
Abstract
Background
Miller-Fisher syndrome (MFS) is a rare variant of Guillain-Barré syndrome (GBS), characterized by the classic triad of ataxia, areflexia, and ophthalmoplegia. Atypical presentations involving severe headache and transient loss of consciousness (LOC) are extremely rare, posing a significant diagnostic challenge.
Case presentation
We present a 38-year-old Yemeni female with hypertension, type II diabetes mellitus, and recurrent deep vein thrombosis, with no history of recent infection. She presented with an acute, severe headache lasting three days, followed by transient LOC. By the fourth day, the classic MFS triad emerged. In a resource-limited setting, diagnosis was based on the clinical triad and evidence of demyelinating polyneuropathy on nerve conduction studies (NCS), in the absence of anti-GQ1b antibody testing and normal brain neuroimaging. After receiving intravenous immunoglobulin (IVIG), the headache resolved significantly by the third dose. The patient showed improvement in ophthalmoplegia and ptosis, achieving complete neurological recovery within two months.
Conclusion
Acute severe headache and transient LOC can be an early, atypical manifestation of MFS. The clinical triad and electrophysiology findings remain the cornerstones of diagnosis when antibody testing is unavailable. Early intervention with IVIG led to a successful outcome.
Keywords
Introduction
Miller Fisher Syndrome (MFS) is a rare subtype of Guillain-Barré Syndrome (GBS), characterized as an acute, immune-mediated neuropathy that primarily affects the peripheral nervous system. 1 MFS typically presents with the classic diagnostic triad of ophthalmoplegia, ataxia, and areflexia. 2 While first described by Collier in 1932 and later formally characterized by Miller Fisher in 1956, its pathogenesis is largely attributed to molecular mimicry involving anti-GQ1b antibodies. The global incidence of MFS is 1–2 per 100,000.1,3,4 Although MFS often follows a prodromal infection, atypical presentations can emerge idiopathically, manifesting as unusual symptoms that challenge standard diagnostic criteria.3,5,6 The occurrence of acute severe headache and transient loss of consciousness (LOC) is extremely rare in MFS and often misleads clinicians toward vascular or central nervous system pathologies, such as stroke or hypertensive emergencies. Here, we report a case of a 38-year-old female whose initial presentation was dominated by severe headache and LOC. This case highlights the importance of clinical vigilance in identifying MFS when it is masked by atypical neurological symptoms, particularly in resource-limited settings where specialized serological testing may be unavailable.
Case Presentation
A 38-year-old Yemeni female presented to the Emergency Department on December 20, 2025, with an acute onset of severe headache for three days
