
Abstract
Introduction
The airway microbiome plays a pivotal role in lung cancer development, but the microbiome characteristics in upper and lower respiratory tract of non-small cell lung cancer (NSCLC) patients remains unclear.
Methods
This was a prospective case-control study. The study included 60 samples from NSCLC patients and non-cancer controls: 23 sputum (SP) samples (14 NSCLC, 9 controls) and 37 bronchoalveolar lavage fluid (BALF) samples (21 NSCLC, 16 controls). Metagenomic sequencing was performed to characterize microbial composition and diversity, differential taxa, inter-kingdom networks, and functional profiles for bacteria and fungi.
Results
For bacterial community, BALF samples from NSCLC tend to show higher alpha diversity than that of non-cancer controls (Shannon p = 0.046, Simpson p = 0.089), whereas SP samples from NSCLC show a trend toward lower alpha diversity (Shannon p = 0.053, Simpson p = 0.033). For fungal community, alpha diversity shows no significant difference between NSCLC and non-cancer groups in either SP (Shannon p = 0.250, Simpson p = 0.480) or BALF (Shannon p = 0.800, Simpson p = 0.700) samples. Beta diversity exhibits differences in bacterial community composition between NSCLC and non-cancer controls in both SP (p = 0.018) and BALF samples (p = 0.015), while fungal communities appear relatively stable (p = 0.611 for SP; p = 0.611 for BALF). LEfSe and Random Forest analyses identify bacterium Porphyromonas SGB2015 and fungus Psilocybe cubensis significantly enriched in BALF samples from NSCLC, whereas no species is enriched in SP samples. Cross-kingdom network indicates increased complexity and connectivity in NSCLC-associated microbial communities. Functional analysis shows the enrichment of biosynthetic pathways in SP samples and metabolic pathways in BALF samples from NSCLC.
Conclusion
These findings suggest that NSCLC may be associated with compositional, structural, and functional alterations of the airway microbiome, with potentially distinct patterns between upper and lower respiratory tract.
Keywords
Introduction
Lung cancer remains the leading cause of cancer morbidity and mortality worldwide, with an increasing incidence observed in recent years. 1 According to histological classification, lung cancer is primarily categorized into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), with NSCLC accounting for approximately 85% of all lung cancer cases.2,3 The principal treatment methods for NSCLC include surgical resection, chemotherapy, immunotherapy, and targeted therapy, these interventions have significantly improved the prognosis for NSCLC patients. 4 However, the overall cure rate and survival rate for NSCLC remain low, particularly as many patients with advanced disease develop resistance to standard treatments. 5 Hence there exists a critical need to elucidate the underlying mechanisms involved in NSCLC development and progression. Such advancements will contribute to find new biological markers and therapeutic targets that facilitate accurate diagnosis and therapy of lung cancer.
Increasing evidence suggests that the pulmonary microbiome, encompassing both bacterial and fungal communities, plays a pivotal role in lung carcinogenesis, not only shaping the tumor immune microenvironment but also driving tumor development.6-12 Current investigations of the lung microbiome primarily rely on two types of specimens: sputum (SP) and bronchoalveolar lavage fluid (BALF).13,14 SP is non-invasive and easily accessible, and its microbial signatures have been proposed as potential biomarkers for early detection and subtyping of NSCLC. 15 In contrast, BALF more accurately reflects the local ecological status of the lower respiratory tract and is therefore widely used in lung cancer-related studies. The microbial metabolite D-phenylalanine has been shown to promote lung cancer metastasis through epithelial-mesenchymal transition, suggesting its important role for tumor dissemination. 16 Furthermore, airway dysbiosis in lung cancer patients has been linked to tumor progression and poor clinical outcomes, with Veillonella parvula identified as a key driver taxon. 17 Enrichment of oral commensals such as Streptococcus and Veillonella in the lower airways has also been associated with activation of the ERK and PI3K signaling pathways, which is further validated in vitro by the exposure of airway epithelial cells to Veillonella, Prevotella, and Streptococcus. 18 Beyond bacteria, fungal communities also contribute to NSCLC pathogenesis. Several studies have reported markedly increased fungal diversity and more complex fungal community structures in patients with lung cancer. Notably, Alternaria arborescens is enriched in tumor tissues and shows a significant association with NSCLC, suggesting a unique role for fungi in carcinogenesis. 19 Furthermore, several studies have attempted to compare bacterial microbiome profiles across different specimen types in NSCLC patients. For instance, Huang et al. found that BALF may better reflect the microbiome of lung cancer tissue than SP. 20 Another study profiled the upper and lower respiratory tract microbiome and metabolome in healthy volunteers without respiratory diseases, uncovering topographical differences in microbial function. 21 Nevertheless, most of these investigations remain confined to a single microbial kingdom. The bacterial and fungal microbiome of SP and BALF, associated with lung cancer, has yet to be fully elucidated and remains controversial.
In this study, we recruited NSCLC patients and non-cancer controls to systematically characterize the bacterial and fungal microbiome in SP and BALF samples. Shotgun metagenomic sequencing was employed to achieve high-resolution taxonomic profiling at the species, along with functional pathway annotation and cross-kingdom co-occurrence network analysis. 22 Furthermore, we applied machine learning models to identify key microbial species associated with NSCLC. By directly comparing SP and BALF samples within the same analytical framework, our study aims to elucidate the microbiome characteristics in upper and lower respiratory tract of NSCLC patients.
Methods
Ethics Approval
The design of this study was approved by the Ethics Committee of the Hefei Cancer Hospital, Chinese Academy of Sciences, on December 15, 2023 (Approval No. SL-PJ2023-102). All procedures performed in this experiment undertaken were in accordance with the ethical standards of the Helsinki Declaration (1975), as revised in 2024.
Study Design and Participants
This was a prospective case-control study. The reporting of this study conforms to the STROBE guidelines. 23 All participants signed the informed consent form. All participants’ data were de-identified prior to analysis to ensure confidentiality. Participants were consecutively enrolled from 2023 to 2025 year. For SP samples, the patients who show a pulmonary mass by CT imaging were recruited in the wards of a cancer hospital (Hefei Cancer Hospital, Chinese Academy of Sciences). Based on subsequent diagnostic results, patients diagnosed with NSCLC were assigned to the NSCLC group, while the remaining participants were classified as non-cancer controls. The non-cancer control group included patients diagnosed with pneumonia and benign lesion. In addition, 4 healthy volunteers were also recruited into non-cancer control group.
For BALF samples, the patients who shows a pulmonary mass by CT imaging were recruited in the endoscopy rooms of a cancer hospital (Hefei Cancer Hospital, Chinese Academy of Sciences). Based on subsequent diagnostic results, patients diagnosed with NSCLC were assigned to the NSCLC group, while the remaining participants were classified as non-cancer controls. The non-cancer control group included patients diagnosed with pneumonia, lung abscess, and benign lesion.
Exclusion criteria were as follows: (i) patients diagnosed with small cell lung cancer. (ii) participants who had received chemotherapy, radiotherapy, or antibiotic treatment within 14 days prior to sampling. (iii) participants with incomplete clinical information.
After quality control, a total of 60 samples were included in the final analysis, including 23 (non-cancer control = 9, NSCLC = 14) SP samples and 37 (non-cancer control = 16, NSCLC = 21) BALF samples. The clinical information of the participants is shown in Supplemental Table 1.
