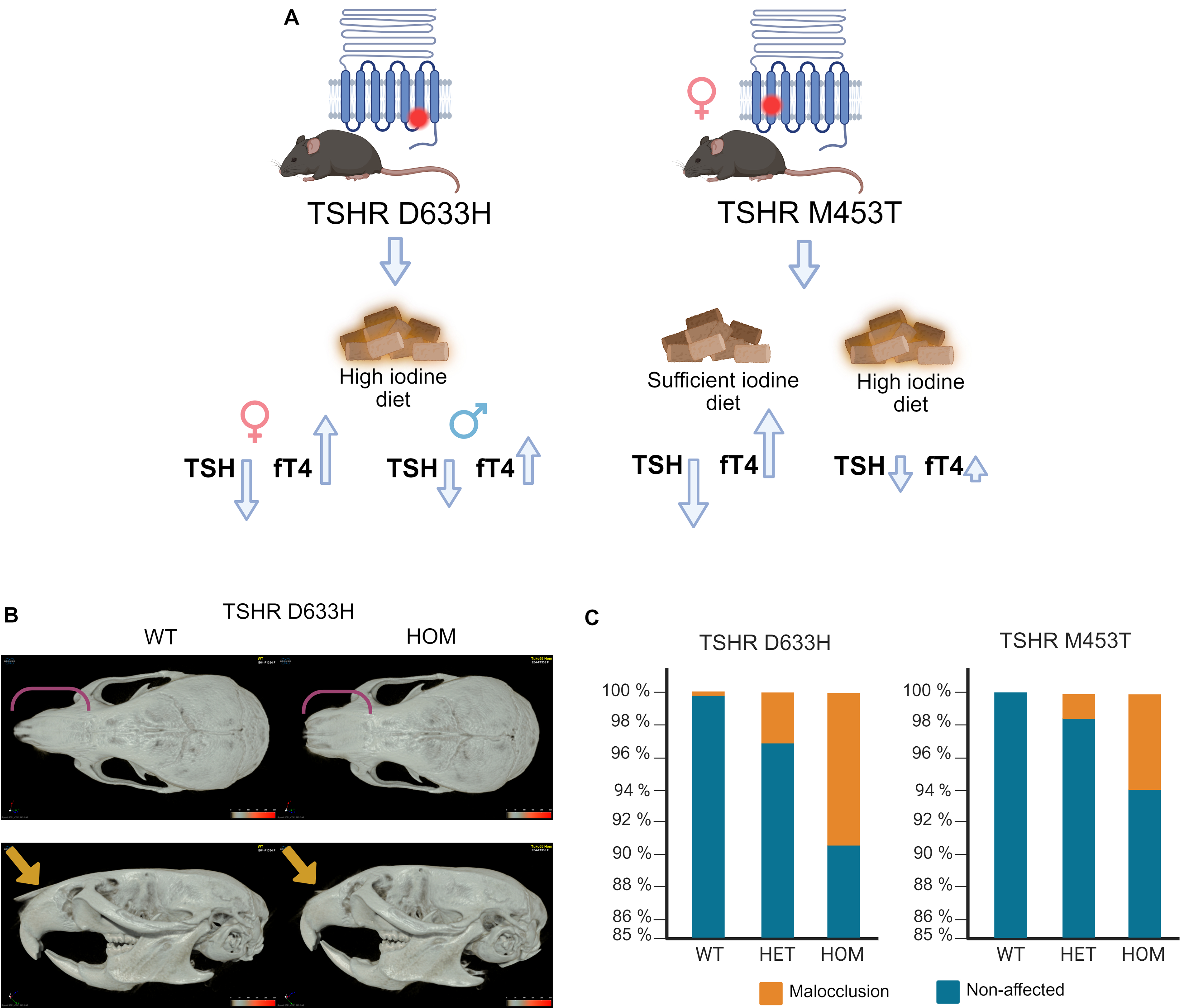
Description of the TSHR constitutively active mutation (CAM) models, craniofacial alterations, and malocclusion rate.