The patient had been clinically stable until three days prior to ICU admission, when she developed a throbbing occipital headache that was refractory to high-dose analgesics (Paracetamol Extra 3000 mg/day). On the third day, she experienced a transient LOC lasting 15 minutes. Notably, there was no post-ictal confusion, and the patient achieved a Glasgow Coma Scale (GCS) of 15/15 immediately upon regaining consciousness, which strongly argued against a prolonged post-ictal state or hypertensive encephalopathy.
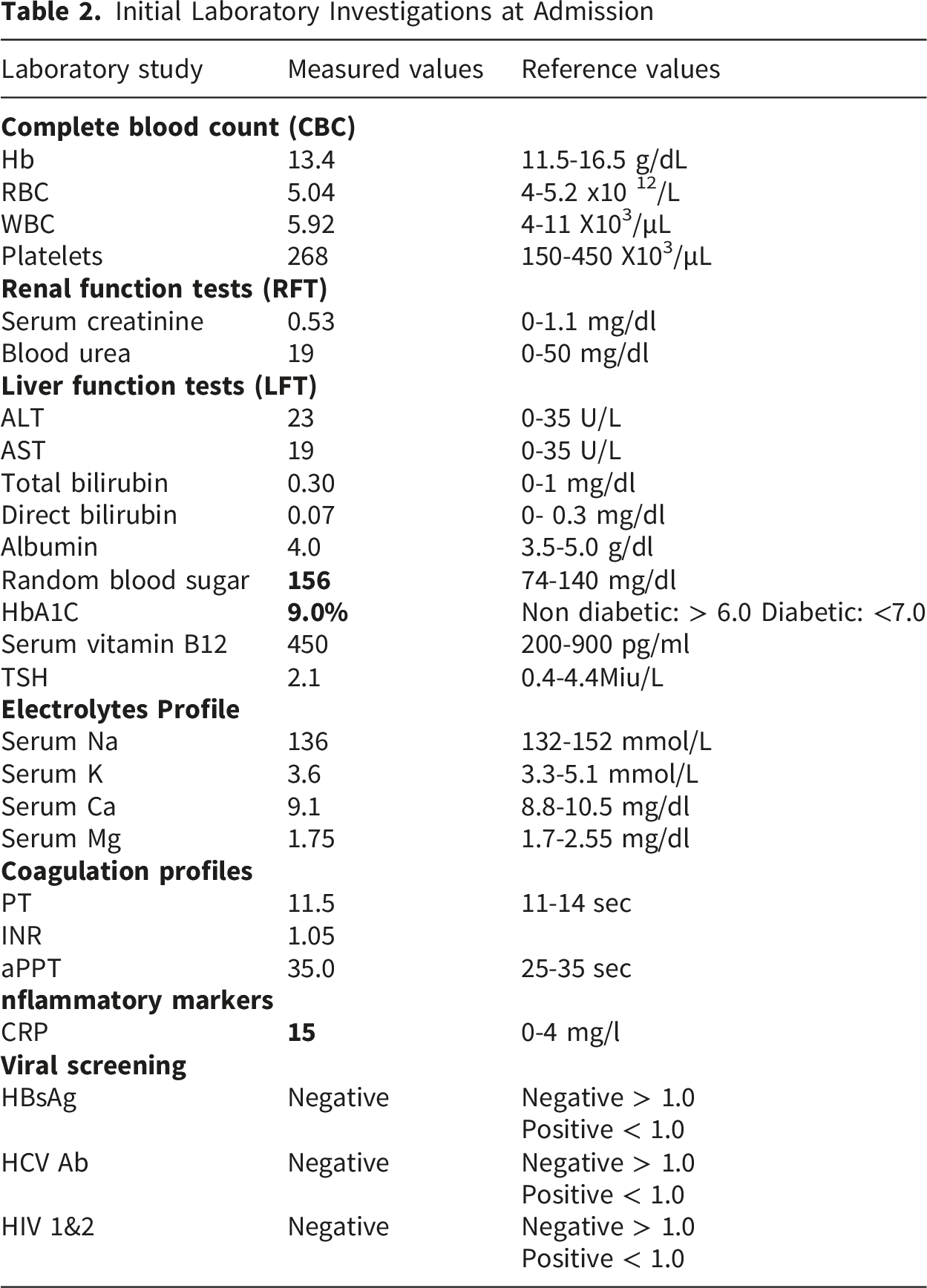
Initial assessment at the primary healthcare center revealed significant hypertension (200/110 mmHg), tachycardia (120 bpm), tachypnea (25 breaths/min), and a temperature of 37.5°C. Upon regaining consciousness, the patient presented with acute painful bilateral ophthalmoplegia, bilateral complete ptosis, and blurred vision. Brain MRI and MRV detected no evidence of acute or chronic venous sinus thrombosis, with no filling defects identified. Furthermore, the absence of intracranial or orbital lesions effectively ruled out space-occupying lesions or vascular causes for the patient’s symptoms. The absence of T2/FLAIR hyperintensities in posterior circulation ruled out Posterior Reversible Encephalopathy Syndrome (PRES) despite the hypertensive urgency. Echocardiography (Echo) showed concentric left ventricular hypertrophy (LVH) with impaired diastolic filling; however, the ejection fraction (EF) was preserved (59%). There was no evidence of mural thrombi, vegetations, or segmental wall motion abnormalities. Although a lumbar puncture (LP) was indicated, the patient initially refused and left the hospital against medical advice.
By the fourth day of disease course, her condition deteriorated significantly, progressing to neck extensor weakness “dropped head sign”, acute ascending motor paralysis, and distal paresthesia sensations. Given the rapid clinical decline and the high risk of respiratory failure, the patient was urgently admitted and transferred to Intensive Care Unit (ICU) for stabilization and definitive management. Upon ICU admission, physical examination revealed profound quadriparesis (Motor power: 2/5 in the upper limbs, 1/5 in the lower limbs), distal paresthesia, and complete bilateral ptosis. Generalized areflexia was confirmed in all four limbs, while plantar responses flexor (negative Babinski signs), localizing the pathology to peripheral nervous system (PNS) and making a central brainstem lesion unlikely. Due to severe quadriparesis, cerebellar testing, such as the finger-to-nose and heel-to-shin tests, could not be performed. Despite stable O2 saturation (≥94%), she developed mild respiratory distress and oropharyngeal dysphagia. Overflow urinary incontinence was also observed, suggesting autonomic instability, a recognized feature of the GBS spectrum.