Sample Collection
To minimize contamination from oral microbiota, SP donors were instructed to thoroughly rinse their mouths with sterile saline before sample collection. SP sample was then expectorated directly into sterile containers. The BALF sample used in this study consisted of residual clinical specimens collected during routine bronchoscope procedures. BALF was obtained from the lung region showing the most pronounced radiographic abnormalities. Bronchoalveolar lavage was performed under sterile conditions by instilling and aspirating pre-warmed 0.9% NaCl through a bronchoscope. BALF and SP specimens were processed within two hours of collection.
DNA Extraction and Metagenomic Sequencing
Microbial DNA was extracted using the FastPure Host Removal and Microbiome DNA Isolation Kit (DC501-01, Vazyme, China). Sequencing libraries were prepared with the UltraClean Universal Plus DNA Library Prep Kit (UND637-01, Vazyme, China). Library quality was assessed by Qubit fluorometer (Thermo Fisher Scientific, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Libraries were sequenced on an Illumina NovaSeq 6000 platform (paired-end, 150 bp), targeting an average 6 Gb data per sample. Negative controls (PBS, extraction reagents, and library preparation reagents) were included to monitor environmental contamination, with no detectable microbial DNA observed.
Metagenomic Data Processing
Raw sequencing reads were quality-filtered using FastQC and TrimGalore (v0.4.5) to remove adapters, low-quality bases (Q < 30), ambiguous nucleotides (“N”), and short fragments (<35 bp). Host-derived sequences were excluded by mapping to the human reference genome (hg38) with Bowtie2. Taxonomic profiling of bacteria was performed with MetaPhlAn4 using the database (mpa_vJun23_CHOCOPhlAnSGB_202403). Functional profiling was conducted with HUMAnN3. Fungal taxonomic assignment and abundance estimation were performed using Kraken2 (v2.1.3) followed by Bracken. A fungi-specific database was constructed using the Kraken2 build pipeline by downloading fungal reference genomes from the NCBI RefSeq database along with taxonomy information. Only fungal sequences were included in the database to improve the specificity of fungal taxonomic classification. The database was built with default parameters (k-mer length = 35, minimizer length = 31). Bracken was subsequently applied to estimate species-level relative abundances based on Kraken2 classification results.
Sequencing Depth and Coverage Assessment
Species-level count data for bacteria and fungi were obtained from metagenomic profiling. The total number of sequencing reads per sample was calculated by summing species-level read counts across all bacterial and fungal species. Total reads were log10-transformed for visualization. Boxplots were generated to display the distribution of species richness and total reads across the four groups. Individual data points were overlaid using jittered points. For total reads, the y-axis was scaled to log10(TotalReads).
Sequencing depth and diversity coverage were evaluated using rarefaction curves based on taxonomic profiles derived from metagenomic sequencing. For each sample, sequencing depth (x-axis) was defined as the cumulative number of sequencing reads, and subsampling was performed to generate rarefaction curves. Species richness was estimated using the Chao1 index. The Chao1 index was calculated based on the presence-absence and inferred count information of annotated species. The number of observed species per sample was determined from detected taxa. Rarefaction curves were constructed by repeatedly subsampling reads and recalculating species richness, allowing assessment of sequencing sufficiency. Curves approaching a plateau indicated adequate sequencing depth to capture the majority of microbial diversity. Rarefaction curves for sequencing depth assessment were generated using the vegan package.
Microbial Species Composition
Microbial community composition was characterized at the species level based on metagenomic relative abundance profiles. Taxonomic abundance tables for bacteria and fungi were sorted by total abundance across all samples. The top 15 most abundant species in each dataset were selected, and the remaining taxa were grouped into an “Other” category. Stacked bar plots were generated to illustrate the relative abundance of dominant taxa across samples and groups. In addition, group-level comparisons were performed by aggregating species abundances within each group and calculating the proportion of each species relative to the total abundance of that group.
Statistical Analysis
Microbial Diversity Analyses
Alpha diversity was quantified using the Shannon-Wiener and Gini-Simpson indices based on relative abundance data. Beta diversity was calculated using Bray-Curtis dissimilarities after Hellinger transformation of relative abundance data. Principal coordinate analysis (PCoA) was used for ordination. Homogeneity of group dispersion was evaluated using permutational analysis of multivariate dispersions (PERMDISP).
Alpha diversity differences were evaluated using the Wilcoxon rank-sum test, while beta diversity differences were assessed using permutational multivariate analysis of variance (PERMANOVA) with 9,999 permutations.
Identification of Differential Taxa
Species-level biomarkers distinguishing NSCLC from non-cancer control were identified using LefSe, following the standard LEfSe workflow, with an additional Latent dirichlet allocation (LDA) threshold applied to control false positives (thresholds: p < 0.05, LDA > 2.5). LEfSe analysis was performed using relative abundance data, as implemented in the microeco package.
In addition, a Random Forest classifier was constructed using species-level relative abundances as predictors and clinical group as the outcome. Feature importance was ranked by MeanDecreaseGini, and the top 10 discriminatory taxa were selected.
The discriminatory ability of the selected taxa between NSCLC and non-cancer control was evaluated using multivariable logistic regression model. Receiver operating characteristic (ROC) curve analysis was performed, and the area under the curve (AUC) with 95% confidence intervals was calculated.
Microbial Co-occurrence Network Analysis
To characterize inter-kingdom associations, bacterial-fungal co-occurrence networks and hub co-occurrence subnetworks were constructed by relative abundance profiles. Low-abundance taxa were filtered (present in < 70% of samples). Pairwise correlations were computed using Spearman’s rank correlation, followed by multiple-testing correction with the Benjamini-Hochberg method. Significant associations (|r| ≥ 0.7, FDR < 0.05) were retained. Network topology was analyzed with the igraph and ggraph packages.
Centrality measures, including degree, betweenness, and closeness, were calculated to identify hub species, defined as nodes exceeding the mean + 2 SD threshold for any of these metrics. Community detection was performed with the Louvain algorithm. Nodes were annotated at the species level and further classified by feature type (bacterial or fungal taxa). Cross-domain interactions were quantified by counting edges connecting nodes from different feature categories. Networks were visualized using the Fruchterman-Reingold force-directed layout, with positive correlations in red and negative correlations in blue.
Functional Annotation and Pathway Enrichment
Functional annotation was performed at the KEGG Orthology (KO) level based on relative abundance profiles. Differential KOs between groups were identified using the Wilcoxon rank-sum test, with fold change, log2 fold change, and multiple-testing correction (Benjamini-Hochberg). KOs with p < 0.05 were retained. Significant KOs were mapped to KEGG pathways using clusterProfiler. Pathways were considered biologically relevant only if they met both enrichment significance (p.adjust < 0.05) and pathway abundance differences (FDR < 0.05).
All statistical analyses were performed using R (version 4.2.2). Differences between groups were assessed using non-parametric tests, including the Kruskal-Wallis test for multiple-group comparisons and the Wilcoxon rank-sum test for pairwise comparisons. P values were adjusted using the Benjamini-Hochberg method where applicable. All tests were two-sided, and a P value < 0.05 was considered statistically significant.