Craniofacial alterations and malocclusion in TSHR CAM mice
After 2 months of age, TSHR D633H mice displayed a visibly shortened snout compared with WT controls (Fig. 1B and Supplementary Fig. S2). Linear measurements of defined cranial landmarks confirmed these alterations (Fig. 2). At 6 months, TSHR D633H HOM mice had significantly shorter snout and cranium lengths. Male HOM mice also had wider nasal bones, whereas females showed no significant changes in nasal width. At 2 months, male HOM D633H mice had significantly wider skulls, while other cranial metrics were unchanged (Fig. 2).
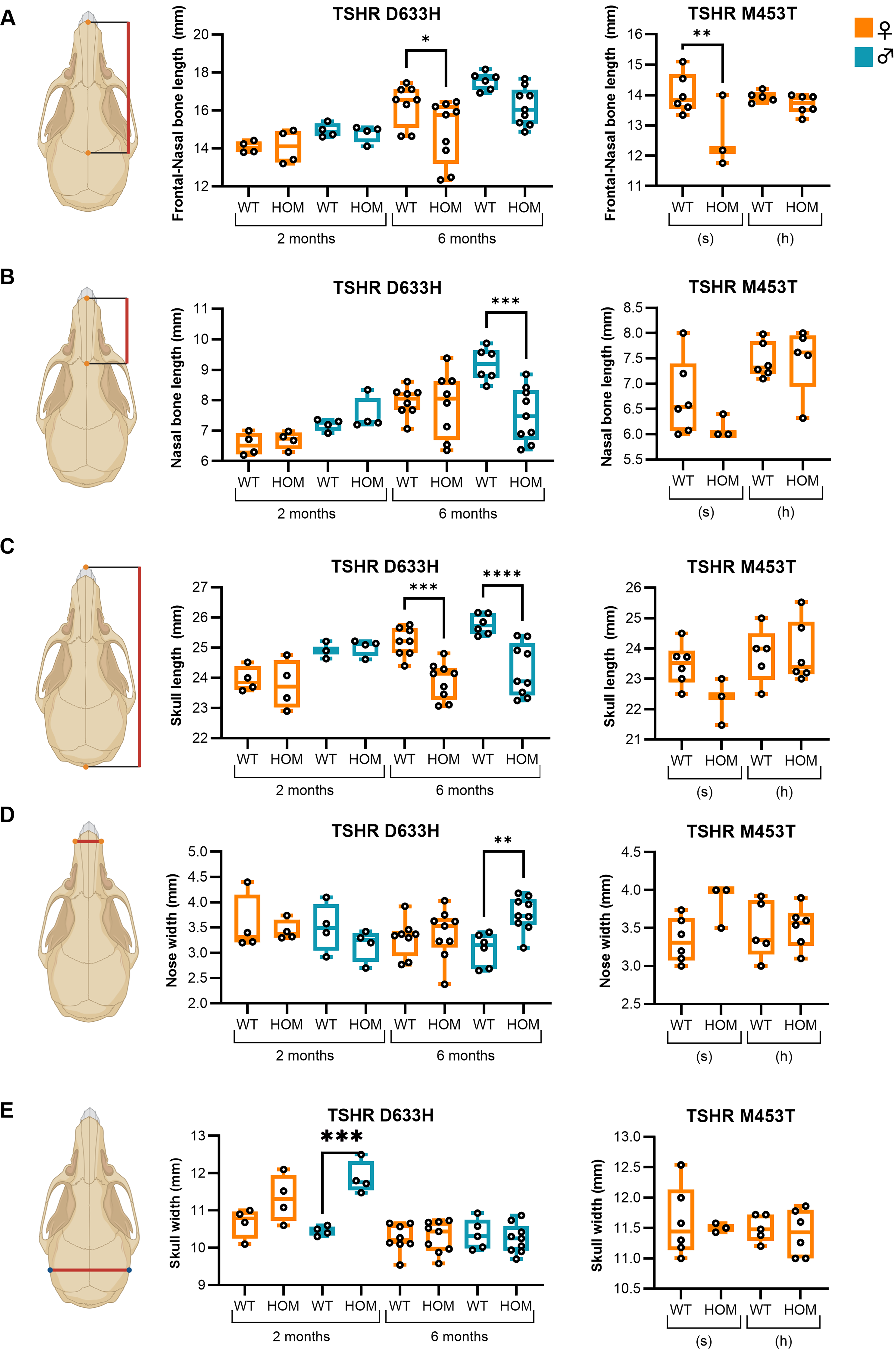
Altered craniofacial morphometry in TSHR constitutively active mutation mouse models. Linear cranial measurements were obtained from the dorsal aspect of the skull using predefined landmarks as shown on the left.