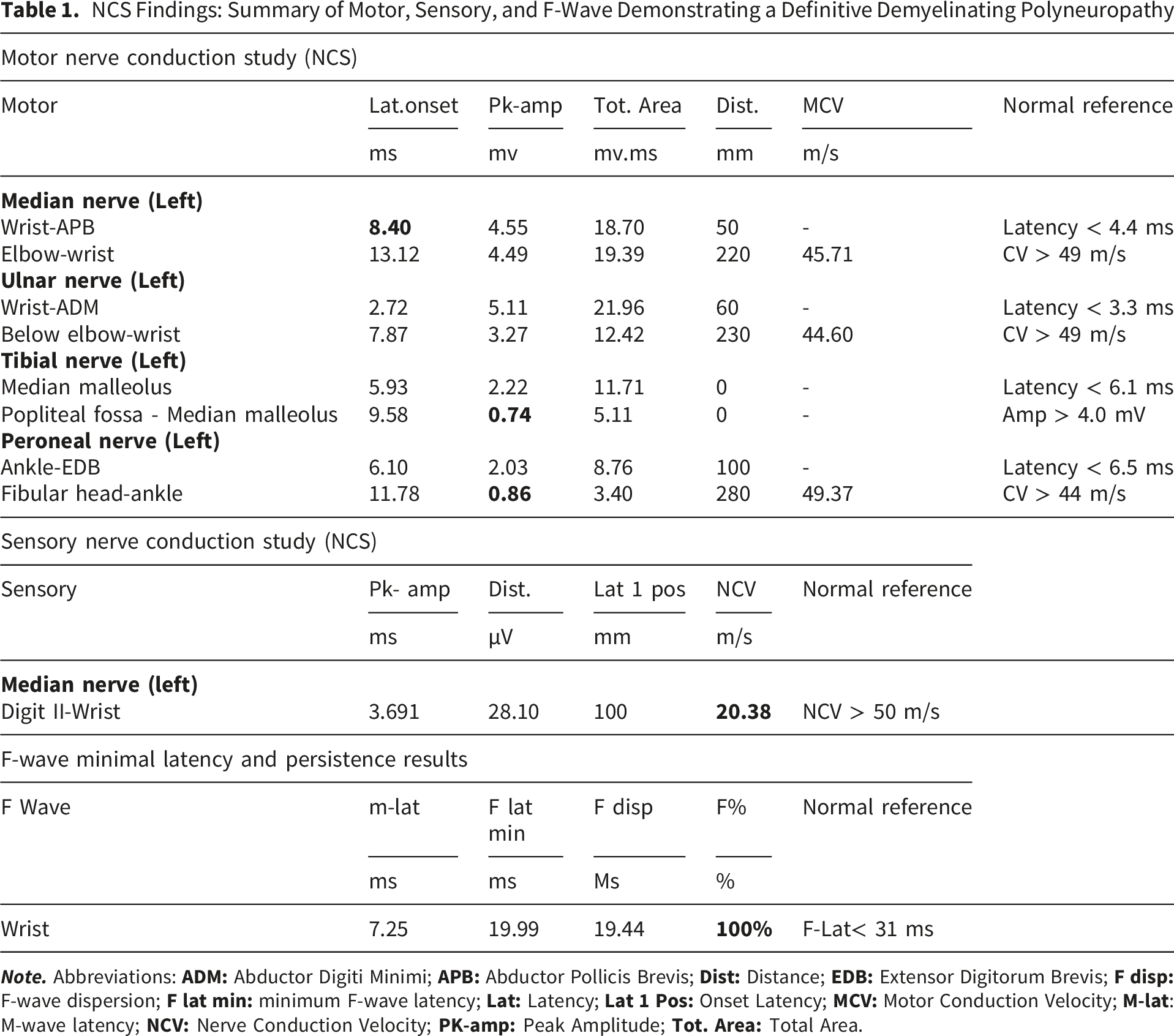
Following an urgent neurological consultation, the classic MFS triad (ophthalmoplegia, ataxia, and areflexia) was identified. Comprehensive nerve conduction studies (NCS) were performed. The NCS of the left upper and lower extremities showed a definitive pattern of demyelinating polyneuropathy. Specifically, the motor NCS of the left median nerve exhibited a normal compound motor action potential (CMAP) and CV but with prolonged distal motor latency (DML) and temporal dispersion pattern (Figure 1A). In the lower limbs, the left peroneal nerve showed reduced CMAP amplitude with a marked temporal dispersion (Figure 1B), and the left tibial nerve exhibited a reduced CMAP amplitude with an absent proximal response. Furthermore, the left median nerve SNAP showed reduced amplitude, slowed conduction velocity (CV), and an absence of F-waves (Figure 1C) in initial trials, followed by persistent F-waves. Collectively, prolonged distal latency and dispersion in the median nerve, reduced peroneal CMAP with temporal dispersion, and the reduced tibial proximal response —electrophysiologically confirm a primary demyelinating