
Abstract
The incidence of antibiotic resistance is critically jeopardizing the public health of the new millennium, affecting both clinical and therapeutic outcomes. Typhoid perforation in low- and middle-income countries (LMICs) is mostly caused by Salmonella typhi. The development of antibacterial therapies is drawn to the enzyme 3-dehydroquinate dehydratase type 1 (DHQ1), which is an important enzyme of the shikimate pathway in bacteria. Its role in the synthesis of chorismite, a natural precursor of the route leading to the synthesis of aromatic amino acids, validates it as an important therapeutic target. Therefore, the current work is based on the investigation of bioactive compounds that inhibit the crucial DHQ1 enzyme. Structure-based virtual screening (SBVS) and docking studies were done using AutoDock Vina tools to retrieve potential hit molecules with high binding affinity against DHQ1 from several ligand repositories. From a total of 551 compounds, 206 were filtered out for physicochemical properties, the Lipinski rule and subjected to molecular docking. A total of 10 ligands were selected with the highest binding affinities ranging from −9.8 to −9.1 (kcal/mol), ie, Cabotegravir, Imatinib, Prulifloxacin, Limonin, Silibinin, Atovaquone, Betamethasone Valerate, GSK1324726A, Isavuconazole, and Raltegravir. In addition, ADMET analysis, bioactivity prediction, and pharmacokinetic property prediction were carried out as prediction of activity spectra for substance (PASS) analysis to further prioritize the best inhibitory compound. Molecular dynamic simulations were used to verify the stability of the protein “DHQ1” docked with 3 hit ligands Cabotegravir, Limonin, and Silibinin by calculating root mean square fluctuation (RMSF), root mean square deviation (RMSD), radius of gyration (Rg), H-bond interaction, and molecular mechanics/generalized Born surface area (MM/GBSA) scores. Based on molecular dynamic (MD) simulations, it is reported that Cabotegravir and Limonin showed the optimal binding features with bacterial DHQ1 enzyme and can be deemed as a repurposed drug candidate as compared to Silibinin, which did not show favorable interactions with DHQ1 and needs to be explored further against typhoidal Salmonella.
Introduction
There is a global uproar regarding typhoid fever, particularly among medical professionals and authorities, while 93% of typhoid fever epidemics occur in Asia. 1 According to data from 2018, Pakistan has the high-rise rate of typhoid fever among South Asian regions, reported 493.5 cases per 100 000 patients. 2 One of the significant public health problem facing the globe today is extensively drug-resistant Salmonella typhi (XDR S typhi), which poses a danger to future global economic growth.3,4 Typhoid fever is a systemic febrile, fecal-oral transmissible sickness caused by Salmonella enterica subspecies enterica serovar Typhi (S typhi) which is a gram-negative bacterium.5-7 Humans are the only species which serve as a host and reservoir for typhoid pathogens via the intake of infected food and water.8,9 The hallmark symptoms of typhoid fever include step ladder-high-grade fever, gastrointestinal distress, myalgias, nausea, abnormal bowel movements, and bradycardia.10,11
Therapeutics that target bacterial virulence and essential metabolic enzymes create an in vivo schema similar to vaccination with an attenuated live bacterial strain, in which bacterial cells are killed by the host immune system.12,13 The shikimate pathway is important in this context since it is only found in microorganisms, plants, fungi, and apicomplexan parasites; humans do not possess it, which makes it an important target for drug research. 14 In certain species, the shikimate pathway uses around 20% of the carbon obtained from the breakdown of carbohydrates; as a result, it plays a significant role in their metabolism. 15 The pathway synthesizes all the essential aromatic compounds and metabolites through chorismate. 16 In contrast, animals derive their aromatic compounds from their diet. 17 The scope of creating broad-spectrum antibiotics that take advantage of harmful bacteria’s shikimate pathway is also confirmed by genomic research.18,19
The shikimate pathway in bacteria contains 7 enzymes, which catalyze the conversion of erythrose 4-phosphate and phosphoenol pyruvate to chorismite. 20 Chorismate is a precursor of aromatic amino acids (tryptophan, phenylalanine, tyrosine), the production of folic acid, iron-scavenging siderophores, p-aminobenzoic acid, vitamin K, naphthoquinones, and ubiquinone. 21 The Dehydroquinase enzyme (DHQ, 3-dehydroquinate dehydratase) is the third enzyme in the biosynthetic pathway of aromatic amino acids. 22 The DHQase catalyzes the reversible dehydration reaction of 3-dehydroquinic acid to 3-dehydroshikimic acid. There are 2 classes of DHQase with distinct structure known as type 1 (DHQ1) and type II (DHQII), that catalyze the elimination of H2O by completely different mechanisms. The DHQ1 is an aldolase (dehydratase) enzyme found in several critically pathogenic bacterial cells such as Escherichia coli, S typhi, and Staphylococcus aureus. Type 1 DHQase (DHQ1) is a distinct, monofunctional enzyme harboring a dimeric 28 kDa subunit. 23 It is assumed that DHQ1 may act as a virulence factor in vivo after deletion of the aroD gene (which synthesizes DHQ1 in S. typhi and Shigella flexneri), which has been shown to act as a satisfactory live oral vaccines22,24 used the strategy of repurposing existing drugs against DHFR-S typhi (metabolic enzyme) suggesting Duvelisib, Amenamevir, Lifitegrast, and Nilotinib as bactericidal agents through molecular and docking studies. 24
Several studies have identified that DHQ1 (EC 4.2.10) as a promising antivirulence target for drug design due to the following reasons: (1) DHQ1 behaves as a virulence factor in vivo after deletion of the aroD gene, that codes this enzyme, and has proved to afford satisfactory live oral vaccines;25-28 (2) the aroD mutation renders bacteria auxotrophic, defects cell wall and outer membrane integrity, and inhibits the biofilm formation; 29 (3) DHQ1 enzyme is overexpressed in small colony variants which facilitates the recurrent infections;30,31 (4) DHQ1 is found in several pathogenic bacteria, ie, Clostridium difficile, E coli, and S aureus; and (5) no mammalian counterpart of DHQ1 has been identified, which makes it an attractive target for developing bactericidal agents. 32
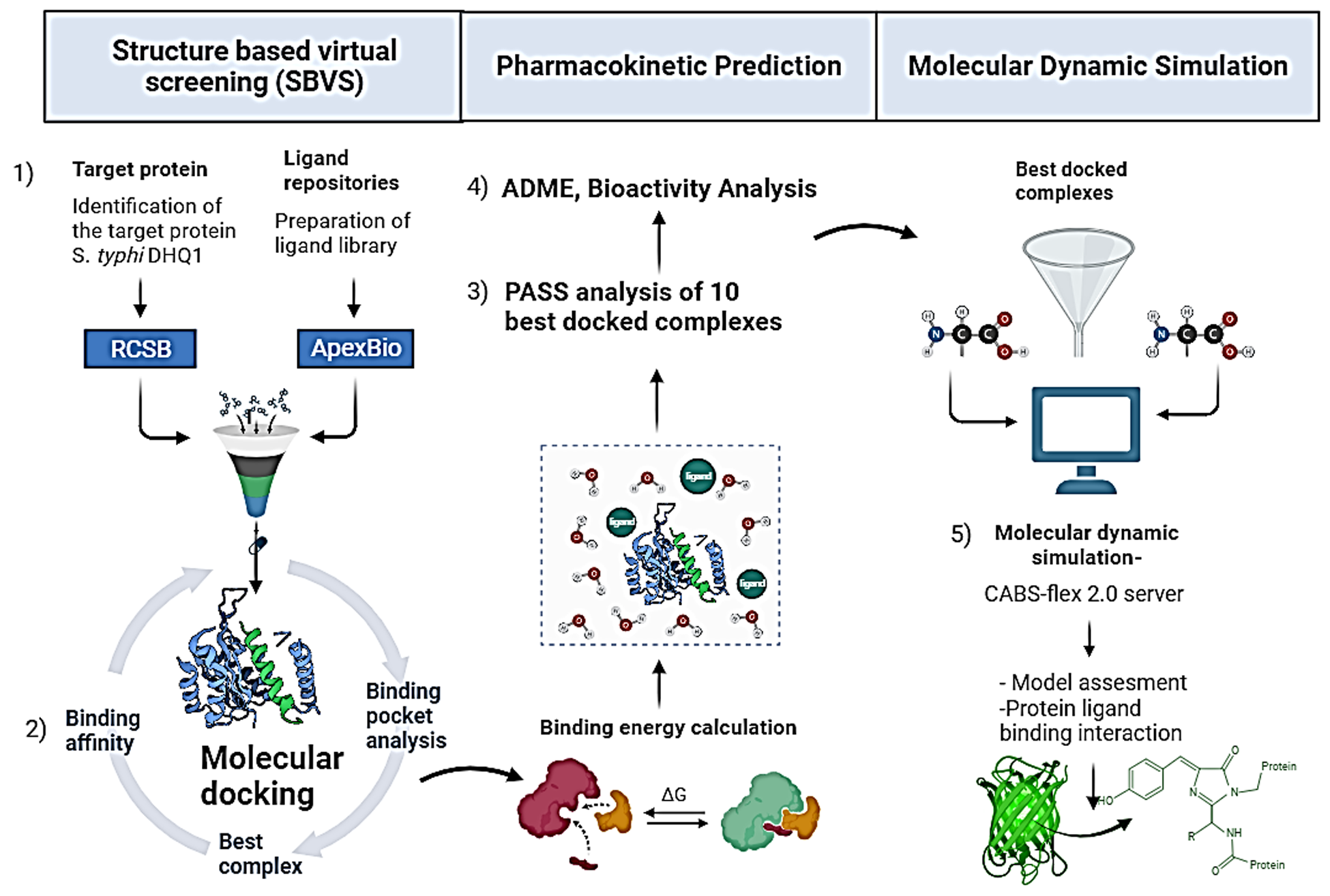
