
Abstract
Introduction
Symptoms of ADHD may significantly affect the quality of life of children and adults diagnosed with the disorder, as well as their families (Agarwal, Goldenberg, Perry, & IsHak, 2012; Cappe, Bolduc, Rouge, Saiag, & Delorme, 2017; Klassen, Miller, & Fine, 2004; Peasgood et al., 2016). In addition to facing more academic and occupational difficulties, it was recently shown that children with ADHD experience greater morbidity than those without the disorder (Dalsgaard, Leckman, Mortensen, Nielsen, & Simonsen, 2015). Pharmacological treatment can significantly reduce the impact of ADHD symptoms and improve the lives of these individuals (Agarwal et al., 2012; Banaschewski et al., 2014; Dalsgaard et al., 2015). Stimulant medications, such as methylphenidate and amphetamine, are recognized as first-line pharmacologic treatments for ADHD (Rabito-Alcon & Correas-Lauffer, 2014). Due to the variability in responses to different therapies, lifestyle requirements, and the degree of impairment of patients with ADHD, treatment should be individualized to achieve optimal outcomes (reviewed in Hodgkins, Dittmann, Sorooshian, & Banaschewski, 2013; Lopez & Leroux, 2013). Individualization includes finding a stimulant formulation and dose that provides symptom reduction with minimal side-effect burden and is convenient for the patient (Wolraich et al., 2011).
An extended-release amphetamine oral suspension (AMP XR-OS) has recently been developed. The AMP XR-OS uses a cation-exchange resin that is loaded with amphetamine to form a drug–resin complex. A portion of the drug-resin complex is coated with an acid-resistant polymer tailored to produce a delayed-release profile, and another portion is left uncoated to provide immediate release of the amphetamine. AMP XR-OS contains
We present the results of a Phase 1 study assessing the rate of absorption and oral bioavailability of AMP XR-OS compared with an equivalent oral dose of a commercially available extended-release mixed amphetamine salts reference product, Adderall XR® (MAS ER; Shire US, Inc., Wayne, PA), in healthy adult volunteers under fasted conditions. The studied drug doses were 15 mL of AMP XR-OS and a 30 mg MAS ER capsule, both containing 18.8 mg of amphetamine base with the
Method
Study Design
This single-dose, open-label, randomized, two-period, two-treatment crossover study was approved by an institutional review board (IntegReview, Austin, TX). Written informed consent was obtained from all participants before performing study procedures. The study was conducted in accordance with the clinical research guidelines established by the U.S. Investigational New Drug regulations (CFR section 21 parts 50, 56, and 312), the International Council for Harmonisation, and the latest guidelines of the Declaration of Helsinki.
Eligible participants were males or females greater than or equal to 18 years of age with baseline body mass index between 18 and 30 kg/m2 (inclusive), minimum weight of 50 kg, heart rate of 40 to 100 bpm, systolic blood pressure (BP) of 90 to 145 mm Hg, and diastolic BP of 50 to 95 mm Hg. Female participants were included if they were not pregnant, were postmenopausal for at least 2 years prior to dosing, or agreed to use an acceptable form of birth control during and up to 14 days after completion of the study.
Participants with any history or presence of suicidal ideation or behavior as assessed by the Columbia-Suicide Severity Rating Scale at baseline or check-in were excluded. Participants were also excluded if, prior to the first dose of study medication, they used over-the-counter medications or supplements within 7 days or prescription medications (except for hormonal contraceptive or replacement therapy) within 14 days, smoked or used tobacco products within 60 days, were on an abnormal diet within the preceding 4 weeks, participated in a clinical trial as a randomized participant in the past 30 days, or donated blood or plasma within the past 30 days. Other exclusion criteria were allergy or adverse reaction to amphetamines and related drugs or any medication with a sulfonamide component; history of intolerance to amphetamine or amphetamine-like medications; history or presence of substance abuse, urine test positive for cotinine or drugs of abuse, clinically significant physical or psychiatric disease (including depression, mania, bipolar disease, and psychosis), tics/Tourette’s syndrome, marked anxiety, tension, or agitation; currently receiving any psychotropic medication; and previously treated or positive for hepatitis B, hepatitis C, or HIV.
During the study, participants could not take supplements or over-the-counter or prescription medication, with the exception of approved birth control or hormonal therapy. The consumption of foods or beverages containing alcohol, grapefruit, or caffeine/xanthine and the use of tobacco products were prohibited. Participants were not permitted to donate blood or plasma, or to engage in strenuous exercise.