Results
Participants and Metagenomic Sequencing Data
A total of 79 samples were collected, and after stringent quality control, 60 participants were retained for metagenomic analysis. Among these participants, the 23 SP samples from 14 NSCLC patients and 9 non-cancer controls were subjected to metagenomic sequencing; the 37 BALF samples from 21 NSCLC patients and 16 non-cancer controls were subjected to metagenomic sequencing (Figure 1A and Supplemental Table 2). The baseline clinical characteristics of these participants were summarized in Table 1. The overview of metagenomic sequencing in the study. (A) The scheme of the study design. (B) Distribution of reads counts for bacterial and fungal species in SP and BALF samples. (C) Number of bacterial and fungal species in SP and BALF samples. (D) Microbiota rarefaction curve generated based on Chao 1 diversity Clinical Characteristics of the Enrolled Participants Age: Median (interquartile range), SP: sputum, BALF: bronchoalveolar lavage fluid.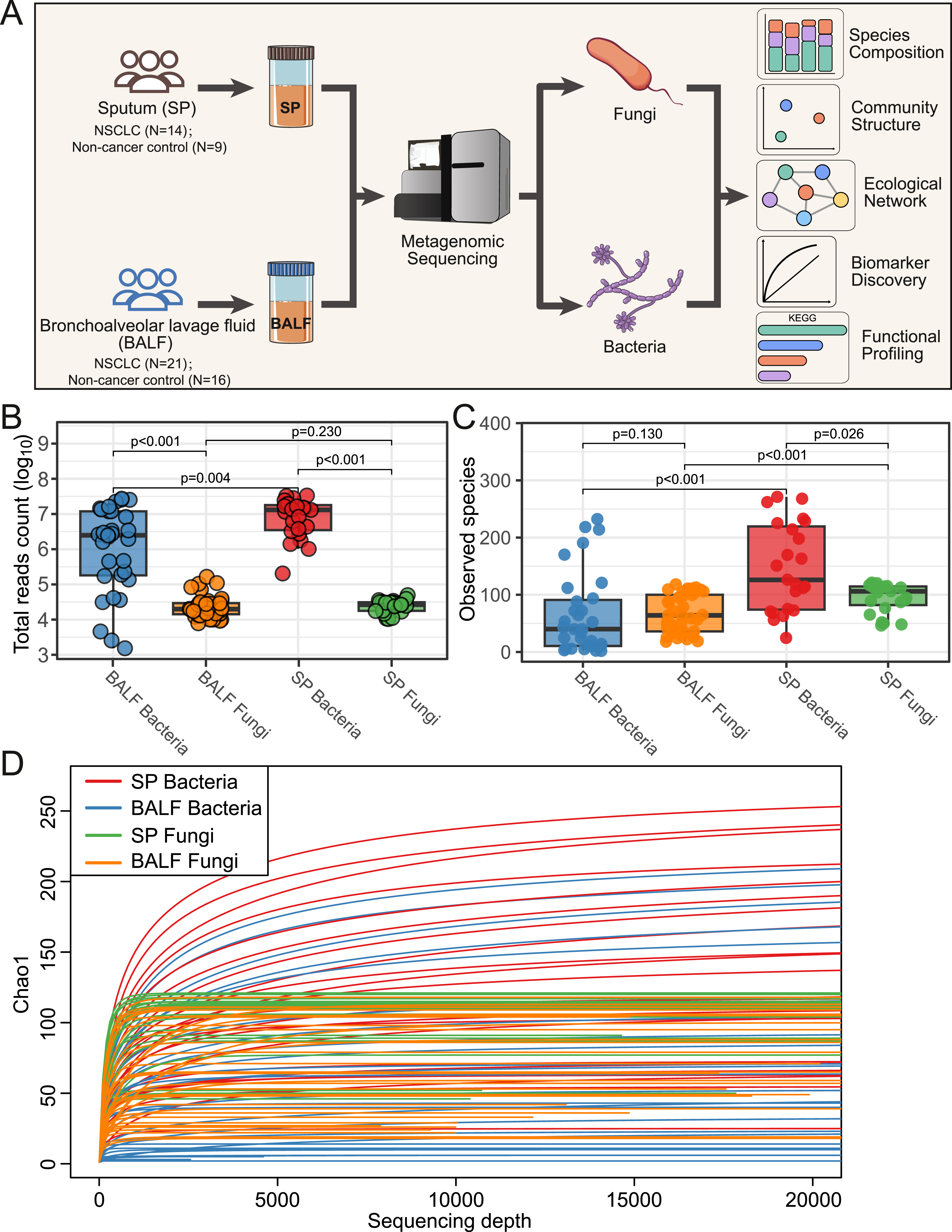
Metagenomic sequencing data indicated that reads mapped to bacterial were substantially higher than reads mapped to fungi. In BALF samples, the median reads counts were 2.48×106 mapped to bacteria, and 1.99×104 mapped to fungi. In SP samples, the median reads counts were 1.31×107 mapped to bacteria, and 2.65×104 mapped to fungi (Figure 1B). Regarding species richness, in SP samples bacterial diversity was higher than fungal diversity, while in BALF samples no difference was observed between bacterial diversity and fungal diversity (Figure 1C). Rarefaction curves indicated that sequencing depth was sufficient for downstream diversity and compositional analyses (Figure 1D).
Dominant Microbial Species Composition in SP and BALF
To investigate the differences in species level between NSCLC patients and non-cancer controls, we analyzed the top 15 most abundant bacterial and fungal species in SP and BALF samples (Supplemental Figure 1 and Supplemental Table 3). Distinct microbial community compositions were observed across the four groups defined by disease status and sampling site, revealing the differences between SP and BALF samples as well as between NSCLC and non-cancer controls.