Virtual screening (VS) is an approach that is complementary to high-throughput screening (HTS). The SBVS is a computational technique employed in the early stage of drug discovery process in order to search for the bioactive compound library for novel molecules against a specific drug target. The SBVS is preferred in computational biology and pharmaceutical research because its success rate is 400 times higher, less costly, and less laborious than the traditional in vitro screening techniques. 33 The SBVS provides prediction of the likely binding conformations as well as an easy method of ranking the docked compounds based on the scoring functions. The SBVS strategies follow a particular sequence of processes such as (1) molecular target selection and preparation, (2) compound database/library generation, (3) ligand preparation, (4) molecular docking, and (5) analysis of results/interpretation. 34 This virtual technique is deemed as the in silico equivalent of in vitro methods like HTS, which filters the potential ligand on the basis of its pharmacokinetics, molecular weight (MW), number of hydrogen donors and acceptors, etc. By exploiting the 3D structure of a protein target, this approach is specifically screened libraries of natural compounds, focusing on a plant source to develop inhibitor compounds against a target protein/receptor. 35
Molecular docking is a computational method that tells the placement and position of ligands in the binding site of the target protein. It provides an estimation of favorable binding modes and affinities of ligands with the protein. 36 AutoDock Vina v1.5.6 estimates the binding energy and fixes the binding poses by employing the Lamarckian genetic algorithm. 37 Docking was completed to achieve all the possible conformation and orientation for ligands at the binding site and binding scores. It estimates the binding energy of the protein-ligand complex by considering the van der Waals forces, electrostatic, and hydrogen bond interactions. 38
Current investigations propose that DHQ1 enzyme behaves as a virulent element in vivo, is conserved, and does not have any counterpart in human cells. A groundbreaking strategy amid the prevalence of antibiotic resistance era would be a stratagem that disables the bacterial capacity to cause infection. 39 Considering this information, the development of a new arsenal of selective small molecular potent inhibitors against such essential enzymes is the key concept to curb the problem of typhoid fever. In the given research work, various in silico approaches are utilized to explore the antagonists of DHQ1 protein, binding hypothesis and molecular basis of interactions with the protein DHQ1, and validation through MD simulations.
Materials and Methods
Protein preparation and docking grid generation
The 3D structure of DHQ1 from S typhi (PDB ID:4CNN) was obtained from the RCSB protein data bank (PDB) (https://www.rcsb.org/) as given in Table 1 and Figure 1. The crystal structure 4CNN exhibited a high resolution of 1.00 Å obtained by the X-ray diffraction method. It has 2 chains A and B; both chains are mirror images of each other. 17 The protein DHQ1 structure was downloaded in PDB format. The preparation of protein structure was performed using AutoDock Vina software v1.5.6. The attached ligands C6-H8-O7, Cl, and Na ions were removed from the original structure of DHQ1 using the BIOVIA Discovery Studio v2021. 40 Then, DHQ1 standardization involved Gasteiger charges, addition of polar hydrogens, Kollman charges, removal of heteroatoms, and removal of crystallized water molecules. The transformation of file format from the PDB format to the PDBQT format was done using AutoDock Tools v1.5.6. 41
X-ray crystal structure of protein 4CNN with high resolution retrieved from protein data bank.


The 3-dimensional structure of bacterial protein DHQ1 after removing irrelevant ions, ligands, and water molecules.
Ligand library preparation
The ligand library was retrieved by searching for natural and chemical bioactive compounds from the ApexBio database. From a total of 551 Food and Drug Administration (FDA)-approved bioactive anti infectious compounds, 206 were selected from screening library based on MW and the Lipinski rule. Compounds with MW less than 500 Da were selected for docking because less MW shows the better bioactivity (Absorption, metabolism) in the body. 43 Filtered out ligands (small molecules) were passed through docking experiments. Out of 206, the top 10 docked ligands, with the highest binding affinity, were further evaluated through prediction of activity spectra for substance (PASS), 44 bioactivity score (BAS) prediction, pharmacokinetic prediction, and toxicity potential prediction. 45 After passing through these predictions’ filters, 3 hit ligands were selected for MD simulations out of 10 ligands. 46 Filtered ligands were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The SDF formats were then converted into PDB formats using BIOVIA Discovery Studio Visualizer 2021. 47 The ligand PDB files were prepared in AutoDock Vina v1.5.6 for the docking procedure in PDBQT format. 48
Docking study
The docking of ligands was carried out using AutoDock Vina v1.5.6.49,50 The AutoDock Vina software automatically calculates the dimensions of the grid boxes and provides accurate binding energy scores. 36 AutoDock Vina v1.5.6 estimates the binding energy and fixes the binding poses by employing the Lamarckian genetic algorithm. 38 Docking was completed to achieve all the possible conformations and orientations for ligands at the binding site and binding scores. In the present study, the center of grid box was fit to (11.463, 0.454, 29.442) Å as central coordinates (center x, center y, center z). Meanwhile, the size of the grid box was defined as 40 Å in each dimension; the energy range was set to 4, while the exhaustiveness was set to 8 as shown in Table 2. A bash script file was encoded to execute the docking process. A script file contained the Vina program and certain parameters essential for the Vina program with input and output parameters. 51 The software can automatically implement the Vina program and execute the docking procedure. 52 For all ligand molecules, a configuration file containing grid box parameters was generated and saved in .txt format. The docking protocol followed was consistent with the previous studies.51,53,54 For 3D visualization of the protein-ligand interactions, the PDB structure of protein-ligand complexes was saved in the PDB format using PyMol v3.1.6.1. The molecular interaction profiles of the best-fit model from the docking experiments were visualized in 2D and 3D in Discovery Studio v2021 for evaluation. 47
Grid sizes and grid centers used in AutoDock Vina 1.5.6 for docking study.

