
Abstract
Background:
Recent research has suggested that rituximab is not non-inferior to ocrelizumab in the treatment of relapsing multiple sclerosis (RMS). We compared the effectiveness of rituximab versus ocrelizumab in RMS.
Methods:
Individuals with RMS treated with rituximab or ocrelizumab with ⩾12 months of clinical follow-up and/or post-rebaseline magnetic resonance imaging (MRI) data were propensity-scored matched 1:1 and included.
Objectives:
Primary outcomes were annualized relapse rate (ARR), ARR ratio and cumulative hazard of relapses. The secondary outcome was the proportion of each group demonstrating post-rebaseline radiological activity.
Results:
In total, 538 individuals were included in the primary (clinical) outcome analysis and 350 individuals were included in the secondary (radiologic) outcome analysis. Over a median 2-year follow-up, very low ARR was observed in both groups (rituximab: 0.05 (95% CI: 0.03–0.08) versus ocrelizumab: 0.03 (95% CI: 0.02–0.05)). The adjusted ARR ratio for rituximab compared with ocrelizumab was elevated at 1.76 (95% CI: 0.70–4.59), and the adjusted cumulative hazard of relapses was also elevated for rituximab-treated individuals (HR = 2.17 (95% CI: 0.91–5.1)).
Conclusion:
Although our results suggest that rituximab may be inferior to ocrelizumab for relapse suppression, the rarity of relapses underpowered comparisons, and no definitive conclusions could be drawn.
Introduction
Anti-CD20 monoclonal antibodies causing peripheral B-lymphocyte depletion have emerged as highly effective treatments in relapsing multiple sclerosis (RMS). 1 Rituximab and ocrelizumab are two medications in this class with similar effects on B-lymphocyte depletion, but preferentially greater CD4 and CD8 T-lymphocyte depletion has been described with ocrelizumab. 2 Nonetheless, both agents have benefit on clinical and radiological outcomes over placebo in phase II studies,3,4 and over active comparators in phase III clinical trials.5,6 Consequently, the use of anti-CD20 therapies for RMS continues to increase globally.7–9
Both rituximab and ocrelizumab are commonly used in British Columbia (BC), Canada, allowing for a unique opportunity to observe clinical responses in a similar patient population. Rituximab is routinely used globally, but the practice of parallel utilization of rituximab and ocrelizumab is uncommon at other centres outside of BC.
There are no head-to-head randomized clinical trials comparing the effectiveness of rituximab to ocrelizumab in multiple sclerosis (MS), although several non-inferiority trials are in progress.10–13 Recent observational research combining data from MSBase and Danish MS Registries suggested treatment with rituximab is associated with a higher annualized relapse rate (ARR) ratio and increased cumulative hazard of relapses when compared to ocrelizumab. 14 However, the study population included few patients with newly diagnosed MS, and magnetic resonance imaging (MRI) data were only partially available. Understanding the difference between these therapies is critical to make decisions about cost-effectiveness and/or the need for advocacy to improve access to ocrelizumab.
We performed a multicentre retrospective observational study comparing the effectiveness of rituximab and ocrelizumab on clinical relapses and radiological activity in people living with MS in British Columbia, Canada.
Methods
Standard protocol approvals, registrations and patient consents
The study protocol was approved by the University of British Columbia Clinical Research Ethics Board (study ID H24-02237) and the University of Northern British Columbia Research Ethics Board (study ID 2025-008). Informed consent was waived due to the retrospective nature of the study. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines.
Participants
Patient data were abstracted via chart review of electronic health records from two MS clinics: the University of British Columbia MS Clinic (Vancouver, British Columbia, Canada) and the Northern Health Regional MS Clinic (Prince George, British Columbia, Canada).