process. The detailed quantitative parameters are summarized in (Table 1). NCS of the left Extremities: (A) Motor Nerve Conduction Study (NCS) of the Left Median Nerve showing prolonged distal latency (8.40 ms) with a prominent dispersion pattern (black arrow). (B) Left peroneal motor NCS demonstrating marked amplitude reduction and severe dispersion (red arrow). (C) Left median SNAP and F-waves study illustrating initial absent F-waves, followed by persistent F-waves with prolonged latency (blue arrows) NCS Findings: Summary of Motor, Sensory, and F-Wave Demonstrating a Definitive Demyelinating Polyneuropathy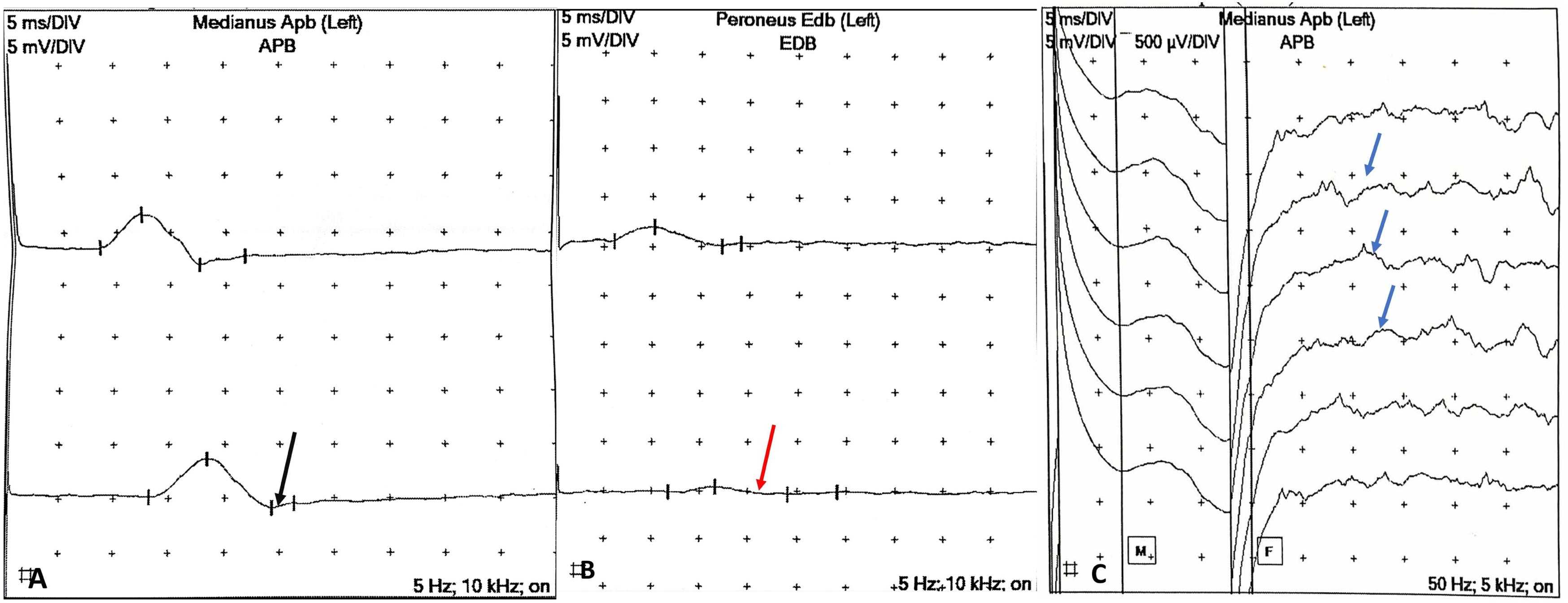
Initial Laboratory Investigations at Admission
Upon ICU admission, intravenous immunoglobulin (IVIG) therapy (0.4 g/kg/day for five days) was initiated. The treatment regimen also included daily IV methylcobalamin, oral thioctic acid (600 mg twice daily), and intensive physical therapy. To mitigate the high thrombotic risk-given the patient’s history of recurrent DVT and the potential for IVIG-induced hyperviscosity-she was maintained on prophylactic enoxaparin (4,000 IU/day) throughout the course.