Prediction of activity spectra for substance analysis
As an alternate to clinical experiments, PASS analysis estimates the biological activity of compound including physiochemical properties, pharmacological features, bioactivity, drug-likeness, and drug side effects based on the structure-activity relation of the ligand molecule. In this study, PASS analysis was performed for the ligands. Molinspiration v2023 mentioned in the Lipinski rule section was used for the prediction of physiological properties. while pharmacological and toxicological properties were assessed through SWISS-ADME software and ProTox-3.0 which are discussed in sections “Pharmacokinetic property prediction” and “Toxicity potential assessment.”55,56
Lipinski rule
The Lipinski rule estimates the drug-likeness and physiochemical properties of ligand molecules based on their pharmacokinetic features such as absorption, metabolism, distribution, and excretion. 57 The rule is critical for drug development and repurposing while using cheminformatics approaches. The drug-likeness of 10 ligand molecules was assessed by the Lipinski rule. 58 The criteria for a drug to be a potential candidate were the following: (1) lipophilicity (clogP), (2) MW, (3) number of hydrogen donor sites, (4) hydrogen bond acceptor sites, (5) Topological Polar Surface Area (TPSA), and (6) the number of rotatable bonds. 47 This was achieved through an online software Molinspiration v2023.08 (https://www.molinspiration.com/). Molinspiration calculated the parameters such lipophilicity (clogP ⩽ 5), MW (⩽500), number of hydrogen donors (NOHNH ⩽ 5), hydrogen bond acceptor sites (NON ⩽ 10), topological polar surface area (TPSA ⩽ 140 Å2), and the number of rotatable bonds (⩽ 10). To maintain the ligands bioavailability, an orally active drug should not possess more than 1 Lipinski violation. 59
Bioactivity score prediction
The BAS predicts the potential of a compound to be a potent drug candidate. 56 Molinspiration v2023.08 (https://www.molinspiration.com/) was employed again to provide comprehensive data regarding BAS of the prioritized ligands with respect to human receptors like G protein-coupled receptors (GPCRs), kinases, proteases, ion channels, enzymes, and nuclear receptors. According to rule, the higher the BAS, the more the possibility of the compound being active. If the BAS value is more than 0.0, then the compound is active; if the value lies between −5.0 and 0.0, then the ligand is deemed moderately active, and if the BAS displays values less than −5.0, then it is reasoned to be idle/ inactive. 60 The BAS scores table is given in the Supplementary Data.
Pharmacokinetic property prediction
The ADME parameters (absorption, distribution, metabolism, excretion descriptors) were systematically predicted by SWISS-ADME software (http://www.swissadme.ch/). 61 The pharmacokinetic properties like blood-brain barrier (BBB), drug distribution, gastrointestinal (GI) absorption, and metabolism (which rely on P-glycoprotein [P-gp] substrate), cytochrome P450s such as CYP1A2, CYPC19, CYP2D6, and CYP3A4 inhibitor, as well as lipophilicity for the plasma membrane absorption for the 10 best compounds 62 have been given in Table 5. According to Lipinski et al, the compounds should adhere to the minimum range of 3 physiochemical attributes out of 5 to be considered as a potential drug candidate. 63
Toxicity potential assessment
Toxicity assessment is a crucial step in the drug designing and development process. Computational toxicity estimation does not only save time but also reduces the step of animal testing. Here, in the study, we employed ProTox-3.0 web server (https://tox.charite.de/protox3/), a freely online available tool that takes 2D chemical compound input to generate the possible toxicity profile of the compound. It incorporates machine learning and molecular similarity to predict numerous toxicity endpoints such as immunotoxicity, tumorigenicity, carcinogenicity, mutagenicity, hepatotoxicity, and reproductive effects. 64 Compounds with high risks of undesired properties like carcinogenicity or poor intestinal absorption are displayed in red, while a green color shows a drug’s positive role. The input files in the ProTox-3.0 web server were submitted via PubChem name or canonical SMILES.
Molecular dynamic simulations and molecular mechanics/generalized Born surface area calculation
Protein-ligand interactions were observed using molecular dynamic (MD) simulations. The docked solution of 3 ligands; Cabotegravir, Limonin, and Silibinin, with protein DHQ1 showed high binding energies in docking and passed through toxicity filters were subjected to validation. The MD simulations were used to estimate the potential energies among atoms present in a closed system with periodic boundary conditions. 65 For this purpose, parameters for ligands were calculated by AnteChamber Python Parser Interface 2022.3.11 (ACPYPE) 66 using General Amber Force Field 2 (GAFF2). 67 For protein, parameters were created using AMBER99SB-ILDN. 68 A triclinic box was established around protein-ligand complex with a distance of at least 1 nm from all sides of protein-ligand complexes. The triclinic box was filled with Transferable Intermolecular Interaction Potential 3 Points (TIP3P) water models and Cl− ions for creating a neutral aqueous virtual environment. 64 The whole system was subjected to energy minimization for 100 ps or till the system achieved <10.0 kJ/mol energy compared to initial configuration. The simulations were performed using the Number of particles, Pressure, and Temperature (NPT) ensemble, with 300 K temperature and 1 atm pressure. 69 The protein-ligand complexes were further subordinated to 100 ps of Number of particles, Volume, and Temperature (NVT) equilibrium parameters before 100 ns of MD simulation runs. All of these steps were performed through a Linux-based tool, GROMACS v2024.
During simulations, the average change in protein structure was observed by calculating the root mean square deviation (RMSD) of protein backbone with reference to the structure in the first frame of simulation. While changes in the position of ligands (Cabotegravir, Limonin and Silibinin) within the binding site of protein (DHQ1) were observed by calculating RMSD of ligand heavy atoms with respect to the protein backbone as a function of time. The contribution of each protein residue in RMSD was observed through root mean square fluctuation (RMSF). The radius of gyration (Rg) was calculated to find out the size and compactness of DHQ1 during simulations.
For assessment of binding forces involved in keeping the ligands within the binding site, hydrogen bonds between DHQ1 and Cabotegravir, Limonin and Silibinin were calculated based upon the mutual distance and angle between donor-acceptor atoms through the hydrogen bond plugin of Visual Molecular Dynamic (VMD v1.9.4a53). 70 Moreover, MM/GBSA calculations were performed to estimate the binding free energies of protein-ligand complexes throughout the MD simulation for 100 ns. 64 Calculation of the binding energies among protein-ligand complexes was performed by gmx_MMGBSA v1.6.4 module. 71
Results
Virtual screening of ligands
A chemical database “Apexbio” was explored in order to narrow down the pool of bioactive compounds for the purpose of starting the structure-based virtual screening (SBVS) procedure as shown in Figure 2. Each ligand (small molecule) was an FDA-approved drug that had been obtained from Apexbio database. Virtual screening was applied to 551 natural and chemical substances in total. Following the installation of the filter, which states that a compound’s Lipinski rule violation should not be more than 1 and MW must be less than 500 kDa to allow for improved absorption, about 206 compounds were filtered out from a total of 551. Hence, the filtered-out 206 small compounds were considered for molecular docking experiments. Docked ligands were further reduced to 10 on the basis of the highest calculated binding affinity. These 10 filtered out ligands were checked for PASS (BAS prediction, pharmacokinetic prediction, and toxicity potential prediction). 46 After passing through these prediction filters, 3 hit ligands were selected for MD simulations from 10 docked ligands. The results of each of the VS steps are described in the following sections.