We first compared dominant taxa between SP and BALF samples. In non-cancer controls, the dominant bacterial composition differed markedly between the two sample types. In BALF samples, the most abundant bacterial species included Neisseria subflava, Prevotella melaninogenica, Streptococcus pneumoniae, Sphingomonas paucimobilis, and Fusobacterium nucleatum. In SP samples, the dominant bacterial species were Prevotella melaninogenica, Streptococcus mitis, Neisseria sicca, Pseudomonas aeruginosa, and Neisseria subflava. In contrast, fungal communities showed relatively consistent dominant taxa across sample types, with Fusarium pseudograminearum, Pyricularia pennisetigena, and Pyricularia grisea being predominant in both BALF and SP samples. A similar pattern was observed in NSCLC patients, where bacterial composition differed substantially between BALF and SP samples. BALF samples were dominated by Neisseria subflava, Neisseria sicca, Prevotella melaninogenica, and Sphingomonas paucimobilis, whereas SP samples were enriched with Neisseria subflava, Streptococcus mitis, Rothia mucilaginosa, Haemophilus influenzae, and Neisseria sicca. In contrast, fungal dominant taxa remained largely consistent across sample types, including Fusarium pseudograminearum, Pyricularia pennisetigena, Pyricularia grisea, and Fusarium fujikuroi (Figure 2A). Microbial community composition in SP and BALF samples of non-cancer control and NSCLC. (A) Relative abundances of the top 15 bacterial and fungal species in SP and BALF samples of non-cancer control and NSCLC. Other taxa were grouped as “Other”. (B) Relative abundances of the top 10 bacterial and fungal species between NSCLC patients and non-cancer controls. Heatmap shows the standardized relative abundances (Z-score) of bacterial and fungal species. The bar plots on the right represent the overall abundance contribution (%) of each species to the total microbial community. (C) Venn diagrams show the common bacterial and fungal species shared in SP and BALF samples of NSCLC patients and non-cancer controls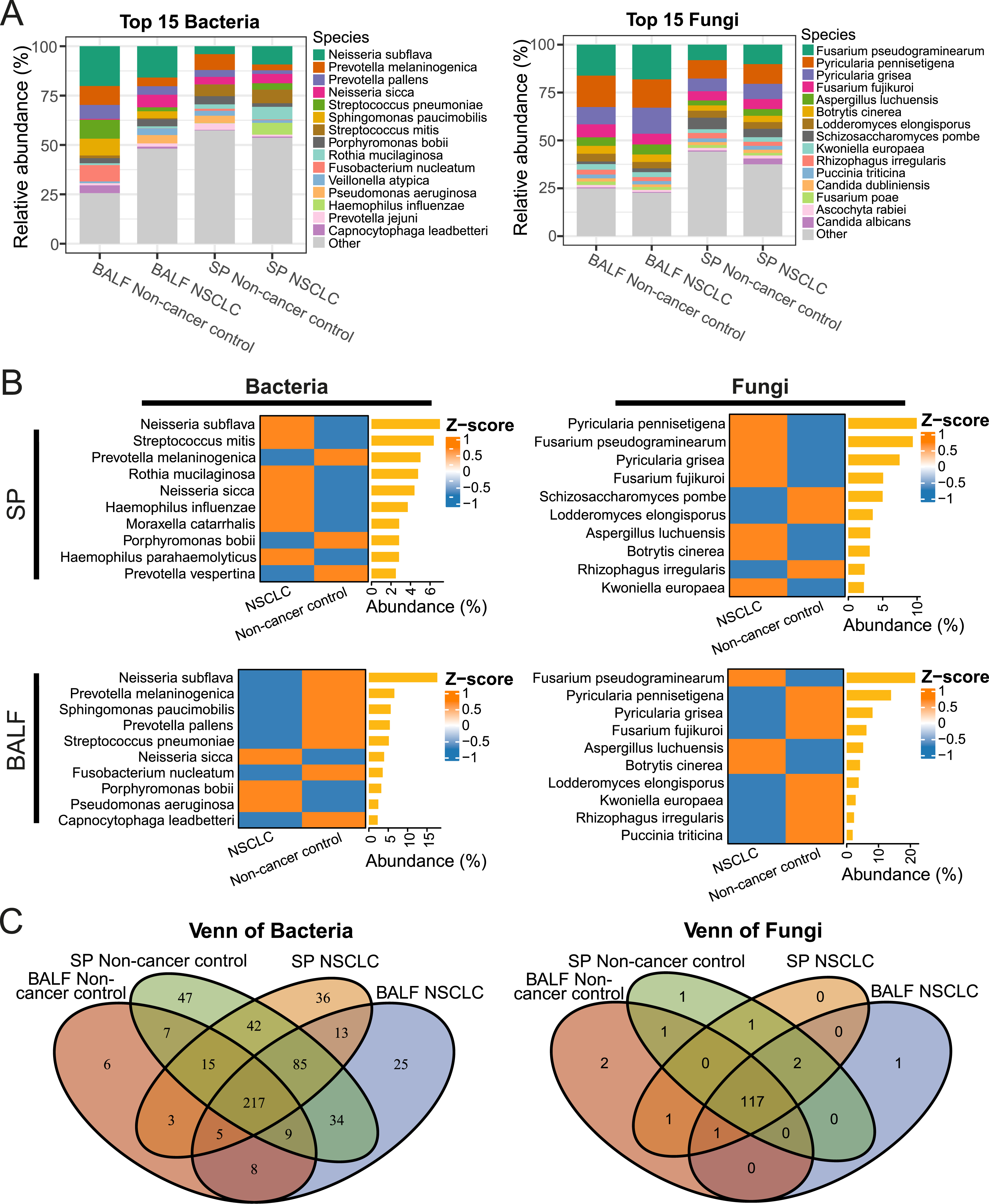
We next examined microbiome alterations associated with disease status. In the SP samples, bacterial taxa such as Neisseria subflava and Streptococcus mitis, together with fungal species including Fusarium pseudograminearum and Aspergillus luchuensis, were enriched in NSCLC. In the BALF samples bacterial species such as Neisseria sicca and fungal taxa including Fusarium pseudograminearum and Aspergillus luchuensis showed higher abundance in NSCLC than in non-cancer control (Figure 2B).
SP samples exhibited higher species richness and diversity than BALF samples, suggesting a more complex upper airway microbiota. 217 bacterial and 117 fungal species were shared in all the four groups (Figure 2C).
Microbial Community Structure and Diversity in SP and BALF
Next, we compared alpha diversities between SP and BALF samples within each group. In non-cancer control participants, bacterial communities in SP samples exhibited significantly higher alpha diversity than those in BALF samples, as indicated by both Shannon and Simpson indices (Shannon p < 0.001, Simpson p < 0.001). In contrast, no significant differences in bacterial alpha diversity between SP and BALF samples were observed in NSCLC patients (Shannon p = 0.440, Simpson p = 0.670) (Figure 3A). For fungal communities, SP samples also showed higher alpha diversity than BALF samples in non-cancer controls (Shannon p = 0.007, Simpson p = 0.057), whereas the differences between SP and BALF samples were less pronounced in NSCLC patients (Shannon p = 0.034, Simpson p = 0.100) (Figure 3B). The comparison of microbial diversity. (A) Alpha diversity for bacterial across the four groups, quantified by Shannon and Simpson indices. Significance was tested using the Wilcoxon rank-sum test with Benjamini-Hochberg correction. (B) Alpha diversity for fungal across the four groups, quantified by Shannon and Simpson indices. Significance was tested using the Wilcoxon rank-sum test with Benjamini-Hochberg correction. (C) Beta diversity for bacterial across the four groups, showed by PCoA based on Bray-Curtis distances. Group differences were assessed using PERMANOVA. (D) Beta diversity for fungal across the four groups, showed by PCoA based on Bray-Curtis distances. Group differences were assessed using PERMANOVA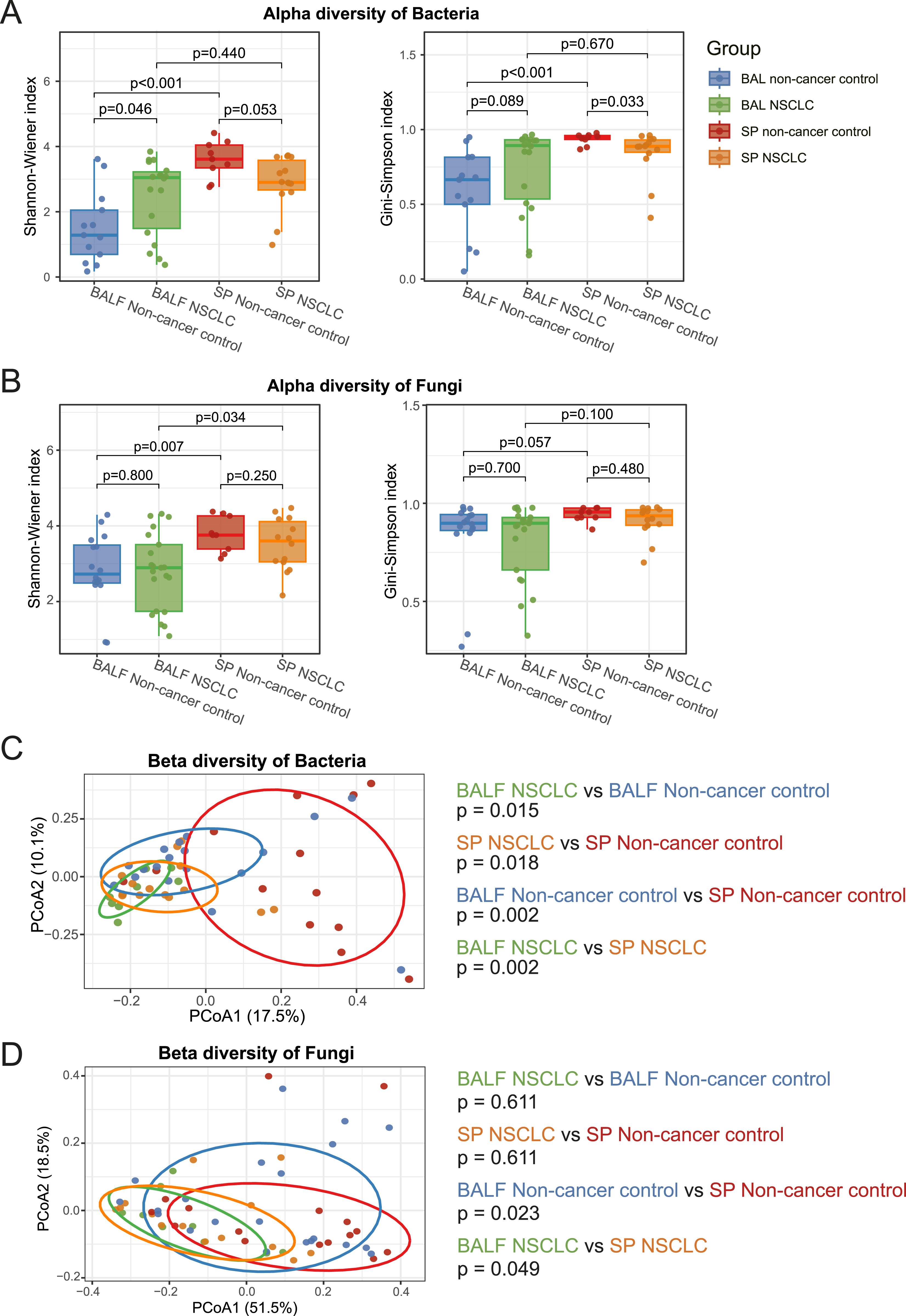
Beta diversity analyses based on Bray-Curtis distances further supported distinct microbial separation between sample types. Bacterial communities in BALF and SP samples were clearly separated in both non-cancer controls (p = 0.002) and NSCLC patients (p = 0.002) (Figure 3C). Similarly, fungal beta diversities differed significantly between SP and BALF samples in both groups (non-cancer controls: p = 0.023; NSCLC: p = 0.049) (Figure 3D).