A dramatic response was observed: the severe headache, previously refractory to all analgesics, resolved completely following the third IVIG dose, supporting an immune-mediated mechanism. Motor power was 3/5 and 1/5 in the upper and lower limbs, respectively. By the completion of the five-day course, motor power improved to 4/5 in the upper limbs and 2/5 in the lower limbs. Swallowing functions and urinary continence were fully restored. By the day of discharge, the patient’s clinical status exhibited a profound neurological recovery. She was able to walk with assistance, with upper limb strength reaching 5/5, lower limb strength improving to 3/5. Regained partial head posture control, and a moderate improvement in her bilateral ptosis was noted. Two months later after discharge, the patient showed complete resolution of her motor deficits. She was walking independently without any assistance, with muscle strength having fully reverted to 5/5 in all four limbs. Her bilateral ptosis had complete resolution, accompanied by a marked subjective improvement in vision. Clinical timeline of the case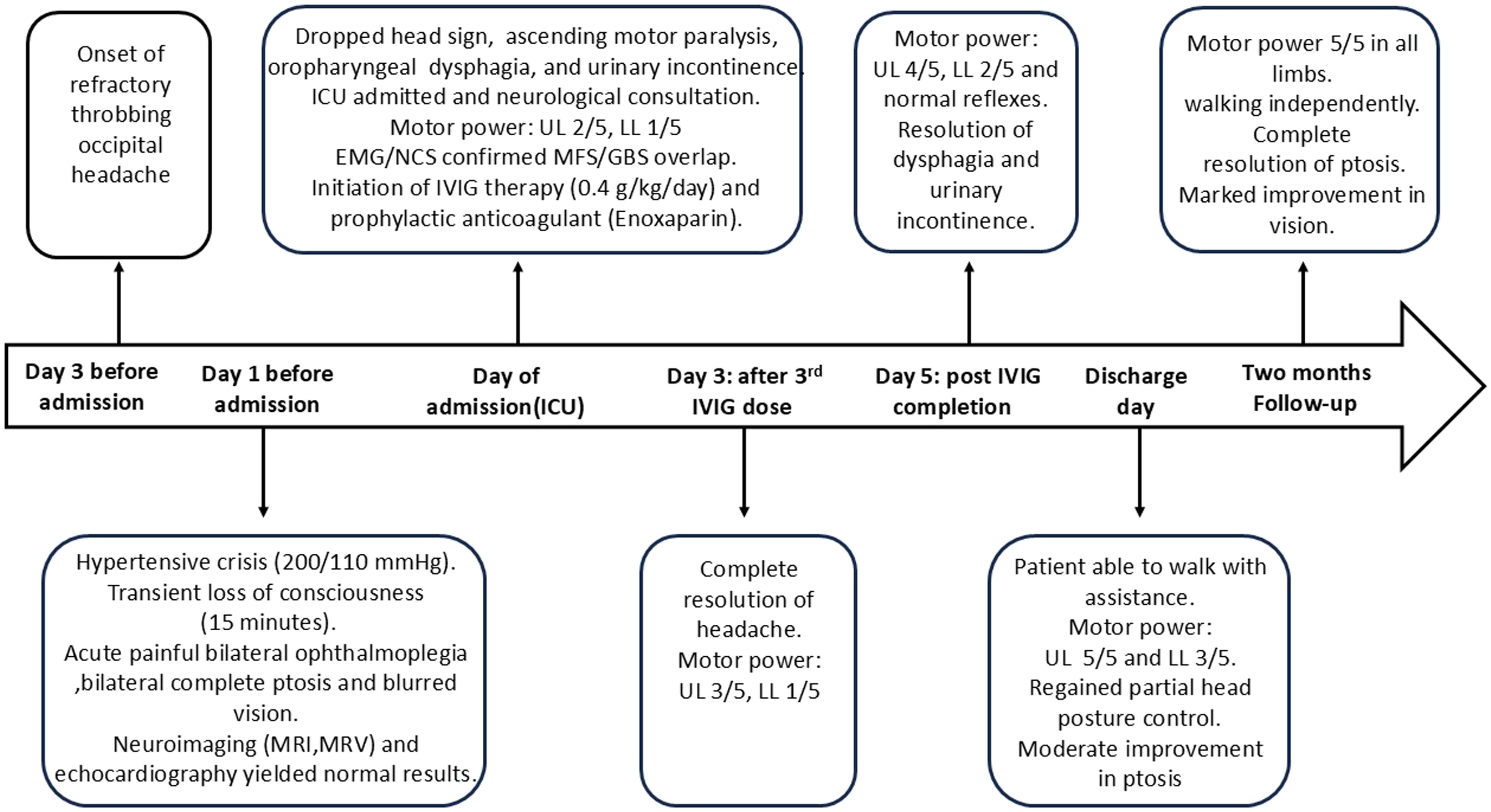
Discussion
MFS is an immune-mediated inflammatory demyelinating disorder of the peripheral nerves, typically diagnosed by the classic triad of ataxia, areflexia, and external ophthalmoplegia. It is a rare GBS variant with a global incidence of 1–2 per 100,000.1,4 The prevalence of MFS is significantly higher in Asian populations (15-25%) compared to Western regions 1-7%. While the reasons for this disparity are not clearly understood, environmental triggers likely play a crucial role.3,4 Although MFS is often preceded by a gastrointestinal or viral infection, its etiology can be idiopathic.2,5,6 Similar to GBS, MFS typically manifests days following a specific infection caused by pathogens such as Campylobacter jejuni, Cytomegalovirus (CMV), and Epstein–Barr virus. Molecular mimicry is identified as the primary mechanism driving the disease. More than 90% of MFS patients have elevated serum anti-ganglioside antibody levels, specifically anti-GQ1b antibodies. 6 Under the 2014 classification, MFS, Bickerstaff’s Brainstem Encephalitis (BBE), and the Pharyngeal-Cervical-Brachial (PCB) variant represent a clinical continuum. 7 These conditions are categorized based on their overlapping clinical features. 7 The complexity of the initial presentation posed a significant diagnostic challenge, and required several differential diagnoses to rule out various neurological and vascular etiologies.
The patient’s history of recurrent deep vein thrombosis (DVT) and severe headache initially suggested Cerebral Venous Sinus Thrombosis (CVST). However, it was ruled out through Magnetic Resonance Venography (MRV) which demonstrated an absence of filling defects. Similarly, despite a history of T2DM, the combination of bilateral painful ophthalmoplegia and generalized areflexia was inconsistent with diabetic mononeuropathy. Diabetic cranial nerve involvement typically spares the pupils and lacks the generalized loss of deep tendon reflexes. 1 Additionally, Myasthenia Gravis (MG) was ruled out as the patient’s ptosis lacked diurnal fluctuation. Furthermore, the progression to ascending palsy and generalized areflexia is inconsistent with neuromuscular junction disorders. 8