Forty-two participants were enrolled in the study. Treatments were administered in two periods separated by a washout of at least 7 days and the sequence of treatments was randomized. In one period, participants received a single 15 mL oral dose of AMP XR-OS and, in the other period, a single 30 mg capsule of MAS ER (both containing 18.8 mg of amphetamine base). Each dose was administered following an overnight fast of at least 10 hr and fasting continued for 4 hr postdose. Except for water consumed with administration of MAS ER, no water was consumed for 1 hr prior to and 1 hr post administration of either treatment. Participants were to remain seated for the first 4 hr after dosing unless otherwise directed by clinical staff.
Sample Collection and Pharmacokinetic (PK) Analysis
Blood samples were drawn at 0, 1.0, 2.0, 3.0, 4.0, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 9.0, 10.0, 12.0, 16.0, 24.0, 36.0, 48.0, and 60.0 hr after treatment administration. Human plasma was analyzed for
PK analysis was performed using Phoenix® WinNonlin® software (Version 6.3, Certara L.P. [Pharsight], St. Louis, MO). The following PK parameters were determined: maximum drug concentration in plasma (Cmax), time to maximum concentration (Tmax), observed elimination half-life (T1/2), area under the plasma concentration-time curve to the time of the last quantifiable concentration (AUClast), area under the curve from 0 to 5 hr postdose (AUC0-5), area under the curve from 5 hr postdose to time of the last quantifiable concentration (AUC5-last), and area under the curve from 0 hr extrapolated to infinity (AUCinf).
Statistical Analysis
Statistical analysis was performed using SAS® (Version 9.3, SAS Institute Inc.) software. Natural log-transformed Cmax, AUC0-5, AUC5-last, and AUCinf were compared across treatments using ANOVA tests with a linear mixed-effects model. The geometric mean ratio (AMP XR-OS/MAS ER) and the 90% confidence interval (CI) for the ratio were calculated for each of these parameters. Bioequivalence criteria are met if the 90% CI for Cmax, AUC0-5, AUC5-last, and AUCinf of
Safety Analysis
Safety was evaluated throughout the study period and at the end of the study by the site investigator. Safety assessment included electrocardiograms, vital signs, physical and oral cavity examinations, clinical laboratory evaluations, assessment of suicidal ideation or behavior, and reported or observed adverse events (AEs). BP, pulse rate, respiration rate, and temperature were measured at screening, 0, 36, and 60 hr postdose in each study period. BP and pulse rate were also measured at 4, 8, 12, 24, and 48 hr after each dose of study drug.