We next examined differences between NSCLC patients and non-cancer controls. In BALF samples, NSCLC patients had slightly higher bacterial alpha diversity than non-cancer controls (Shannon p = 0.046, Simpson p = 0.089). In SP samples, however, NSCLC patients showed slightly lower bacterial alpha diversity (Shannon p = 0.053, Simpson p = 0.033) (Figure 3A). For fungal communities, no significant differences in alpha diversity were observed between NSCLC patients and non-cancer controls in both SP and BALF samples (Figure 3B).
Beta diversity analyses revealed significant NSCLC-associated shifts in bacterial communities in both SP and BALF samples (BALF: non-cancer controls vs. NSCLC, p = 0.015; SP: non-cancer controls vs. NSCLC, p = 0.018) (Figure 3C). In contrast, fungal communities did not show significant NSCLC-associated differences in beta diversity in either sample type (BALF: p = 0.611; SP: p = 0.611) (Figure 3D).
Furthermore, we analyzed the effects of gender, age, and smoking status on community diversity. In our cohort, no significant differences in alpha or beta diversity were observed between males and females in either SP or BALF samples, for either bacteria or fungi (Supplemental Figure 2A–D). For the effect of age, no significant differences in bacteria alpha diversity were found between the higher and lower age groups in either SP or BALF samples (Supplemental Figure 3A). For fungi, BALF samples from the lower age group showed lower alpha diversity compared to the higher age group (Shannon p = 0.020, Simpson p = 0.007), while no significant differences in alpha diversity were observed in SP samples (Supplemental Figure 3B). No significant differences in beta diversity were observed between the higher and lower age groups, for either bacteria or fungi (Supplemental Figure 3C–D). With respect to smoking status, no significant differences in alpha or beta diversity were observed between smokers and non-smokers in either SP or BALF samples for bacteria or fungi (Supplemental Figure 4A–D).
NSCLC-Associated Microbial Species
To identify NSCLC-associated microbial species, we performed species-level differential analyses between NSCLC and non-cancer control samples using LEfSe and Random Forest.
In bacterial community of SP samples, LEfSe analysis identified 21 differential species, and Random Forest highlighted the top 10 important species. Among these bacterial species, three overlapped species were Actinomyces sp. ICM47, Porphyromonas endodontalis, and Porphyromonas gingivalis, which were more abundant in non-cancer control samples (Figure 4A). For fungi, in SP samples, LEfSe analysis identified 4 differential species, and Random Forest highlighted the top 10 important species. Among these fungal species, two overlapping species were Puccinia striiformis and Colletotrichum destructivum, which were depleted in the NSCLC group compared with non-cancer control (Figure 4B). Key bacterial and fungal species associated with NSCLC in SP and BALF samples. (A) The differential bacteria species between non-cancer control and NSCLC in SP samples. Differentially bacteria species were identified by LEfSe analysis (α = 0.05, LDA > 2.5). Bars represent LDA scores, with colors denoting enrichment in NSCLC (orange) or non-cancer control (blue). Asterisks indicate statistical significance (* p < 0.05; ** p < 0.01; *** p < 0.001). Random forest analysis was trained on the top 100 most abundant taxa, and the 10 most discriminatory taxa were identified based on the MeanDecreaseGini index. Bubble plot shows the mean relative abundances (%). Bar plot shows the relative importance (MeanDecreaseGini). Venn diagram shows the overlap species identified by Random Forest and LEfSe. (B) The differential fungal species between non-cancer control and NSCLC in SP samples. (C) The differential bacteria species between non-cancer control and NSCLC in BALF samples. (D) The differential fungal species between non-cancer control and NSCLC in BALF samples
In bacterial community of BALF samples, LEfSe analysis detected 26 differential species, including three overlapped species with the top 10 important species identified by Random Forest. Alloprevotella sp. Lung230 and GGB1144_SGB1469 were enriched in non-cancer control, while Porphyromonas SGB2015 was enriched in NSCLC (Figure 4C). In BALF sample, LEfSe analysis detected 3 differential fungi, including one overlapped species with the top 10 important species identified by Random Forest. The overlapped species Psilocybe cubensis was enriched in NSCLC (Figure 4D).