Inclusion criteria were as follows: (1) a diagnosis of RMS according to 2017 McDonald Criteria 15 including patients with secondary progression, (2) age 18–65 years (inclusive) at initiation of either rituximab or ocrelizumab, (3) initial dose of rituximab or ocrelizumab received on 1 January 2017 or later, (4) standard dosing of rituximab (1000 mg at weeks 0 and 2, then 1000 mg every ~6 months) or ocrelizumab (300 mg at weeks 0 and 2, then 600 mg every ~6 months) – alternative dosing schedules were included (e.g. extended interval dosing every 9 months), provided doses were not adjusted, (5) treatment with rituximab or ocrelizumab with initial two induction doses, and at least one maintenance dose, and (6) data from at least one clinical follow-up available at 12 months or later following rituximab or ocrelizumab initiation. As an illustrative example, a patient who received induction doses at weeks 0 and 2 and a 6-month maintenance dose would be excluded if their last documented follow-up occurred at 9 months post induction (due to insufficient duration of follow-up). For inclusion in MRI data analysis, the above had to be fulfilled in addition to (1) at least one MRI brain (with or without cervical and/or thoracic spine) at a minimum of 90 days or greater following first infusion of rituximab or ocrelizumab to act as a rebaselined MRI standard, (2) one or more MRIs following rebaselined MRI without interruption of rituximab or ocrelizumab treatment and (3) MRI interpretation/report available on electronic medical record from a radiologist or neurologist.
Exclusion criteria were as follows: (1) treatment with two different anti-CD20 medications at any time point (including patients switching from one anti-CD20 agent to another), (2) concurrent treatment with immunosuppressive or immunomodulatory therapy simultaneously with rituximab or ocrelizumab, excluding oral or intravenous steroids less than or equal to 7 days for relapse management, (3) previous allogenic or autologous bone marrow transplantation and (4) diagnosis of primary progressive MS or alternative central nervous system demyelinating or inflammatory disorder (e.g. neuromyelitis optica spectrum disorder, myelin oligodendrocyte glycoprotein antibody disease, etc.).
Procedures
Retrospective patient data were screened from disease-modifying therapy (DMT) tracking lists as recorded in routine clinical practice at our centres. All patient data were assigned a unique patient identification number for anonymization. Patient data were screened for inclusion and exclusion criteria by N.Y.C., D.J.H. and J.M., and if appropriate, an electronic chart review was undertaken to abstract data. Demographic information gathered included the following: date of birth, sex, postal code, MS centre, date of MS onset (or if unknown, date of MS diagnosis), treatment with rituximab or ocrelizumab (no discrimination was made between rituximab and its biosimilars), Expanded Disability Status Scale (EDSS) score before initiation of anti-CD20 therapy, date of first infusion of anti-CD20 therapy, date of last clinical assessment, relapse(s) in the 12 months before anti-CD20 therapy initiation and prior DMTs before anti-CD20 therapy (acceptable prior DMTs include but were not limited to platform injection therapy (glatiramer acetate, interferon-beta), teriflunomide, dimethyl fumarate, fingolimod, alemtuzumab, natalizumab, cyclophosphamide, mitoxantrone, azathioprine, mycophenolate and cladribine). As a measure of socioeconomic status, neighbourhood income quintiles before tax standardized to national averages were derived from postal code information using the Postal Code Conversion File Plus (Version 7E) 16 using SAS (version 9.4, SAS Institute Incorporated, Cary, United States), which is a programme developed by Statistics Canada linking postal code to Canadian census data, as described previously. 17 Follow-up data were abstracted up to 30 June 2024, inclusive.
Study outcomes
The primary study outcomes were ARR, ARR ratio and cumulative hazard of relapses between rituximab- and ocrelizumab-treated patients. The secondary endpoint was the occurrence of radiologic activity on post-rebaseline MRI (new T2 hyperintense lesions and/or T1 gadolinium-enhancing lesions).
Relapses were defined as new neurological symptoms with subjective and/or objective changes from baseline typical of MS, with a duration of at least 24 hours, and in the absence of fever and/or infection. 15 Relapses and MRI activity were recorded based on documentation by the treating neurologist/radiologist. Relapses and MRI activity on anti-CD20 therapy were counted if occurring ‘on treatment’ with rituximab or ocrelizumab. ‘On treatment’ was operationally defined at a minimum of 30 days or greater following initial infusion of anti-CD20 therapy, based on previous evidence demonstrating robust suppression of clinical/radiological activity within that time frame. 18 If treatment was discontinued, clinical relapses and/or MRI activity were considered on therapy within 6 months of the last infusion. New MRI activity also required a previous on-treatment MRI established at least 90 days or greater following initial anti-CD20 initiation. Disagreements between reviewers (N.Y.C., D.J.H. and J.M.) were mediated by the senior author (A.J.S.).