In the TSHR M453T model, HOM females on a sufficient-iodine diet showed reduced frontonasal bone length at 2 months, whereas no significant changes in cranial morphometry were observed in mice on a high-iodine diet (Fig. 2).
The prevalence of malocclusion was markedly increased in both D633H and M453T HOM and HET mice, independent of sex. In the D633H model, malocclusion occurred in 9.3% of HOM (n = 58/623) and 3.1% of HET mice (n = 31/995) compared with 0.2% in WT controls (n = 2/883) (p < 0.005, Fisher’s exact test). Similarly, in the M453T model, malocclusion affected 5.9% of HOM (n = 6/102) and 1.6% of HET mice (n = 4/258) compared with 0% in WT controls (n = 0/156; p < 0.005, Fisher’s exact test; Fig. 1C). Created with Biorender.com.
Altered bone structure in TSHR D633H and M453T CAM models
µCT analysis of tibiae from 2-month-old TSHR D633H mice revealed no significant differences in BMD (Fig. 3A) or cortical bone parameters compared with WT controls (Supplementary Table S1, Supplementary Fig. S3). However, male D633H HOM mice showed reduced trabecular bone volume (Tb.BV) and trabecular tissue volume (Tb.TV; Fig. 3), with no other trabecular parameters altered.
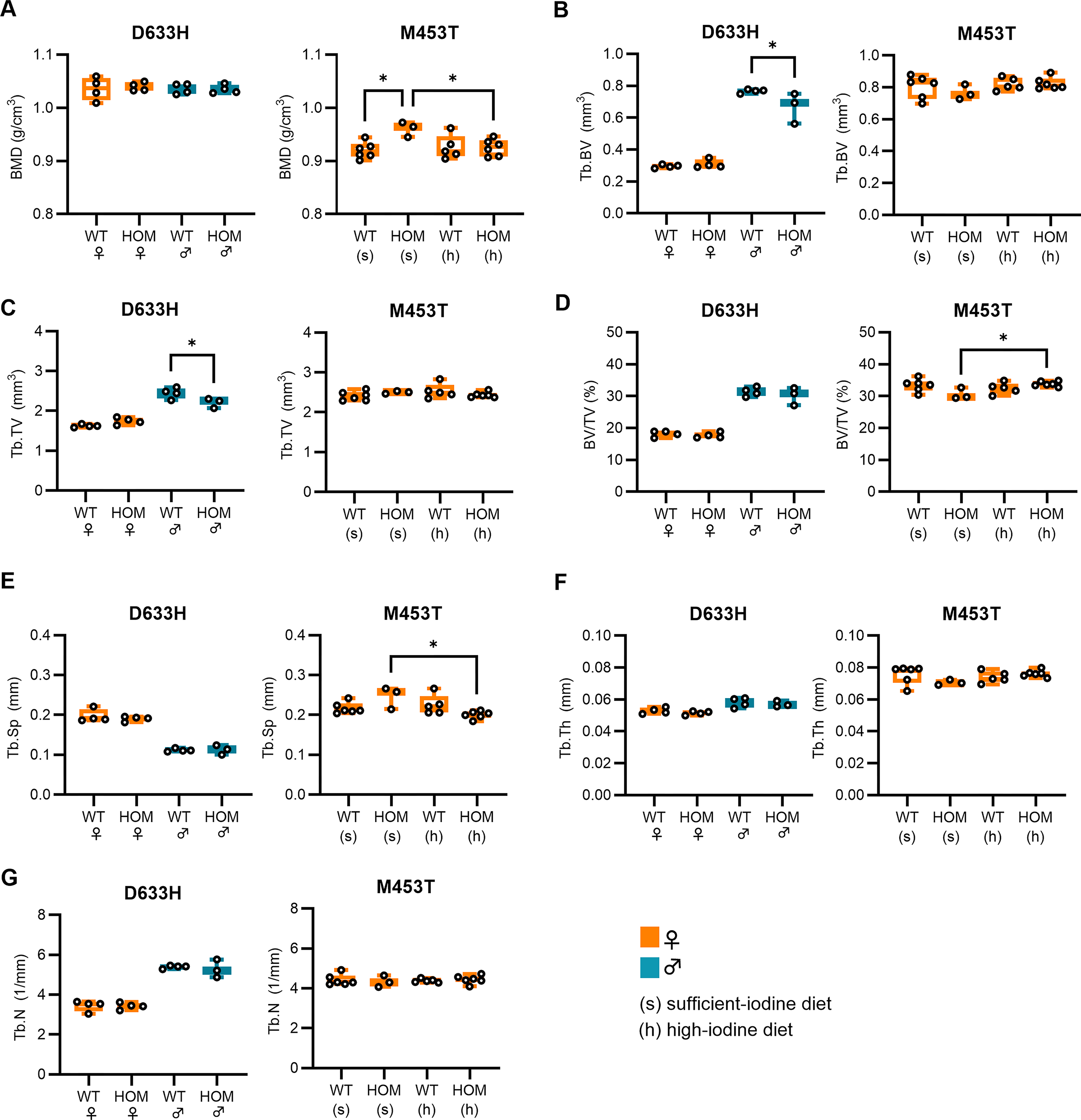
µCT analysis of tibiae in TSHR D633H and TSHR M453T mouse models.
In the M453T model, HOM females on a sufficient-iodine diet had significantly higher BMD than both WT controls on the same diet and M453T HOM mice fed a high-iodine diet, which suppresses hyperthyroidism (Fig. 3A). When comparing HOM and WT mice within the same diet group, no significant differences in trabecular or cortical bone parameters were observed (Fig. 3, Supplementary Table S1 and Supplementary Fig. S3). However, when comparing M453T HOM mice across diet groups, those on the iodine-sufficient diet (hyperthyroid) showed increased trabecular bone volume fraction (Tb.BV/TV) and reduced Tb.Sp compared with those on the high-iodine diet (suppressed hyperthyroidism). These findings indicate that the hyperthyroid state in M453T mice alters trabecular microarchitecture in an iodine-dependent manner.
Three-point bending tests of femora showed no significant differences in stiffness, Young’s modulus, or ultimate load in 2-month-old TSHR D633H mice compared with WT controls (Supplementary Table S2).