Virtual screening of 551 ligand compounds (natural, chemical) were filtered out based on the molecular mass, the Lipinski rule violation, molecular docking, and pharmacokinetic properties into potentially 3 hit compounds.
Lipinski rule
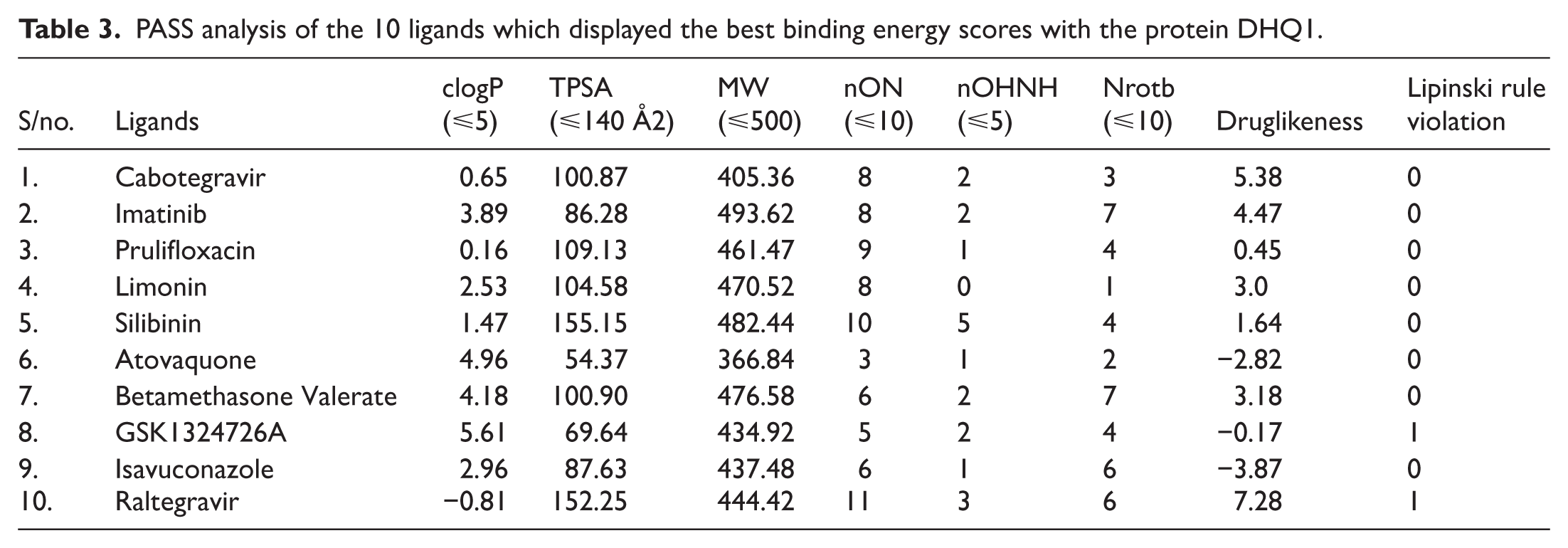
The physicochemical properties of 10 ligands who had the highest binding scores were assessed by PASS analysis given in Table 3 by employing the Lipinski rule. GSK1324726A and Raltegravir exhibited 1 Lipinski violation. GSK1324726A displayed a clogP value of 5.6, greater than the threshold of 5, and Raltegravir analysis revealed 11 nON (hydrogen bond acceptors), as opposed to the standard value of 10. The rest of the compounds displayed no violation. As the ideal lead compound must not exhibit any violation, 8 of 10 ligand compounds had successfully met the criteria. Cabotegravir, Imatinib, Prulifloxacin, Limonin, Silibinin, Betamethasone, Valerate, and Raltegravir showed drug-likeness scores greater than 1 (5.38, 4.47, 0.45, 3.0, 1.64, 3.18, and 7.28, respectively); it means they can be potential drug candidates, while the remaining 3 ligands—Atovaquone, GSK1324726A, and Isavuconazole—displayed negative values of −2.82, −0.17, and −3.87, respectively, thus showing poor drug-likeness (Table 3).
PASS analysis of the 10 ligands which displayed the best binding energy scores with the protein DHQ1.
Bioactivity Score prediction
With the exception of Raltegravir, which was only moderately active, all of the ligands were biologically active as GPCR ligands, according to the bioactivity prediction. All ligands exhibited ion channel modulators with moderate biological activity, except atovaquone, which exhibited strong activity. Nuclear receptor ligands and kinase inhibitors with modest activity included Cabotegravir, Isavuconazole, Raltegravir, and Limonin. It was shown that Imatinib, Prulifloxacin, and Silibinin were effective kinase inhibitors. Because the result was below the threshold value, ie, 0. Betamethasone and Valerate were inactive (−0.74). As protease and enzyme inhibitors, it was shown that Cabotegravir, Limonin, Silibinin, Atovaquone, Betamethasone Valerate, and Raltegravir were physiologically active, while the remaining ligands had only modest action. The highest values for both protease inhibitor (ie, 0.78) and enzyme inhibitor (ie, 0.77) were displayed by Betamethasone and Valerate. It was predicted that Atovaquone functions as a nuclear receptor ligand, kinase inhibitor, ion channel modulator, GPCR ligand, and inhibitor of proteases. The detail of the BAS prediction of understudy ligands is given in Figure 3.

Heatmap showing bioactivity score of 10 selected ligands, generated by Molinspiration an online tool.