Statistical analysis
All statistical analyses were performed using RStudio version 4.4.0. Propensity score matching was used to achieve covariate balance between treatment groups. A directed acyclic graph was used to pre-define potential confounders of treatment assignment (rituximab or ocrelizumab) and MS disease activity for inclusion in a propensity score model using expert knowledge. Baseline propensity scores for the predicted probability of treatment assignment were estimated using main-effects logistic regression incorporating baseline: age, sex, income quintile, MS centre, MS duration since symptom onset (or if unknown, date of MS diagnosis), EDSS, occurrence of relapse(s) in the prior 12 months and most recent DMT. A categorical variable was generated for most recent DMT, with groups including ‘no prior DMT’, ‘low efficacy DMT’ (platform injectables, teriflunomide, dimethyl fumarate, azathioprine, daclizumab, mycophenylate mofetil and docetaxel), ‘immune reconstitution therapies’ (cladribine and alemtuzumab), ‘lymphocyte sequestering therapies’ (natalizumab and fingolimod/siponimod) and ‘potent immunosuppressants’ (cyclophosphamide and mitoxantrone). The positivity assumption was evaluated with overlayed propensity score density plots by treatment group.
To achieve covariate balance between treatment groups, 1:1 greedy nearest neighbour matching without replacement (calliper 0.2) was performed using the MatchIt package; individuals treated with rituximab were paired to individuals treated with ocrelizumab by similar propensity score. Initial post-match balance and overlap between treatment groups were visualized using jitter, density and love plots. Standardized mean differences (SMDs) were calculated for each covariate to assess for post-match imbalances between treatment groups.
To account for differential follow-up duration, primary analyses were restricted to a common follow-up window of 2 years. Treatment differences in the ARR were estimated with a negative binomial model testing with and without adjustment for calendar year. Treatment differences in time to first relapse were evaluated using Cox proportional hazards with and without adjustment for calendar year. A cluster term was used to account for matched pairs. The study start date was 30 days after the first anti-CD20 infusion. Individuals were right censored at (1) time of relapse, (2) 6 months after the last anti-CD20 infusion, (3) last follow-up within a 2-year window or (4) at 2 years (whichever was earliest). The proportionality assumption was evaluated using the Schoenfeld global test. Time-to-event data were visualized using Kaplan–Meier plots.
For the secondary outcome of radiologic disease activity, the original cohort was restricted to individuals who had at least one MRI brain following radiologic rebaseline. Recalculation of baseline propensity scores was performed for this cohort, followed by 1:1 greedy nearest neighbour matching without replacement using the procedures described above. Evidence of new radiologic disease activity was defined as any new or enhancing lesion observed on MRI of the brain or spinal cord. The proportion of individuals in each treatment group with new radiologic activity was reported along with 95% confidence intervals (95% CI) calculated using a binomial distribution.
Results
The study period spanned from 1 January 2017 to 28 June 2024. From the screened cohort of 1640 individuals who received ocrelizumab or rituximab, 692 individuals (409 ocrelizumab; 283 rituximab) with MS fulfilled study inclusion criteria (Figure 1). The most common reason for study exclusion was having a non-MS diagnosis (21.7%). The proportion of individuals excluded for having less than 12 months of clinical follow-up was slightly higher for those treated with rituximab (n = 92, 11.6%) relative to ocrelizumab (n = 32, 3.8%), as was the proportion of those excluded for not completing a 6-month maintenance dose (rituximab, n = 10, 1.3%; ocrelizumab, n = 3, 0.4%). The median duration of clinical follow-up was 3.6 years (interquartile range [IQR]: 2.0–5.1), and the median duration of post-rebaseline radiologic follow-up was 3.5 years (IQR: 2.2–4.8) across all patients. A substantially larger proportion of individuals were started on ocrelizumab earlier in the study period, with more participants initiated on rituximab in later years (2021 onwards) (eFigure 1 in Supplement 1).