Results
A total of 42 participants completed both study periods. Baseline characteristics of the study population are summarized in Table 1. A single dose of AMP XR-OS or MAS ER administered under fasted conditions produced maximum plasma concentrations of
Participant Demographics.

Note. BMI = body mass index.
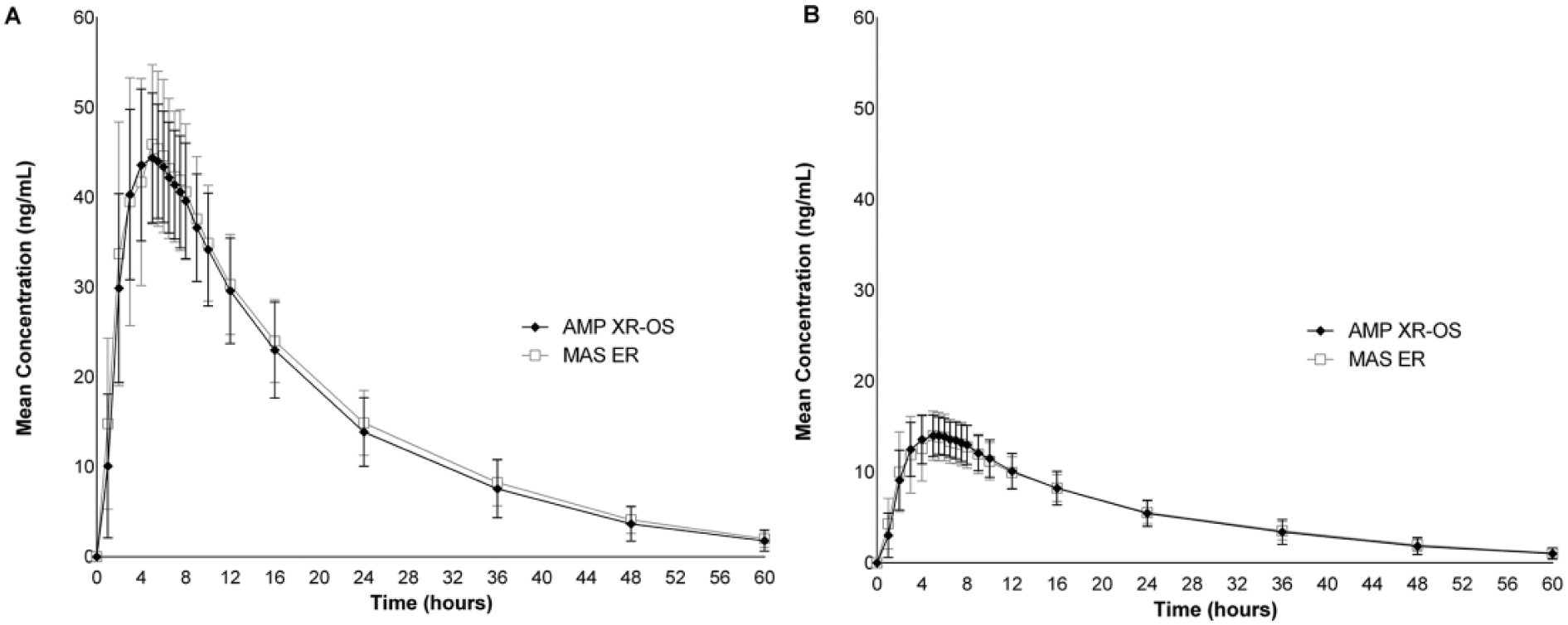
Mean concentration over time of (A)
Pharmacokinetic Parameters of

Note. AMP XR-OS = amphetamine extended-release oral suspension; MAS ER = extended-release mixed amphetamine salts; Tmax = time to peak plasma level; Cmax = peak plasma level; AUC0-5 = area under the concentration-time curve from time 0 to 5 hr; AUC5-last = area under the concentration-time curve from 5 hr to the time of the last quantifiable concentration; AUClast = area under the concentration-time curve from time 0 to the time of the last quantifiable concentration; AUCinf = area under the concentration-time curve from time 0 extrapolated to infinity; T1/2 = elimination half-life.
All parameters reported as M ± SD, except for Tmax, which is reported as median (range).
Statistical Analysis of Amphetamine Enantiomer Exposure After Administration of AMP XR-OS or MAS ER.

Note. AMP XR-OS = amphetamine extended-release oral suspension; MAS ER = extended-release mixed amphetamine salts; CI = confidence intervals; Cmax = peak plasma level; AUC0-5 = area under the concentration-time curve from time 0 to 5 hr; AUC5-last = area under the concentration-time curve from 5 hr to the time of the last quantifiable concentration; AUCinf = area under the concentration-time curve from time 0 extrapolated to infinity.
Data are presented as least squares geometric means.
Ratio % = geometric mean AMP XR-OS / geometric mean MAS ER × 100.
90% CI within the 80% to 125% acceptance limits indicate bioequivalence.
A total of 12 AEs were reported by eight participants. Six participants (14.3%) reported an AE following AMP XR-OS, and five participants (11.9%) reported an AE following MAS ER (Table 4). All AEs were classified as mild by the investigator, and all except one (muscle spasms unrelated to study treatment, treated with a heat pack) resolved without intervention. The most commonly reported AEs were nausea and decreased appetite (Table 4). No AEs were related to abnormal laboratory evaluations, and there were no clinically significant abnormalities in vital signs or physical exams.
Treatment-Emergent AEs.