Pharmacokinetic property prediction
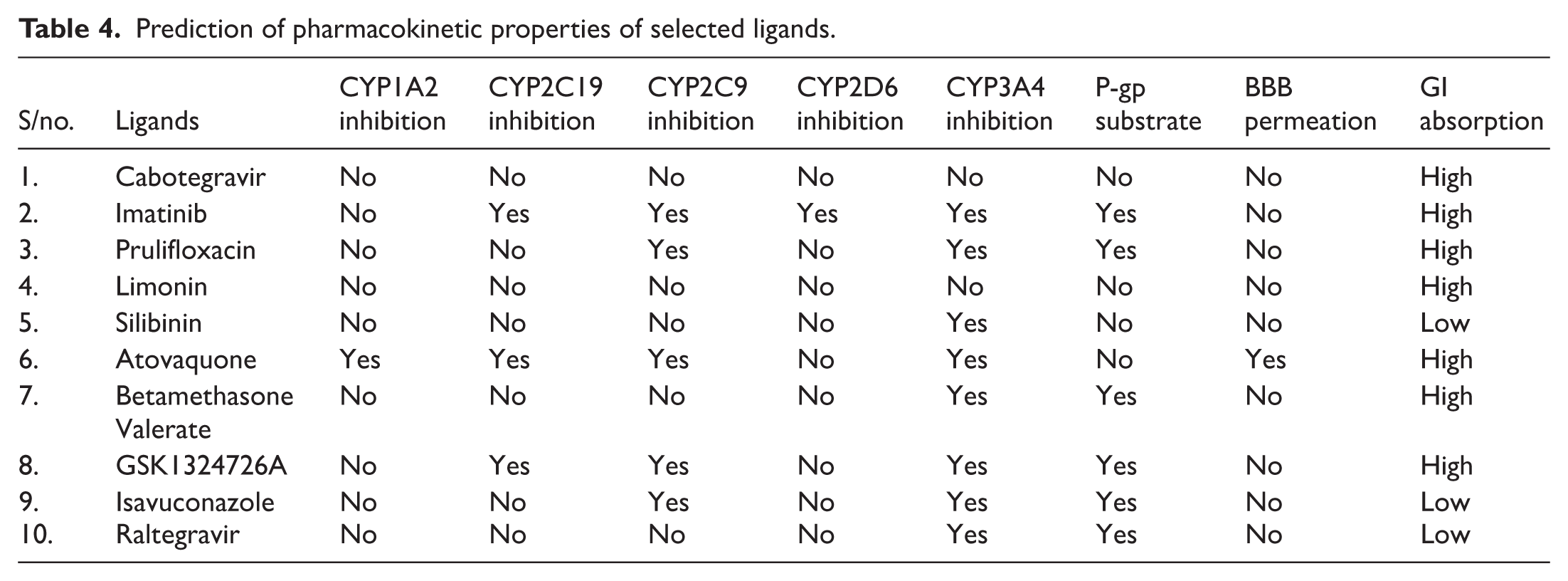
The ADME analysis of the selected ligands suggested that only Atovaquone was able to cross the BBB. Imatinib, Prulifloxacin, Betamethasone Valerate, GSK1324726A, Isavuconazole, and Raltegravir were found to be positive as P-gp substrates, while the remaining ligands were negative. The results suggest that being non-P-gp substrates, the pharmacokinetic effectiveness of the ligands will be enhanced since they will stay in the cells for a longer period. All the ligands were reported to show high gastrointestinal absorption except for Silibinin, Isavuconazole, and Raltegravir. Based on incessant plasma concentration and elevated bioavailability, the selected ligands were anticipated to inhibit the 5 classes of cytochromes P450, ie, CYP2C9, CYP1A2, CYP2D6, CYP2C19, and CYP3A4. Cabotegravir showed no inhibition to any of the cytochrome P450. Whereas Imatinib showed inhibition against all the P450 classes except for CYP1A2. Limonin, Prulifloxacin, and Silibinin did not show inhibition to CYP1A2, CYP2C19, and CYP2D6. Atovaquone showed inhibition to all the P450 classes except for the CYP2D6. All the ligands were revealed to inhibit CYP3A4 except Limonin and Cabotegravir. Table 4 demonstrates the detailed pharmacokinetic properties of the ligands.
Prediction of pharmacokinetic properties of selected ligands.
The BOILED-Egg analysis represented by Figure 4 showed an intuitive, reproducible, and statistically reliable method to determine the human intestinal absorption (HIA) and BBB permeation as a graphical model for drug discovery and development. The assessment of the relation between the lipophilicity and the polarity of the selected ligands (1-10) was carried out using BOILED-Egg method as a predictive model. Only molecule 6 (Atovaquone) lies in the yolk region (yellow) which denotes a high possibility of brain penetration. While the rest of the molecules 1, 2, 3, 4, 7, and 9 lie in the egg white region, which denotes a high chance of passive absorption by the GI tract. Moreover, the blue dots in Boiled-Egg indicate that they are actively effluxed by P-gp (PGP+), whereas red dots predict that they are non-substrate of P-gp (PGP−). The “BOILED Egg” analysis reveals that most of the compounds have high HIA and do not cross BBB, which is a good attribute of a drug to be a potential candidate.

Graph of the dependence of lipophilicity on the polarity of the studied molecules 1 to 10, determined by the BOILED-egg method.
Toxicity potential assessment
The toxicity risk predictions of selected ligands have been shown in Figure 5 revealed that Cabotegravir, Limonin, Silibinin, and Betamethasone valerate showed no toxicity toward mutagenicity, hepatotoxicity, cytotoxicity, cardiotoxicity, carcinogenicity, reproductive toxicity, and irritant. Prulifloxacin showed cardiotoxicity, Atovaquone displayed reproductive toxicity, and GSK1324726A showed mutagenic effect, whereas Imatinib, Isavuconazole, and Raltegravir showed hepatotoxic effects.

Heatmap showing the toxicity risk assessment of 10 studied ligands, predicted by ProTox-3.0 an online web server. Red color shows the toxic nature of ligand while green color shows the ligand is non-toxic against a range of parameters such as Mutagenicity, Hepatotoxicity, cytotoxicity, Cardiotoxicity, Carcinogenicity, Irritant and Reproductive effects. Based on toxicity assessment results, the ligands which showed no toxicity were selected for further analysis (MD simulations).
Molecular docking experiments
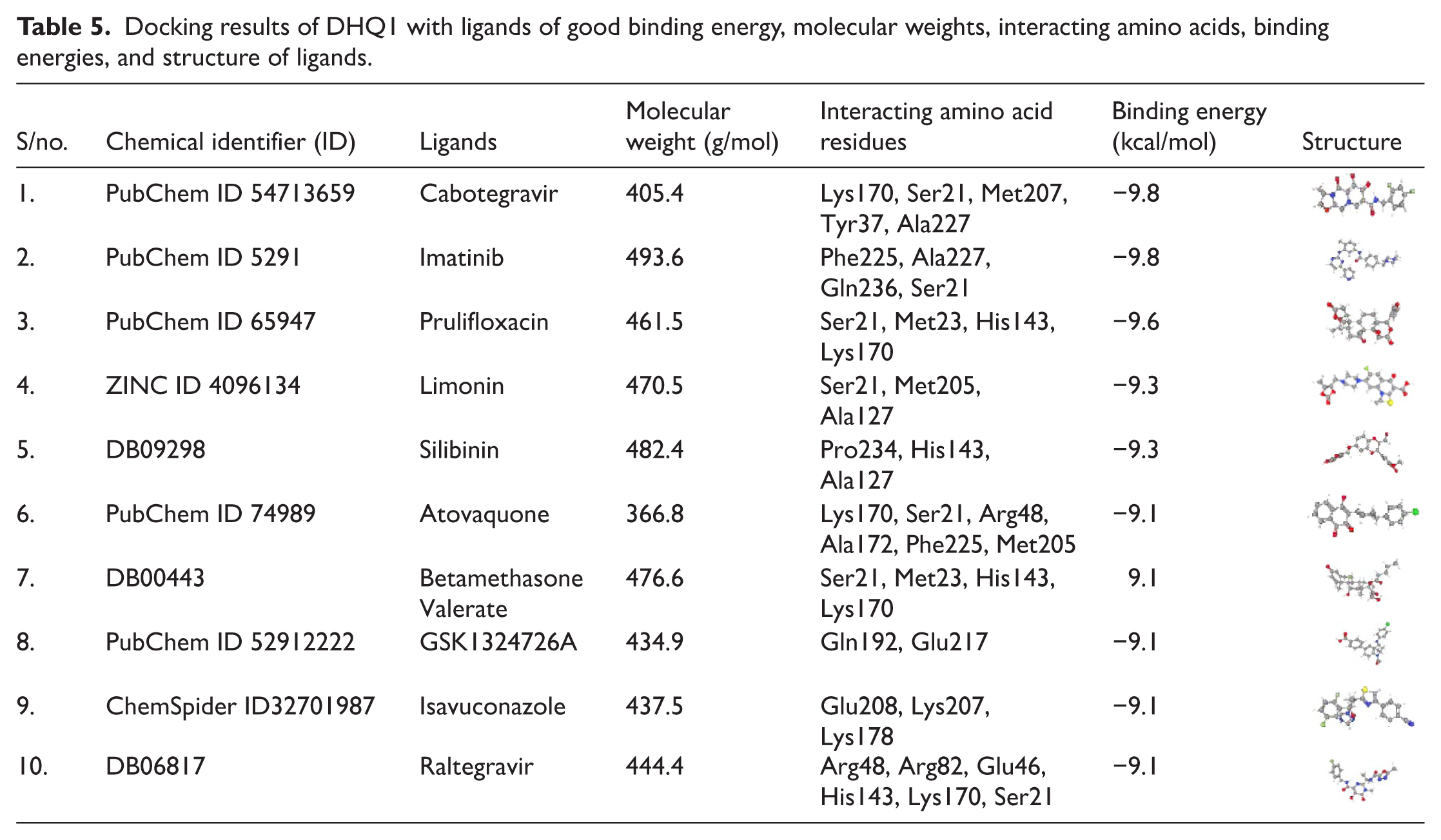
The binding affinity scores of the 206 ligands against the DHQ1 bacterial protein were estimated using AutoDock Vina v1.5.6. The ligands that showed strong binding (high binding affinity) were further selected for MD simulations. Varying interactions between the protein DHQ1 and ligands (Cabotegravir, Imatinib, Prulifloxacin, Limonin, Silibinin, Atovaquone, Betamethasone valerate, GSK1324726A, Isavuconazole, Raltegravir) and binding site residues revealed varying binding energies as represented in Table 5. These findings showed that binding energies of DHQ1 ranging from −9.8 kcal mol−1 to −9.1 kcal mol−1. However, from 10 potential ligands, Cabotegravir and Imatinib exhibited the highest binding affinity with the binding energies of −9.8 kcal mol−1, followed by Prulifloxacin with −9.6 kcal mol−1 and Limonin and Silibinin showed −9.3 kcal mol−1, while rest of the ligands had −9.1 kcal mol−1 binding value.