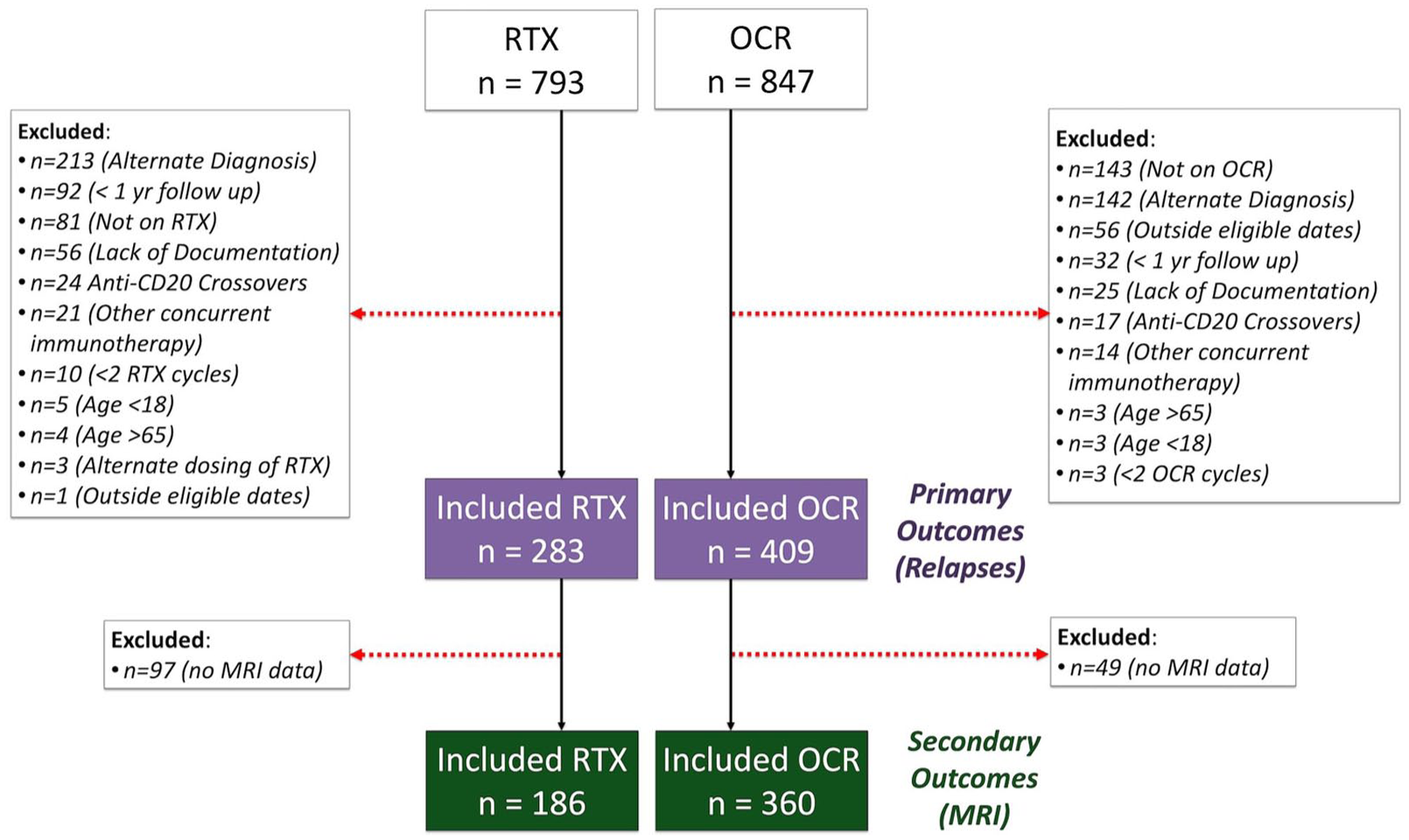
Patient flow diagram. RTX = rituximab; OCR = ocrelizumab.
The probability of being treated with rituximab versus ocrelizumab was calculated using a logistic regression model (eTable1 in Supplement 1). In comparison to those prescribed ocrelizumab, a larger proportion of individuals prescribed rituximab were male and low income (Table 1). Disability and disease activity were slightly higher in the rituximab group, as indicated by higher baseline EDSS and a larger proportion having had a relapse within the last year (Table 1). A larger proportion in the rituximab group were DMT naïve (Table 1).
Participant characteristics before and after propensity score matching.

DMT: disease-modifying therapy; SMD: standardized mean difference.
Propensity score matching decreased the sample size from 692 to 538 individuals for the primary outcome (clinical relapse) and from 546 to 350 for the secondary outcome (MRI activity) (Table 1). Propensity score matching improved balance between the matched groups (eFigure2 in Supplement 1); after matching for the primary outcome, all SMDs were less than 0.1 (Table 1). Individuals treated with rituximab who were not propensity score matched tended to be older and male, with a higher baseline EDSS and recent treatment with an immune reconstituting therapy or potent immunosuppressant (eTable2, Supplement 1). As propensity score matching with inclusion of calendar year drastically increased the number of unmatched and discarded pairs (141 matched pairs with inclusion of calendar year vs. 269 matched pairs without) and demonstrated persistent covariate imbalance (SMD = 0.188), it was not included as a covariate during matching and instead adjusted for in subsequent analysis.