The initial presentation of hypertensive crisis (200/110 mmHg) and transient LOC initially suggested an acute cerebrovascular event. However, the spontaneous resolution of LOC within 15 minutes, Glasgow Coma Scale (GCS) of 15/15, and the absence of focal neurological deficits made a permanent stroke unlikely. The transient LOC in our patient was likely a neurocardiogenic response to the agonizing “thunderclap” headache or a manifestation of acute autonomic dysfunction, rather than primary brainstem lesions, as evidenced by the rapid return to baseline cognition. Furthermore, normal renal function tests and the absence of acute kidney injury ruled out a typical hypertensive emergency. 9
Moreover, the absence of vasogenic edema on MRI FLAIR sequences, despite the severe hypertension, effectively excluded Posterior Reversible Encephalopathy Syndrome (PRES). 10
Bickerstaff’s Brainstem Encephalitis (BBE) was initially suspected; however, the absence of prolonged impairment of consciousness, hyperreflexia, Babinski’s sign, sensory impairment, along with the normal brain MRI, made BBE unlikely.11,12 By physical examination revealed absent limb reflexes and ataxia while NCS finding indicated demyelinating polyneuropathy. 13 In line with Nayak et al, our case aligns with documented seronegative BBE-MFS overlap presentations. Furthermore, the patient’s prompt response to IVIG, despite the unavailability of anti-GQ1b antibody testing, reinforces that clinical and NCS finding remains the gold standard in resource-limited settings. 14
The patient also demonstrated features of pharyngeal-cervical-brachial (PCB) variant of GBS. PCB is a rare variant of GBS often characterized by axonal neuropathy. PCB presented with progressive oropharyngeal weakness, neck extensor muscle weakness “dropped head sign”, areflexia in the upper limbs, and minimal or no lower limb involvement.15-17
Approximately 60% of patients with acute GBS have a subtype with autoantibodies against different glycolipids of gangliosides, which correlate with different subtypes of GBS. 18 GQ1b and GT1a are the most antibodies associated with MFS where GT1a associated with PCB variant of GBS. Clinicians must maintain a high suspicion for bulbar involvement. Detailed history taking and physical examination are essential to identify potential overlaps as early as possible. Serum anti-GQ1b antibodies have been taken as a key biological marker for MFS. The GQ1b antigen is highly expressed in the oculomotor, trochlear, and abducens nerves, which are the major cause of ophthalmoplegia. 19 When anti-GQ1b antibody tests are negative, it remains possible that pathogenic antibodies are present in low titers. Even at low titers, these antibodies can cause severe neuropathic damage which typical clinical manifestations occur despite negative serological result. 20