We then used ROC analysis to evaluate the performance of biomarker panels composed with these overlapping species in discriminating NSCLC. In SP, the bacterial panel comprising the overlapping species (Actinomyces sp. ICM47, Porphyromonas endodontalis, and Porphyromonas gingivalis) achieved an AUC of 0.889; the fungal panel (Puccinia striiformis and Colletotrichum destructivum) reached an AUC of 0.801. In BALF, bacterial panel (Alloprevotella sp. Lung230, GGB1144_SGB1469 and Porphyromonas SGB2015) had an AUC of 0.839; Fungal panel (Psilocybe cubensis) had an AUC of 0.704 (Figure 5A). Integrating bacterial and fungal features improved the discrimination. In SP, combined panel reached an AUC of 0.976; in BALF, combined panel reached an AUC of 0.877 (Figure 5B). ROC curves showing the performance of identified common microbial markers by LEfSe and random forest in distinguishing NSCLC from non-cancer control. (A) ROC curve of bacteria markers in SP (Red line), AUC=0.889; ROC of bacteria in BALF (Blue line), AUC=0.840; ROC of Fungi in SP (Green line), AUC=0.802; ROC of Fungi in BALF (Orange line), AUC=0.704. (B) ROC of combined bacterial-fungal panels in SP cohort (purple line), AUC=0.976; ROC of combined bacterial-fungal panels in BALF cohort (pink line), AUC=0.877
Cross-Kingdom Microbial Network and Hub Species
Next, we constructed cross-kingdom co-occurrence networks to investigate potential bacteria-fungi interactions.
Compared with BALF samples, SP samples in non-cancer controls exhibited a greater number of nodes and edges, indicating higher species richness and more co-occurrence relationships. In addition, SP networks showed a larger diameter and a lower clustering coefficient, suggesting a less compact and less modular network structure. A similar pattern was observed when comparing SP and BALF samples in the NSCLC group (Figure 6A B, Supplemental Table 4). Cross-kingdom microbial co-occurrence networks. (A) Co-occurrence networks in SP samples, (B) Co-occurrence in BALF samples. Species that appear in at least 70% of the samples are included in the analysis. Spearman’s rank correlations were calculated from species-level abundance profiles, followed by false discovery rate (FDR) correction. Nodes represent microbial taxa, with node size proportional to degree centrality. The resulting community structure, as revealed by the Louvain algorithm, is visualized through distinct color assignments. Edges represent significant correlations, where gray denotes positive associations and red denotes negative associations, and edge width is scaled by correlation strength. The network layout was generated using the Fruchterman-Reingold force-directed algorithm. (C) Hub co-occurrence networks in SP samples. (D) Hub co-occurrence networks in BALF samples. Hub species were identified based on degree, betweenness, and closeness centrality. Nodes represent microbial species, with their size proportional to degree centrality, their color indicating the taxonomic domain, and their labels showing the species names. Edges indicate significant associations, where red edges represent positive correlations and blue edges represent negative correlations, with edge thickness scaled by correlation strength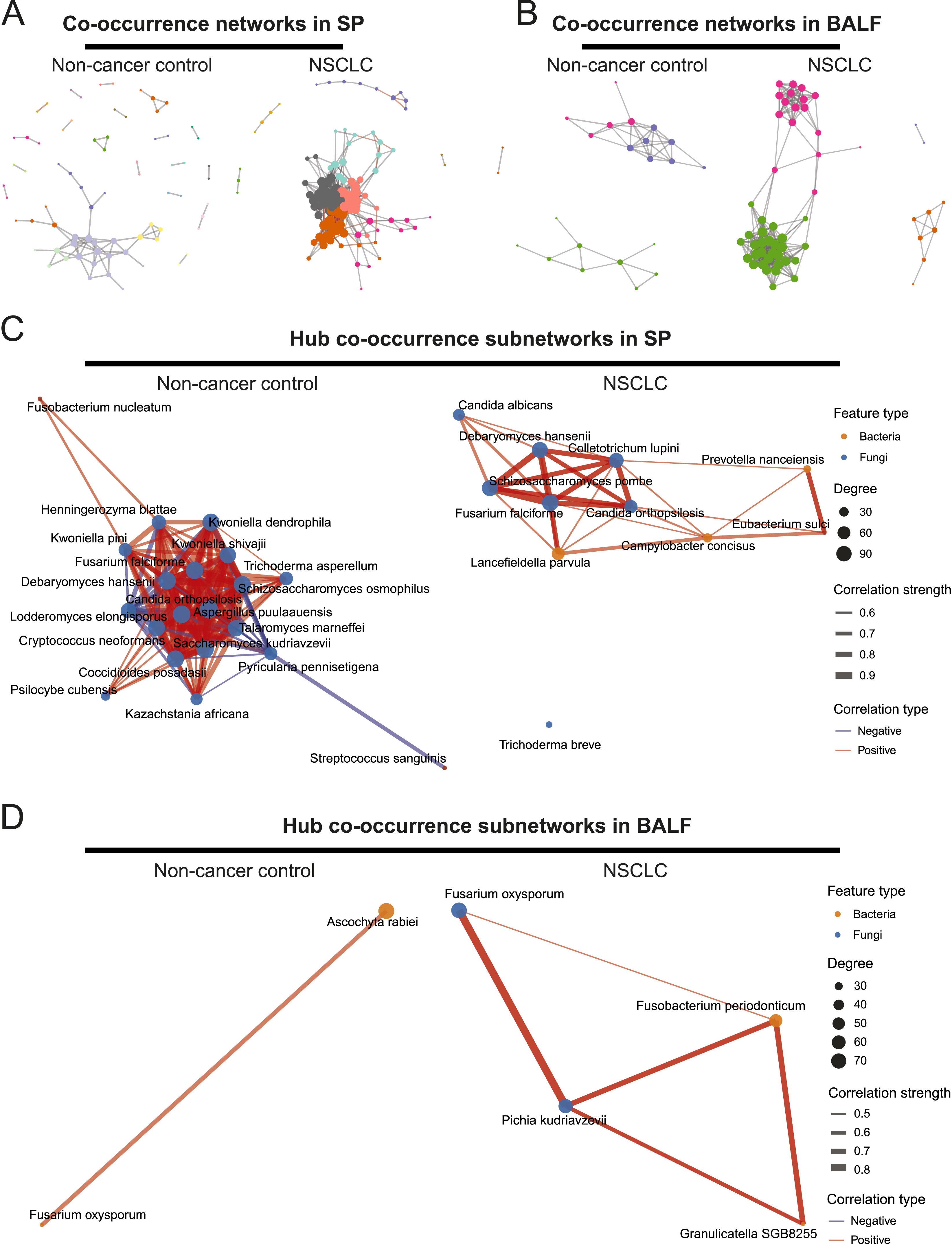
Compared to non-cancer control, NSCLC networks were larger and more complex in both SP and BALF samples. Specifically, in SP samples, microbial networks included 72 nodes and 86 edges in non-cancer control, and expanded to 114 nodes and 736 edges in NSCLC. The network diameter decreased from 8 in non-cancer control to 6 in NSCLC, suggesting a more compact network structure. The average path length increased from 1.80 to 2.40, indicating that a greater number of intermediate species are required for transmission associations between species. The clustering coefficient was similar between the two groups, suggesting a comparable level of network modularity. In BALF samples, microbial networks expanded from 23 nodes and 44 edges in non-NSCLC to 55 nodes and 351 edges in NSCLC. The network diameter increased from 4 to 5, while the average path length decreased from 3.20 to 2.46, indicating a more densely connected network structure in NSCLC. The clustering coefficient remained similar between groups (Figure 6A B, Supplemental Table 4).