Docking results of DHQ1 with ligands of good binding energy, molecular weights, interacting amino acids, binding energies, and structure of ligands.
The 2D interactions and 3D visualization between target protein DHQ1 and 3 hit ligands (Cabotegravir, Limonin, Silibinin) have been given in Figure 6. Cabotegravir showed interacting residues Lys170, Arg48, and Ser21 forming hydrogen bonds, π-cation interaction with Met205, Met207, Tyr37, and Val228 showed Pi-Alkyl interactions. Limonin revealed hydrogen bonding with Ala238 and pi-alkyl interactions with His143 and Met205. No interacting residues forming hydrogen bonds in Silibinin were observed, while it showed pi-alkyl interactions with Val128, Val138, His143, and Ile113.

Docking poses of bacterial protein DHQ1 with selected 3 ligands (A) Cabotegravir, (B) Limonin, and (C) Silibinin represented as 3D models alongside their respective 2D structures.
Molecular dynamics simulations
The simulation was carried out for 100 ns using GROMACS software. Three ligands were selected for MD simulation. The overall simulation results of selected docked complexes were represented in the form of RMSD, RMSF, Rg, hydrogen bond interactions, and MM/GBSA analysis. The RMSD was used to measure the average variations between the sampled structures during simulation with the initial structure as reference. Figure 7 shows the RMSD values for 3 selected ligands. The RMSD of Cabotegravir with respect to protein DHQ1 showed minor deviations between 10 and 70 ns and remained almost stable from 1 to 1.25 nm up to 70 ns (Figure 7A). However, at 70 to 95ns, the ligand showed deviations from 1 to 1.6 nm, but again at 100 ns, the ligand gained stability. The second ligand, Limonin, revealed very little fluctuations from 0.4 to 0.6 nm and remained stable throughout the simulation runtime of 95 ns (Figure 7B). It showed fluctuations from 0.4 to 0.7 nm at 95 ns to onward. The third ligand Silibinin displayed the highest fluctuation wavelength of 16 nm (Figure 7C). The ligand had minor stability from 2 to 7.5 nm between 20 and 55 ns runtime.

RMSD Plots of MD simulations of ligands with backbone of protein (DHQ1) (A) DHQ1-Cabotegravir, (B) DHQ1-Limonin, and (C) DHQ1-Silibinin.
Figure 8 shows the RMSF plots of Cabotegravir, Limonin, and Silibinin complexed with DHQ1. The RMSF plot of Cabotegravir-bound protein revealed higher fluctuations of 2.7 nm at residue number 225 compared to other residues (Figure 8A). Limonin showed flexibility at residues 125 and 225 (Figure 8B). For Silibinin, a sharp peak was reported at residue 225 (Figure 8C). The RMSF describes the average movement of residues (flexibility) throughout the simulation, assigning the flexibility contribution of each amino acid in protein structure.

RMSF plots of MD simulation of DHQ1 complexed with ligands (A) Cabotegravir, (B) Limonin, and (C) Silibinin.
Based on the Rg plots with respect to time, the Rg variations during the 100 ns MD simulation were probably caused by the binding site of the target and ligand. The Rg indicates protein stability during simulation. A higher value of Rg shows greater dispersion of atoms and a bigger molecule. Figure 9 shows the Rg plots of the ligands complexed with DHQ1. The binding of Cabotegravir and Limonin produced conformational shifts in protein, thus causing change in protein radius. While, in the case of Silibinin, no change in the protein radius was observed due to very little interactions between DHQ1 and Silibinin.

Radius of gyration (Rg) plot of ligands Cabotegravir, Limonin, and Silibinin complexed with DHQ1 protein.
The hydrogen bond plot (Figure 10) showed a maximum of 3 hydrogen bonds between ligand and protein in DHQ1-Cabotegravir as well as DHQ1-Limonin complexes. However, more consistent hydrogen bonds were observed among DHQ1 and Cabotegravir compared to DHQ1 and Limonin. Asp167 and Asp197 contributes significantly to hydrogen bonding between DHQ1 and Cabotegravir complex as represented by the occupancy graph (Asp 67 and Asp197 interact with the Cabotegravir 49.5% and 38.5% of the simulation time, respectively) (Figure 10A). On the contrary, DHQ1-Limonin complex showed a higher number of residues forming H-bonds with Limonin from 40 to 100 ns of simulation runtime with 11.5% of Ala131 and 17.5% of Asn101H-bond occupancy (Figure 10B). The third ligand “Silibinin” showed no hydrogen bonding, suggesting that the ligand might bind to a different position on this protein or it may not be stable at binding positions.

Representing the hydrogen bonds between protein and ligands. (A) The number of hydrogen bonds between Complex 1 (DHQ1 and Cabotegravir) along with percentage H_bond occupancy and (B) the number of hydrogen bonds between complex 2 (DHQ1 and Limonin) along with percentage H_bond occupancy.