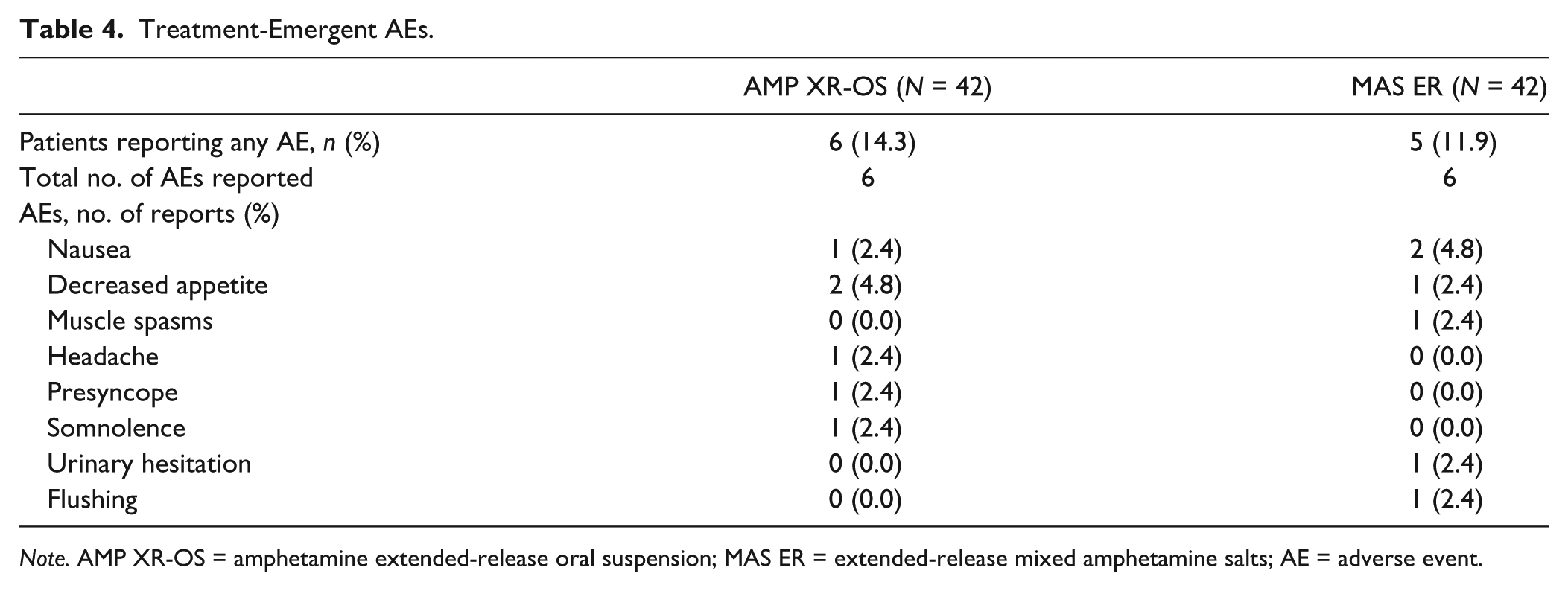
Note. AMP XR-OS = amphetamine extended-release oral suspension; MAS ER = extended-release mixed amphetamine salts; AE = adverse event.
Discussion
We compared the PK parameters of amphetamine after administration of a single dose of AMP XR-OS with that of the reference amphetamine formulation, MAS ER, both delivering an equivalent amount (18.8 mg) of amphetamine base in healthy, fasted adults. The amphetamine concentration-time profiles for the liquid suspension, AMP XR-OS, and capsule, MAS ER were highly similar with a comparable rate and extent of absorption. Statistical analysis of peak plasma concentrations and the exposure parameters AUC0–5, AUC5-last, and AUCinf established that AMP XR-OS was bioequivalent to the reference product as the 90% CIs were well within the 80% to 125% range. Both study drugs were generally well-tolerated with an AE profile similar to other extended-release amphetamine formulations (Shire US Inc., 2015b; Stark, Engelking, McMahen, & Sikes, 2016a; Tris Pharma Inc., 2015). The PK profile of AMP XR-OS is consistent with a once-daily dosing regimen.
Five extended-release amphetamine products are currently approved for ADHD treatment: an extended-release orally disintegrating tablet (AMP XR-ODT), three capsule formulations, and one oral suspension. AMP XR-OS uses the same extended-release delivery technology as the once-daily AMP XR-ODT. The technology platform provides similar amphetamine concentration profiles for both formulations when administered in a dose equivalent to 30 mg MAS ER (18.8 mg of amphetamine base) to healthy, fasted adults; the Cmax and Tmax of AMP XR-OS, shown in Table 2, are comparable with those of AMP XR-ODT 18.8 mg (Cmax M ± SD for
For individuals with swallowing difficulties or the need for treatment flexibility, a liquid formulation, such as AMP XR-OS, may provide a useful alternative to solid oral formulations.
Footnotes
Author Contributions
All authors contributed to the writing, provided critical evaluation at all stages of the manuscript, and approved the final article. Dr. Jeffrey Stark performed the PK and statistical analysis. Medical writing assistance was provided by Nicole Seneca, PhD, of AlphaBioCom, LLC, and funded by Neos Therapeutics, Inc.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dorothy Engelking is an employee of Neos Therapeutics, Inc. with stock options. Russ McMahen is an employee of Neos Therapeutics, Inc. with stock options. Dr. Carolyn Sikes is an employee of Neos Therapeutics, Inc. with stock options, and stock in Pfizer. Dr. Jeffrey Stark is an employee of Worldwide Clinical Trials.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Neos Therapeutics, Inc.