Our clinical observations align with the recent findings of Huang et al regarding the atypical presentation of Miller-Fisher Syndrome. A key contribution of our report is the correlation between our NCS findings and the serological evidence presented in their report. While Huang et al confirmed their diagnosis using anti-GQ1b antibody titers and CSF analysis, we established the diagnosis using NCS in a resource-limited setting. 1 The present of demyelination polyneuropathy in our patient (Table1 and Figure 1) provide a neurophysiological counterpart to the albuminocytological dissociation and antibody positivity reported by Huang et al. This synergy between different diagnostic modalities—serological in their report and NCS in ours—demonstrates that both approaches can reliably identify the same underlying neuropathic process. This is particularly evident when presenting with atypical symptoms, such as severe headache, and transient LOC.
Although our patient experienced a severe headache, this remains a rare and atypical manifestation of MFS.
However, Jung et al and Koga et al documented headache in 16% and 22% of cases, respectively, during the acute phase of MFS.21,22 While the exact etiology of the headache remains unclear, however, several potential mechanisms have been suggested. (1)
Diagnostic Limitations and Diagnostic Certainty
While our patient presented with the classic triad and electrophysiological evidence that firmly establishes the diagnosis of MFS within the GBS spectrum, the features suggestive of a Pharyngeal-Cervical-Brachial (PCB) or Bickerstaff’s Brainstem Encephalitis (BBE) overlap remain hypothesized. We acknowledge significant diagnostic limitations in our resource-limited setting. The patient’s refusal to undergo a lumbar puncture prevented CSF analysis, which limits our ability to exclude other infectious or inflammatory CNS processes. Furthermore, the unavailability of anti-GQ1b and other anti-ganglioside antibody testing dictates that our diagnosis inherently rests on the combination of the classic clinical triad, characteristic demyelinating features on NCS, and the exclusion of vascular mimics via neuroimaging.
Despite these limitations, fulfilling Level 2 of the Brighton Collaboration Criteria justified the prompt initiation of IVIG, which ultimately led to the patient’s full recovery.
Conclusion
Acute severe headache and transient loss of consciousness can be early, atypical manifestation of MFS. When these symptoms are immediately followed by acute ophthalmoplegia and supported by characteristic NCS findings, a diagnosis of MFS or its overlap variants should be strongly considered, even in the absence of anti-GQ1b antibody testing or CSF analysis. Although atypical MFS generally carries a favorable prognosis, this case highlights that early initiation of IVIG therapy and intensive rehabilitation are critical to preventing respiratory complications and ensuring a rapid return to baseline functions. Clinicians should maintain high vigilance for such neurological mimics in an emergency setting.
Methods
This work has been reported in line with the SCARE criteria. 27
Footnotes
Acknowledgment
We hope to thank SMSR Team Lab. for their efforts and bringing our team together.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying clinical data. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Author Contributions
SB. wrote a part of the manuscript. NA. wrote a part of the manuscript. A.A. wrote a part of the manuscript. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data supporting the findings of this case report are included within the article.