To further identify key microbial taxa driving these network structures, we extracted hub species based on the centrality measures (degree, betweenness, and closeness; threshold = mean + 2 SD) from each co-occurrence network. The number of hub species identified in SP samples was higher than that in BALF samples in both NSCLC and non-cancer control groups. Notably, no overlapping hub species were observed between the two sample types (Figure 6C D, Supplemental Table 4).
We further compared the changes in hub species between the NSCLC group and non-cancer control group. In SP samples, 21 hub species were identified in the non-cancer control group. Among them, Aspergillus puulaauensis (degree = 61) showed the highest connectivity, while Trichoderma asperellum (betweenness = 456.20) and Kazachstania africana (betweenness = 405.42) exhibited the highest betweenness centrality, indicating their strong topological influence on network connectivity. In contrast, 12 hub species were detected in NSCLC, with Fusarium falciforme (degree = 119), Schizosaccharomyces pombe (degree = 112), and Debaryomyces hansenii (degree = 111) showing the highest degrees, while Prevotella nanceiensis (betweenness = 341.19), Lancefieldella parvula (betweenness = 300.79), and Candida orthopsilosis (betweenness = 289.72) acted as key bridges linking distinct network modules (Figure 6C and Supplemental Table 4).
In BALF samples, two hub species were identified in the non-cancer control network based on degree centrality, namely Ascochyta rabiei (degree = 14) and Fusarium oxysporum (degree = 17). The NSCLC network contained four hub species identified according to betweenness centrality, with Fusobacterium periodonticum (betweenness = 117.67) and Fusarium oxysporum (betweenness = 111.58) showing the strongest central roles. Although no hub taxa were identified in NSCLC based on degree centrality, the overall degree values of taxa were markedly higher compared with those in the non-cancer control group. No hub species were identified based on closeness centrality in any group (Figure 6D and Supplemental Table 4).
Functional Profiling of NSCLC-Associated Microbiota
We further performed functional profiling of microbial. KEGG pathway enrichment revealed spatially distinct microbial functional alterations. In SP samples, NSCLC-associated microbiota exhibited enrichment in a limited number of pathways. Compared with non-cancer control, several biosynthetic pathways were relatively enriched in NSCLC, including biosynthetic pathways for branched-chain amino acids (BCAAs: valine, leucine, and isoleucine), pantothenate and CoA, lysine biosynthesis, and ribosome components (Figure 7A and Supplemental Table 5). KEGG pathway enrichment analysis of NSCLC-associated microbiota. (A) KEGG pathway enrichment in SP samples of NSCLC and non-cancer control groups. (B) KEGG pathway enrichment in BALF samples of NSCLC and non-cancer control groups. Bars indicate the number of genes associated with each KEGG pathway
In BALF samples, a broader range of pathways showed higher relative abundance in NSCLC, whereas only the PPAR signaling pathway was enriched in non-cancer control. The pathways enriched in NSCLC were predominantly related to metabolic processes, including pyrimidine and nucleotide metabolism, biosynthesis of cofactors, biosynthesis of amino acids, carbon metabolism, the TCA cycle, and pyruvate metabolism (Figure 7B and Supplemental Table 5).
Discussion
In this study, we systematically compared the metagenomic features in SP and BALF samples from non-cancer controls and NSCLC patients. SP and BALF samples exhibited remarkably different microbial features. Moreover, we observed marked differences between NSCLC and non-cancer controls in terms of species composition, cross-kingdom co-occurrence networks, and functional profiles.
Previous studies have reported that lung cancer patients often harbor respiratory microbiota enriched with oral commensals. For example, Tsay et al reported that BALF samples from NSCLC patients were frequently enriched for oral taxa such as Veillonella parvula, and this alteration in the airway microbiota was associated with poorer prognosis. 17 Consistent with these findings, both SP and BALF samples in our cohort were dominated by genera commonly found in the oral cavity, including Neisseria, Streptococcus and Prevotella. Zheng et al similarly observed that bronchoscopy and lung tissue samples often share oral bacteria. 24 Importantly, by microbial compositions analysis, we found that the bacterial species Haemophilus influenzae and Streptococcus mitis, together with the fungal species Candida albicans, were relatively more abundant in the SP samples of NSCLC group. Previous studies have suggested that Streptococcus mitis and Candida albicans may contribute to tumorigenesis through modulation of mucosal immunity or metabolic alterations.25-29 In addition, Haemophilus influenzae and Candida albicans have been reported to participate in respiratory tract infections and immune dysregulation.30-32 Pindling et al reported that elevated Lactobacillales and other Firmicutes were associate with increased lung cancer risk, consistent with the Streptococcus enrichments in SP samples of NSCLC patients in our study. 33 Liu et al performed 16S rRNA sequencing on protected specimen brush samples and similarly observed an enrichment of Streptococcus in NSCLC patients, suggesting a potential pro-carcinogenic role for this genus. 34
Regarding community diversity, in the non-cancer control group, both bacterial and fungal communities in SP samples exhibited higher alpha diversities compared with BALF samples. However, in NSCLC patients, no significant differences in alpha diversities for either bacteria or fungi were observed between the two sample types. For bacteria, NSCLC samples showed higher alpha diversities than non-cancer control in BALF, whereas the opposite trend was observed in SP. For fungi, alpha diversities showed no significant differences between NSCLC and non-cancer control groups in either SP or BALF samples. Previous studies have reported that that lower alpha diversity in oral microbiota correlates with greater lung cancer risk. 33 Likewise, Ma et al found lower intratumoral microbiome diversity in NSCLC patients who later relapsed. 10 In our study, non-cancer controls exhibited higher alpha diversity in SP than BALF (reflecting the richer upper-airway microbiota), but NSCLC samples showed no significant difference between SP and BALF samples, which may be associated with the enrichment of certain microorganisms from the upper respiratory tract into the lower respiratory tract.
For beta diversity, significant differences were observed between the two sample types. Compared with the non-cancer control group, bacterial communities exhibited notable NSCLC-associated shifts, whereas fungal communities showed no significant differences. Both Tsay et al and our analyses show that SP and BALF communities cluster separately, 17 and NSCLC status significantly shifts the bacterial communities. By contrast, we detected no significant cancer-associated changes in fungal beta diversity, consistent with Zhao et al.’s finding of a relatively resilient NSCLC mycobiome. 19 Therefore, the results of both alpha and beta diversity analyses suggest that the bacterial community structure is altered in NSCLC patients, whereas the fungal community appears to remain relatively stable.
Using LEfSe and Random Forest, the species-level differential analyses found that NSCLC was associated with distinct microbial alterations in both sample types. In the SP samples, bacterial species including Actinomyces sp. ICM47, Porphyromonas endodontalis, and Porphyromonas gingivalis, as well as fungal species Puccinia striiformis and Colletotrichum destructivum, were relatively reduced in NSCLC patients. In the BALF samples, bacterial species Alloprevotella sp. Lung230 and GGB1144_SGB1469 were relatively reduced, whereas Porphyromonas SGB2015 and the fungal species Psilocybe cubensis were relatively enriched in NSCLC. No common key taxa associated with NSCLC were identified across both sample types.