Discussion
Normal bone development and homeostasis require balanced TH action. In congenital hypothyroidism, bone maturation is delayed, head circumference is often increased, skull sutures close late, and craniofacial and dental development are impaired. Whereas congenital hyperthyroidism leads to accelerated bone maturation, accelerated tooth eruption, and in rare cases, craniosynostosis.10,23 Bone is a classical TH target tissue, essential for development, linear growth, and adult bone turnover. Thyrotoxicosis is a well-established cause of secondary osteoporosis, and abnormal TH signaling has also been identified as a novel risk factor for osteoarthritis. 8 Beyond these classical TH-mediated effects, extrathyroidal expression of TSHR has been demonstrated in osteoclasts and osteoblasts, suggesting a potential independent role in skeletal development. However, TSHR’s direct contribution to bone physiology has remained unclear. Here, we investigated skull and bone morphology in two KI mouse models harboring TSHR CAMs that differ in their in vitro signaling properties and in vivo hyperthyroidism phenotypes. Our primary focus was on skeletal and craniofacial phenotyping rather than mechanistic investigation. HOM TSHR D633H mice display mild, transient hyperthyroidism (more notable in females) without notable TFT differences under sufficient—or high-iodine—diets as described earlier.17,22 In contrast, 2-month-old female TSHR M453T mice fed a sufficient-iodine diet exhibit overt hyperthyroidism, whereas high iodine suppresses the hyperthyroid phenotype despite the presence of the CAM. 18 Since the high-iodine diet corresponds to standard commercial rodent chow, while the sufficient-iodine diet more closely reflects physiological iodine intake, these dietary conditions were used as a within-genotype comparison to assess the impact of hyperthyroidism severity on skeletal phenotype. With these models, we show that TSHR CAM associates with altered craniofacial morphology and increased malocclusion rate. Furthermore, mild structural changes and altered BMD were observed in the model with more severe hyperthyroidism. These findings highlight the role of TSHR signaling and thyroid status in craniofacial and skeletal development.
Overall, skeletal phenotypes were mild. TSHR D633H mice had normal birth weight, body length, and tail length. TSHR M453T mice exhibited a more pronounced, sex- and iodine-dependent hyperthyroid state. Specifically, HOM females on a sufficient-iodine diet had lower body weight and shorter tail length at 3 weeks of age, which normalized by 4 weeks. No significant growth differences were observed in M453T HOM males or females on a high-iodine diet. 18 Thus, the effect of TH excess on linear growth in our CAM models was subtle. This contrasts with clinical cases that show rapid growth acceleration in children with hyperthyroidism, 8 and after initiation of thyroxine treatment in early-onset hypothyroidism. 24
Despite minimal effects on linear growth, craniofacial changes were evident. Both TSHR CAM models exhibited shortened frontonasal and nasal bone lengths, along with increased prevalence of malocclusion in HOM and HET mice. Similar findings have been reported in Gpd5-overexpressing mice, where enhanced TSHR signaling led to nasal and frontal bone shortening. 25 These bones originate from neural crest cells, unlike the unaffected mesoderm-derived parietal bone, suggesting region-specific sensitivity to TSHR activation. 26 In hyperthyroid Dio3−/−mice 27 in utero thyrotoxicosis caused cranial dysmorphism, including shortened skull and nasal bones, paralleling our findings. In humans, juvenile hyperthyroidism and TSHR CAMs are associated with premature suture fusion, craniosynostosis, advanced dental eruption, and frontal bossing. Premature fusion of cranial synchondroses can alter cranial base angles, leading to retrognathia, midface hypoplasia, cleft palate, or Chiari malformation. 10 Furthermore, a recent case report described a patient carrying a germline TSHR D633H variant who presented hydrocephalus, Chiari malformation, and craniosynostosis. 16 The severity of these outcomes likely depends on disease timing and antithyroid treatment, which may prevent some features.
Interestingly, the TSHR M453T HOM mice on a sufficient-iodine diet showed higher BMD compared with WT controls, despite overt hyperthyroidism. In humans, hyperthyroidism typically reduces BMD, with serum TSH positively and TH negatively correlating with BMD.28,29 The elevated BMD in TSHR M453T mice may reflect direct TSHR-mediated effects via Gs/cAMP signaling on bone microarchitecture, as these mice also exhibited increased Tb.Sp and reduced BV/TV. In contrast, the TSHR D633H model (with different TSHR signaling) did not show significant BMD differences, although male HOM mice had slightly reduced Tb.BV and Tb.TV, likely reflecting their milder and transient hyperthyroidism.