The final study population was ~70% female, with a mean age of 39 years, average MS duration of 8 years and a baseline EDSS of 2.6 (Table 1). Two-thirds of individuals experienced a relapse in the year before starting anti-CD20 therapy (Table 1). Approximately half of the study population had a new diagnosis of MS and had not previously been on DMT (Table 1).
Primary outcome (clinical disease activity)
One relapse occurred in the first 30 days following anti-CD20 initiation. This relapse occurred on the infusion date for an individual in the rituximab group and was excluded from further analysis. Median on-treatment clinical follow-up time in the post-match study population was 4.6 years (IQR: 3.2–5.3) in the ocrelizumab group and 2.2 years (IQR: 1.6–3.0) in the rituximab group, reflecting differential use of ocrelizumab earlier in the study period (eFigure 1, Supplement 1). To account for differences in clinical follow-up duration, subsequent analyses were restricted to a common follow-up window of 2 years. In the ocrelizumab group, 95.9% remained relapse-free, with 15 relapses recorded more than 519.4 person-years of on-treatment clinical follow-up (median 2.0 years of follow-up). In the rituximab group, 92.9% remained relapse-free, with 22 relapses recorded more than 470.2 person-years of on-treatment clinical follow-up (median 2.0 years of follow-up). The estimated ARR was 0.03 (95% CI: 0.02–0.05) in the ocrelizumab group and 0.05 (95% CI: 0.03–0.08) in the rituximab group, with an ARR ratio of 1.60 (95% CI: 0.74–3.52) for rituximab compared with ocrelizumab, increasing to 1.76 (95% CI: 0.70–4.59) after adjustment for calendar year. Similarly, the cumulative hazard of any relapse activity was higher for patients treated with rituximab relative to ocrelizumab (hazard ratio: 1.86 (95% CI: 0.86–4.00)) (Figure 2). The magnitude of this association was further accentuated following adjustment for calendar year (hazard ratio: 2.17 (95% CI: 0.91–5.10)).

Kaplan–Meier plot for time to first clinical relapse by anti-CD20 therapy.
Secondary outcome (radiologic activity)
At least one post-rebaseline MRI was available for 186 individuals treated with rituximab and 360 individuals treated with ocrelizumab (Table 1). After propensity score matching, 175 individuals remained allocated to each treatment group. Propensity score matching improved balance between the matched groups; all SMD were less than 0.1, other than mild imbalance noted for the most recent DMT (SMD = 0.17), where 49.7% of the rituximab group were treatment naïve relative to 43.4% of the ocrelizumab group (Table 1).
The median time to first post-rebaseline MRI was similar between groups (ocrelizumab: 1.8 years, IQR: 1.4–2.5; rituximab: 1.6 years, IQR: 1.2–2.0 years). On first post-rebaseline MRI, 92.6% (95% CI: 87.7–95.6) of those treated with ocrelizumab showed no evidence of new radiologic activity. Of those treated with rituximab, 94.9% (95% CI: 90.5–97.3) showed no evidence of new radiologic activity.
The median duration of total radiologic follow-up was longer in the ocrelizumab group (4.0 years, IQR = 2.8–4.8), with 10.9% (n = 19) noted to have at least one new lesion more than 674 person-years of follow-up. The median duration of radiologic follow-up in the rituximab group was 2.2 years (95% CI: IQR 1.6–3.5), with 7.4% (n = 13) noted to have at least one new lesion more than 480 person-years of follow-up.
Discussion
Clinical effectiveness of anti-CD20 medications for RMS has been a topic of interest since the development of ocrelizumab in the early 2010s to mid-2010s. Rituximab is used as a cost-effective, off-label therapy in RMS based on similar mechanisms of action to ocrelizumab, but there is limited evidence directly comparing clinical outcomes. Utilizing real-world data from a Canadian cohort of RMS patients, we found low rates of clinical and radiologic disease activity in both treatment groups over a median follow-up of 2 years. Although there was a non-statistically significant trend towards lower relapse activity with ocrelizumab, the rarity of relapses and new MRI activity underpowered comparisons and no definitive conclusions could be drawn.