Cross-kingdom network analysis indicates that NSCLC is accompanied by a pronounced reorganization of microbial interaction networks, characterized by increased node degree consistently observed in both SP and BALF samples. The elevated connectivity suggests intensified microbial interactions and greater ecological complexity within the cancer-associated microenvironment. Notably, a recent investigation focusing on the lung mycobiome (fungal community) in NSCLC patients also reported significant network alterations. 19 In that study, Zhao et al observed that NSCLC patients harbored higher fungal diversity and a more complex co-occurrence network compared to non-cancer controls. This suggests that the tumor environment broadly disrupts the normal microbial ecology, consistent with our observation of greater cross-kingdom network complexity in NSCLC. One striking aspect of the NSCLC-associated networks was the shift in hub taxa. In SP samples, fungal organism C. albicans was a hub in NSCLC. C. albicans is an opportunistic yeast commonly overgrowing in immunocompromised settings and chronic lung conditions. Its new hub status in NSCLC SP may indicate cancer-related immune dysregulation. Notably, C. albicans has demonstrated oncogenic potential in other contexts, possibly through chronic inflammation, production of carcinogenic metabolites, or immune suppression.35,36 Although a direct causative role of Candida in lung cancer is not established, its presence as a network hub raises the possibility that it may contribute to a tumor-promoting microenvironment.
Functional profiling suggests that microbial functional patterns in NSCLC may exhibit spatial heterogeneity between the upper and lower respiratory tracts. In SP samples, NSCLC-associated microbiota showed enrichment of a limited number of biosynthetic pathways, including branched-chain amino acids (BCAAs) biosynthesis, lysine metabolism, and pantothenate and CoA biosynthesis. BCAAs serve as essential substrates for protein synthesis and cell proliferation and can activate mTORC1 signaling, potentially promoting tumor growth.37-39 Moreover, lysine metabolism, through acetylation and methylation modifications, plays a central role in epigenetic regulation, the dysregulation of which is a hallmark of cancer.40,41 Concurrently, 2-oxocarboxylic acid metabolism, pantothenate and CoA biosynthesis are critical to metabolic reprogramming in cancer cells. In contrast, the microbiota in BALF samples of NSCLC patients displayed enrichment of pyrimidine and nucleotide metabolism, amino acid biosynthesis, carbon metabolism, the TCA cycle, and pyruvate metabolism, suggesting that local microbes may contribute to tumor growth by providing metabolic intermediates.
This study has several limitations. Firstly, the sample size was relatively small, with 23 SP samples (from 14 NSCLC patients and 9 non-cancer controls) and 37 BALF samples (from 21 NSCLC patients and 16 non-cancer controls). Such modest group sizes limit the statistical power and may reduce the generalizability of the findings. In addition, no formal sample size calculation was performed, and the study should therefore be considered exploratory. Secondly, potential confounding factors, including age, gender, and smoking status, were not adjusted for due to the limited sample size. The non-cancer control group was hospital-based and included individuals with various pulmonary conditions as well as a small number of healthy participants, which may not be representative of the general population. Notably, imbalances existed in certain subgroups, which may introduce bias and affect the interpretation of microbiome differences. Thirdly, due to the lack of follow up data, causal relationships between microbiome alterations and NSCLC development or progression cannot be established, and longitudinal clinical outcomes were not assessed. Future multi-center studies with larger cohorts, longitudinal follow-up, and mechanistic investigations are warranted to validate these findings and further elucidate the role of the airway microbiome in lung cancer.
Conclusion
In this study, by integrating metagenomic sequencing of SP and BALF samples, our study delineates distinct interconnected microbial communities along the upper-lower airway axis in NSCLC. The study indicates that in NSCLC, the dominant bacterial composition differs markedly between SP and BALF samples, whereas fungal dominant taxa remain largely consistent across the two sample types. Compared to non-cancer controls, alpha and beta diversity analyses reveal altered bacterial but stable fungal community structure in both SP and BALF samples of NSCLC patients. In NSCLC, BALF shows enrichment of Porphyromonas SGB2015 and Psilocybe cubensis, while SP shows depletion of several bacterial and fungal species. Cross-kingdom microbial networks indicate that NSCLC is accompanied by a pronounced reorganization of microbial interaction with increased node degree in both SP and BALF samples. Functional analyses show distinct patterns in NSCLC: biosynthetic pathways enriched in SP and metabolic pathways enriched in BALF. These findings suggest that airway dysbiosis and cross-kingdom ecological interactions may contribute to the pathogenesis of NSCLC.
Supplemental Material
Supplemental Material - Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC
Supplemental Material for Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC by Lianxin Deng, Xinyu Gao, Chang Guo, Xing Hu, Jian Qi, Jialiang Wang, Xiang Huang, Yiyong Zhang, Zongtao Hu, Hongzhi Wang and Bo Hong in Cancer Control.
Supplemental Material
Supplemental Material - Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC
Supplemental Material for Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC by Lianxin Deng, Xinyu Gao, Chang Guo, Xing Hu, Jian Qi, Jialiang Wang, Xiang Huang, Yiyong Zhang, Zongtao Hu, Hongzhi Wang and Bo Hong in Cancer Control.
Supplemental Material
Supplemental Material - Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC
Supplemental Material for Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC by Lianxin Deng, Xinyu Gao, Chang Guo, Xing Hu, Jian Qi, Jialiang Wang, Xiang Huang, Yiyong Zhang, Zongtao Hu, Hongzhi Wang and Bo Hong in Cancer Control.
Supplemental Material
Supplemental Material - Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC
Supplemental Material for Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC by Lianxin Deng, Xinyu Gao, Chang Guo, Xing Hu, Jian Qi, Jialiang Wang, Xiang Huang, Yiyong Zhang, Zongtao Hu, Hongzhi Wang and Bo Hong in Cancer Control.
Supplemental Material
Supplemental Material - Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC
Supplemental Material for Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC by Lianxin Deng, Xinyu Gao, Chang Guo, Xing Hu, Jian Qi, Jialiang Wang, Xiang Huang, Yiyong Zhang, Zongtao Hu, Hongzhi Wang and Bo Hong in Cancer Control.
Supplemental Material
Supplemental Material - Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC
Supplemental Material for Structural and Functional Alterations of Microbiome in Upper and Lower Respiratory Tract in Patients With NSCLC by Lianxin Deng, Xinyu Gao, Chang Guo, Xing Hu, Jian Qi, Jialiang Wang, Xiang Huang, Yiyong Zhang, Zongtao Hu, Hongzhi Wang and Bo Hong in Cancer Control.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Number: 81872438), the Collaborative Innovation Program of Hefei Science Center, CAS (Grant Number: 2022HSC-CIP015), and the Program of Clinical Medical Translational Research in Anhui Province (Grant Number: 202304295107020092).
Ethical Considerations
The design of this study was approved by the Ethics Committee of the Hefei Cancer Hospital, Chinese Academy of Sciences, on December 15, 2023 (Approval No. SL-PJ2023-102). All procedures performed in this experiment undertaken were in accordance with the ethical standards of the Helsinki Declaration (1975), as revised in 2024. All participants signed an informed consent form.
Author Contributions
BH, HW, CG and LD designed the study. LD, CG and XG performed the Genomic DNA extraction and library construction. LD, CG, XH (Xing Hu), XH (Xiang Huang), YZ and ZH collected the samples. LD, CG, JQ and JW performed the bioinformatics analysis. LD and XG wrote the first draft of the manuscript. BH and HW revised the manuscript. All authors reviewed the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant Number: 81872438), the Collaborative Innovation Program of Hefei Science Center, CAS (Grant Number: 2022HSC-CIP015), and the Program of Clinical Medical Translational Research in Anhui Province (Grant Number: 202304295107020092).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Supplemental Material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.