The MM/GBSA calculations were carried out to observe the binding interaction energy of DHQ1 and ligands (Cabotegravir, Limonin, Silibinin) during the MD runtime of 100 ns. The ∆G of protein-ligands interactions were calculated by the following equation:

It can be observed that DHQ1-Cabotegravir complex showed overall −39.67 ± 4.36 kJ/mol. The binding energy value for DHQ1-Limonin complex was calculated to be −31.89 ± 4.74 kJ/mol, while in case of complex 3 (DHQ1-Silibinin), the energy value is −0.0389 ± 1.43 kJ/mol. The high free energy (ie, more negative energy value) represents stronger interactions and complex’s stability. Although, in the present study, the values of ∆G follow the order of stability of complexes provided by the RMSD and RMSF analysis, all the complexes have negative binding free energies of interaction, which further validated the complex stability. The stability of the complexes based on the calculated ∆G is in the order: Cabotegravir (−39.67 kcal/mol) > Limonin (−31.89 kcal/mol) > Silibinin (−0.0389 kcal/mol). The binding energy of Silibinin is close to zero. Representing negligible calculated binding affinity. Suggesting that there is no stable binding between DHQ1 and Silibinin at His132 and Val138 binding sites. However, the 2 ligands—“Cabotegravir and Limonin”—could be the potential drug candidates as validated by MD simulation and MM/GBSA analysis.
Discussion
The current work intended to explore possible antibacterial compounds against S typhi’s highly conserved DHQ1. Tizon and his co-workers employed a pro-drug approach to develop potent inhibitors against DHQase. 22 We employed a VS protocol based on successive drug-likeness 72 and toxicity filters to select drug candidates for S typhi DHQ1 as inhibitors. A total of 551 FDA-approved compounds obtained from ApexBio database capable of binding to DHQ1 were subjected to the Lipinski rule. Based on rule MW < 500, 206 ligand compounds were filtered and subjected to molecular docking. The FDA-approved drugs from 2010 to 2020 had an average MW of 452 ± 116. 43 The 10 best-hit compounds based on the binding affinities were found to be Cabotegravir, Imatinib, Prulifloxacin, Limonin, Silibinin, Atovaquone, Betamethasone Valerate, GSK1324726A, Isavuconazole, and Raltegravir. The docking scores of the ligands against DHQ1 can be collated in the following order: Cabotegravir = Imatinib > Prulifloxacin > Limonin = Silibinin > Atovaquone = BetamethasoneValerate = GSK1324726A = Isavuconazole = Raltegravir. Physicochemical (PASS), Pharmacokinetics, and drugability properties of ligands were used to assess the best drug candidate from 10 selected ligands. Docking analysis indicates that prioritized ligands interact with key residues in the catalytic site of protein including Asp167, Gly165, Thr127, Asp129, Arg198, Lys2, Asp197, His132, Ala131, Asn101, Leu164, and Arg198.
Furthermore, the BAS of ligands was predicted using Molinspiration (https://www.molinspiration.com/). While the remaining ligands showed fair-to-moderate biological activity, Atovaquone was shown to be very active as a cation channel modulator, nuclear receptor ligand, GPCR ligand, and protease inhibitor. Bioactivity numbers show a molecule’s overall potential as a lead chemical. It effectively isolates inactive structures from compounds with promising drug-like qualities. 73 Based on the Lipinski rule, which was computed using Molinspiration, the physicochemical characteristics of the ligands revealed that only GSK1324726A and Raltegravir had 1 Lipinski violation. There were no violations observed in other ligand compounds. All of the ligands met the requirements to be considered excellent lead compounds overall. The pharmacokinetic properties were calculated to evaluate the drug-likeness of compounds. 74 Of 10 ligands, only Atovaquone was shown to pass the BBB, according to research conducted using Swiss-ADME. 61 The selected ligands were explored to hamper the cytochromes P450 classes, namely CYP2C19, CYP2D6, CYP1A2, CYP2C9, and CYP3A4, in order to comprehend the constant plasma concentration and medication toxicity. Cabotegravir with the highest binding affinity to DHQ1 did not show inhibition to any of CYP450 enzyme. This denotes a low risk of toxicity and adverse effects. 75 However, Atovaquone showed inhibition to all the P450 classes except for the CYP2D6, which can lead to drug accumulation and intoxication. All the ligands were able to inhibit CYP3A4 except Limonin and Cabotegravir. All ligands showed high gastrointestinal (GT) absorption except for Silibinin, Isavuconazole, and Raltegravir. Whereas Imatinib, Prulifloxacin, Betamethasone Valerate, GSK1324726A, Isavuconazole, and Raltegravir were found to be P-gp substrates as compared to remaining ligands, which were non P-gp substrates. The results suggest that being non-P-gp substrates, the pharmacokinetic effectiveness of the ligands will be enhanced since they will stay in the cells for a longer period. 76
An analysis done by Rankovic 77 showed the relationship between the lipophilicity and the polarity using the BOILED-Egg method to estimate the penetration through the brain or intestines, as an accurate predictive model. The tested 9 of 10 ligands are within the range of good permeability via BBB and the proper binding to HIA. Only Atovaquone lies in the yolk which denotes high probability of brain penetration. 78 The possible reason could be the inability of the general pharmaceuticals, particularly small molecules, to move across the BBB. While Cabotegravir, Imatinib, Prulifloxacin, Limonin, Silibinin, Betamethasone Valerate, GSK1324726A, Isavuconazole, and Raltegravir show a high probability of passive absorption by the GI tract.
The post-docking analysis was performed on the 3 selected hit compounds from molecular docking using Discovery Studio v2021. 47 Based on the interactions of ligands, the active site residues of the S typhi DHQ1, Asp167, Gly165, Thr127, Asp129, Arg198, Lys2, Asp197, His132, Ala131, Asn101, Leu164, and Arg198 were detected to be involved in hydrogen bond formation. 17 It has been previously reported that a modified epoxide ligand causes modification of S typhi DHQ1 by forming a Schiff base with Asp167 while also highlighting the essentiality of Lys2 in catalysis. 22 In a study, quinic acid-base hydroxylammonium derivatives were designed for covalent moderation of lysine active site residue of DHQ1 through direct nucleophilic reaction of the ε-amino group of lysine with the NH2OH, followed by proton removal of the lysine adduct by the histidine residue. 17 In another work, the substitution of His132 by alanine revealed 106 fold reduction in the catalytic activity while stalling the hydrolytic release of product from the active site. Salt-bridges between its carboxylate group and Arg213 are involved in the attachment of substrate to the active site. 79 Imatinib (−9.8 kcal mol−1) interacted with Phe225, Ala227, Gln236, and Ser21, while Atovaquone with binding energy of −9.1 kcal mol−1 showed interactions with residues, ie, Ser21, Arg48, Lys170, Ala172, Phe225, and Met205. GSK1324726A (−9.1 kcal mol−1) displayed π-cation interactions with Lys207 and Val218 showed π-σ bond, while conventional hydrogen bonds have been reported with Thr127 and Gly165. Analysis of the interaction between S typhi DHQ1 and all of the ligands suggest significant contributions from conventional hydrogen bonding, alkyl interactions, Pi-Cation interactions, π-σ bond, and carbon-hydrogen bonding. 80 Hydrogen interactions are the major determinant of ligand-protein structural stability. The number of hydrogen bonds impacts the docking scores, as it maintains the highest negative weighted value among the other variables. 51
To evaluate the residual component’s fluctuation, RMSF was calculated, which signifies the local residual changes along the protein chains. 24 Structural stability and flexibility of the protein-ligand docked complexes was estimated by GROMACS v2024. The RMSD was calculated to estimate variations of protein-ligand complex system and conformational stability of macromolecules during 100 ns simulation time. 40 The selection of ligands (Cabotegravir, Limonin, Silibinin) for MD simulation was based on their pharmacokinetic properties and BAS. The RMSD plot of the DHQ (positive control) suggests the stable nature of protein. The terminal amino acid residues showed a high RMSF value, ie, the hH loop (residues 227-239). hH loop (also referred to as the flexible covering-substrate loop or simply hH loop) is a structural element in bacterial DHQ1. Typically, loops are less constrained by hydrogen bonding and other structure-stabilizing interactions, which allow flexibility to the protein conformation. The freedom of movement of loop regions causes higher RMSF values. 81
Since the Cabotegravir bound to DHQ1, the maximum RMSF value reached 2.6 nm. The MD simulations validated the structural stability of DHQ1/Cabotegravir docking complex. The fluctuation in the RMSF plot at residue Asp54 seems to be caused by a loop region in the protein (Asp54 to Thr58). As Asp-rich regions tend to form relatively unstructured loops while connecting α-helices and β-sheets. 82 The RMSF plot of DHQ1-Limonin docking complex depicted value of 0.34 nm at residue 125. Analysis confirmed the presence of the glycine residues may provide flexibility to change conformation at the enzyme active site. Gly-loop sequences are highly flexible, which provide flexibility to adjacent residues as well. Formation of loop is also significantly faster around glycine residues compared to any other amino acid. 83 Overall, plot values suggested stability of the interacting amino acid residue of the complex.