Supporting a direct role for TSHR, a study using haploinsufficient TSHR mice with 50% reduction in TSHR expression but normal TFTs showed pronounced osteoporosis and osteopenia. These skeletal changes were not restored by TH supplementation, providing the first evidence that TSH levels may be critical for bone volume maintenance. 13 Similarly, in estrogen-depleted ovariectomized rats, it was shown that exogenous TSH preserved bone mass and improved trabecular architecture without altering TH levels. 30 This suggests that TSHR activation may have protective skeletal effects independent of TH.
In humans, the impact of TSHR polymorphisms on BMD and osteoporosis remains debated; Li et al. linked the TSHR D727E variant with increased osteoporosis risk, suggesting impaired osteoclast suppression. 31 Conversely, the Rotterdam study found D727E associated with elevated BMD. 32 Unlike the TSHR CAMs studied here, D727E is associated with hypothyroidism. Larger studies, including our own using clinical data from CAM patients and the FinnGen population cohorts, found no significant associations between TSHR variants and bone phenotypes.18,33 In general, human cases with familial NAH caused by TSHR CAM seem to have a milder phenotype than sporadic TSHR CAM. 34 Notably, bone age advancement and linear growth acceleration cease after thyroidectomy, despite persistent TSHR activation in extrathyroidal tissues. 35 Further detailed analysis of TSHR CAM patients and their bone characteristics, as well as studies on TSHR-targeted therapies, may offer new insights into bone loss prevention. 36
Our study has several limitations. Only two timepoints were analyzed; our cohort was relatively small; we mainly focused on female mice in which the hyperthyroid phenotype appears stronger in both models; and follow-up measurements of thyroid function tests were not performed in this cohort; instead, we relied on the previously published TFT characterization of these models.17,18,22 Although the thyroid function tests differed markedly between the two models, both displayed altered craniofacial morphology, and increased malocclusion was observed in both HET (euthyroid) and HOM animals, suggesting TH-independent effects. However, we cannot exclude the transient or in utero effects of TH as contributing factors. Finally, the molecular mechanisms by which TSHR CAMs act in bone cells remain to be elucidated, and further studies, perhaps using tissue-specific models, are needed to assess the long-term skeletal consequences of TSHR CAMs.
In conclusion, we demonstrate that TSHR CAMs in mice primarily affect craniofacial morphology, including shortened snout length and increased malocclusion, with only a modest impact on skeletal growth and bone density. These findings highlight the role of TSHR signaling and thyroid status in craniofacial and skeletal development, warranting further mechanistic investigation.
Authors’ Contributions
K.M.: Writing—original draft (lead), phenotyping and analysis (lead), and conceptualization (lead). J.M., K.K.I., H.J., and R.P.: Phenotyping and analysis (equal) and conceptualization (equal). K.P., V.M., V.L., J.O., R.R., and H. U.: Phenotyping and analysis (equal). J.K.: Conceptualization (lead), phenotyping and analysis (equal), and writing—original draft (lead).
Footnotes
Acknowledgments
The authors thank the personnel in Dr. Jan Rozman laboratory (Czech Center for Phenogenomics) with INFRAFRONTIER2020 support for the phenotypic characterization, including 3D CT images of the TSHR D633H model, and Nico Moritz for expertise with the three-point bending test.
Author Disclosure Statement
The authors have declared that no conflicts of interest exists.
Funding Information
This study was supported by grants from the Finnish Pediatric Foundation (Jukka Kero no. 190001), the Turku University Hospital (Jukka Kero no. 13527), the Sigrid Juselius Foundation (Jukka Kero 12/2022), and the Novo Nordisk Foundation (Jukka Kero no. 0078329), the Doctoral Programme in Clinical Research (DPCR) (Kristiina Makkonen); and the Drug Research Doctoral Programme (DRDP) (Vladyslav Melnyk) of the University of Turku. There is no funding information to declare for this study for Jorma Määttä, Kaisa Ivaska, Konrad Patyra, Veli Linnossuo, Johanna Ojala, Rowmika Ravi, Holger Jaeschke, Hendrik Undeutsch, and Ralf Paschke.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.