Based on RMSF plot, DHQ1-Silibinin docking complex shows flexibility at residue 225 during 100 ns simulation. This analysis suggested the presence of Gly residue which formed loop region, in the absence of H-bonds, conferring flexibility to protein structure. Gly-loop regions confer flexibility to the protein structure, which generates fluctuations at the atomic level. 84
Based on the Rg plot, the Rg variations during the 100 ns MD simulation were possibly observed at the binding site of DHQ1 and ligand. The binding of Cabotegravir and Limonin produced conformational shifts in protein, thus causing change in protein radius. While, in the case of Silibinin, no change was observed due to unstable interactions between DHQ1 and Silibinin. One study showed the lower the Rg, the higher the compactness and better will be stability. 64 The hydrogen bonding at Asp167 and Asp197 contribute significantly to DHQ1-Cabotegravir stability. While DHQ1-Limonin complex also showed stability due to Ala131 and Asn101 forming H-bonds with 11.5% and 17.5% occupancy with ligand, respectively. The third ligand “Silibinin” showed no hydrogen bonding with S typhi DHQ1. Literature supported the antiviral activity of Silibinin, as Silibinin interacted with many residues on the binding site of Mpro protein and supported the potential activity of this molecule in impeding the SARS-CoV-2 viral replicon. 85 No studies reported the activity of Silibinin against DHQ1. The MM/GBSA was used to calculate the binding energies of DHQ1 and ligands (Cabotegravir, Limonin, Silibinin) during the MD runtime of 100 ns. It calculates the amount of free energy released by the interaction of protein-ligand. 64 In current study, it was observed that the stability of the complexes based on comparative analysis of ∆G is in the order of Cabotegravir (−39.67 kcal/mol) > Limonin (−31.89 kcal/mol) > Silibinin (−0.03.89 kcal/mol). The binding energy of Silibinin is close to zero. It represents negligible calculated binding affinity. It suggests that there is no stable binding between DHQ1 and Silibinin at His132 and Val138 binding sites.
The compound Cabotegravir is an FDA-approved antiretroviral drug which causes virlogic suppression in HIV infection. 86 However, the antibacterial effects of Cabotegravir have been reported in E coli and S aureus strains. 87 In the study, the in vitro toxicity and antibacterial activity suggested the safety of the encapsulated Cabotegravir with Gold nanoparticles. However, no study indicated anti Salmonella activity of cabotegravir. 87 The second compound Limonin belongs to tetracyclic triterpenoids, having high biological activity. It is extracted from plants of Rutaceae and Meliaceae and has been used in traditional Chinese medicines. Limonin is a versatile substance showing pharmacological properties such as anticancer, antibacterial, anti-inflammatory, antioxidation, analgesic, and antivirus. 88 A study also reported the inhibition of S aureus infection as well as markers of the toxins after treatment with Limonin. 89 Several studies have reported the bacteriostatic activity of Limonin on Salmonella, Micrococcus luteus, Bacillus thuringiensis, Bacillus cereus, E coli, S aureus, and Shigella species. Moreover, modification at C-7 position in the A ring of the Limonin resulted in a stronger antibacterial activity. 90 The third compound Silibinin is a major bioactive compound of Silymarin, an extract of milk thistle (Silybum marianum L. Gaernt.), which is also a medically important plant belonging to Asteracea family that has been used to treat liver illnesses for thousands of years. 91 Silibinin also used as antiapoptosis, antifibrotic, antioxidative, antidiabetic, as well as anti-inflammatory and neuroprotective. Milk thistle does not exhibit toxicity in animals and is generally regarded as safe (GRAS). 92 However, there are fewer literature on plant extracts that possess efflux pump inhibition effects against multi-drug resistance (MDR) bacteria, eg, curcumin has inhibitory potential against Pseudomonas aeruginosa, plumbagin against E coli, and thymol against Salmonella enteritidis. Extracts of milk thistle seeds have been reported to block MDR efflux pump STY4874 of S typhi. 93
Limitations
Most applications of chemoinformatic approaches have been made in the field of drug design and development. Keeping in view the significance of this field and the worth of chemoinformatic tools, these applications are continuing to thrive. However, there are surely some limitations that need to be researched.
The conformational flexibility of ligands and, peculiarly, of proteins poses problems in drug repurposing or designing. Other important challenge that is usually faced in drug repurposing is “in vitro and in vivo testing.” Literature showed that sometimes, the potential drugs that are validated through computational tools do not show 100% accurate results in vitro and in vivo testing. The third limitation of this study is the specificity and sensitivity. Drugs designed by chemoinformatic approaches lack sensitivity and specificity.
Conclusion
Herein, to treat rising MDR/XDR typhoid, molecular docking and MD simulation studies were carried out on a series of FDA-approved drugs against DHQ1 shikimate pathway enzyme of S typhi. Among 551, molecular docking, the Lipinski rule, and physiochemical properties screened the 3 high-affinity ligands with preferable pharmacokinetic properties. By exploiting the repurposing methods, we used an efficient strategy to reuse existing and de-risk drugs for surging typhoid predicament. The drug potential of Cabotegravir, Limonin, and Silibinin (binding affinity: −9.8, −9.3, and −9.3 kcal/mol, respectively) was confirmed by molecular docking studies, PASS analysis (Lipinski rule, BAS prediction, pharmacokinetic property prediction), and MD simulations. Hence, the detailed analysis suggests that drug molecules, particularly Cabotegravir and Limonin binding properties, can have bactericidal effects and may be used as a potential therapeutic agent against S typhi infection in the future, but the third ligand Silibinin did not show potential binding sites with interacted DHQ1 pocket, so it can be further explored against S typhi.
Supplemental Material
sj-pdf-1-bbi-10.1177_11779322261418889 – Supplemental material for Chemoinformatic Approaches to Identify Bioactive Inhibitors Against Type I Dehydroquinase (DHQ1) Enzyme of Typhoidal Salmonella
Supplemental material, sj-pdf-1-bbi-10.1177_11779322261418889 for Chemoinformatic Approaches to Identify Bioactive Inhibitors Against Type I Dehydroquinase (DHQ1) Enzyme of Typhoidal Salmonella by Aqsa Ashfaq, Safah Mariyam, Ubair Aziz, Mamuna Mukhtar and Saadia Andleeb in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We acknowledge the support of the Department of Microbiology and Biotechnology at Atta-ur-Rahman School of Applied Biosciences, NUST, for providing the research facilities and funding.
Ethical Considerations
Ethical approval is not applicable for this article.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of Human and Animal Rights
This article does not contain any studies on human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article, and informed consent is not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.