The largest previous observational study comparing rituximab and ocrelizumab using data from the MSBase and Danish MS Registries found that rituximab was not non-inferior compared with ocrelizumab in terms of ARR and cumulative hazard of relapses over time. 14 Although our study was underpowered relative to the Roos et al. study, 14 our cohort had uniform dosing and administration schedules, shorter MS disease duration, a lower degree of baseline disability, a larger proportion of treatment naïve individuals and a larger number of rituximab-treated individuals. Unmeasured confounding was further reduced through the inclusion of socioeconomic status (via income quintile) in baseline propensity score determination. Balance in socioeconomic status between groups is an important consideration when assessing outcomes in disease activity, as increased socioeconomic deprivation in persons with MS is related to more severe disease.17,19
The ARRs in our study (rituximab – 0.05; ocrelizumab – 0.03) are low compared with observational data from MSBase and Danish MS Registries 14 (rituximab – 0.20; ocrelizumab – 0.09). The ARR of rituximab from previous observational data has been closer to our results, ranging from 0.03 to 0.09.20–23 Similarly, the ARR in patients treated with ocrelizumab from real-world evidence has been low, with ARRs from 0.05 to 0.08.23–27 We also observed high rates of relapse freedom with both ocrelizumab and rituximab, comparable to prior studies showing greater than 80% of patients treated with ocrelizumab or rituximab were relapse-free after 2–3 years.24,28 Our ability to adjudicate relapses through review of the original medical record likely reduced inclusion of pseudorelapses and MS-related fluctuations. In addition, on-treatment follow-up time in our study was defined as 30 days after the initial infusion, which served to exclude relapses occurring before therapeutic suppression of MS activity. Finally, the included participants met the 2017 McDonald Criteria, likely resulting in a study population with a less severe course of disease. 29
The inclusion of MRI data as a measure of efficacy in this study is relevant given that MRI is the workhorse of disease surveillance of subclinical radiologic activity. To our knowledge, there are only two other studies directly comparing rituximab and ocrelizumab that included MRI outcomes, and both found no differences between therapies.23,30 The proportion of gadolinium-enhancing lesions was reduced from 0.41 to 0.07 after a median of 14 months in rituximab-treated patients in a Spanish multicentre study; 20 in another Swiss study, at 12 months after rituximab initiation, only 15.8% of patients had new T2 and/or gadolinium-enhancing lesions. 22 Similarly, studies assessing ocrelizumab showed a lack of MRI activity in 88.4%–93.3% of patients at 1 year. 24 Recently published long-term follow-up of patients in the OPERA open-label extension reported robust suppression of gadolinium-enhancing lesions and new/enlarging T2 lesions at 97.9% and 89.6% of patients, respectively, at the 9-year time point. 31 Overall, it appears both rituximab and ocrelizumab have excellent efficacy in the prevention of new MRI activity, which was supported by our results.
The biological similarities and differences between rituximab and ocrelizumab are an important consideration in the context of this study. While both are monoclonal antibodies targeting an overlapping extracellular CD20 epitope, rituximab is a murine-human chimeric antibody with preferential complement-dependent cytotoxicity, while ocrelizumab is a fully humanized antibody with greater relative antibody-dependent cellular cytotoxicity. 32 The differences in antibody structure likely relate to greater immunogenicity as well, with 37% of rituximab-treated RMS patients possessing anti-drug antibodies, 33 compared with only 1% in ocrelizumab-treated patients. 34 However, an association with disease activity in rituximab-treated patients with anti-drug antibodies compared with those without was not observed. 33 Furthermore, a small study of 50 rituximab-treated and 38 ocrelizumab-treated MS patients found greater suppression of CD4 and CD8T-lymphocytes in the ocrelizumab-treated group at months 1, 3, and 6, although there was no association between disease severity and drug treatment. 2
Establishing rituximab and ocrelizumab as clinically equivalent, highly effective medications for MS could have significant implications from a cost savings perspective. A unique aspect of rituximab as an anti-CD20 medication in MS is the existence of biosimilar medications with markedly reduced costs compared with ocrelizumab, ofatumumab and ublituximab, although studies are underway examining ocrelizumab biosimilars.35,36 A study examining Medicare and Medicaid data from 2018 to 2021 calculated cost savings of $1.9 billion and $590 million, respectively, if ocrelizumab was priced similarly to rituximab. 37 Consequently, rituximab could be considered as a cost-effective and accessible alternative without sacrificing efficacy compared with other monoclonal antibody medications in MS.38–40 Unfortunately, this study is unable to provide additional evidence in support of policies allowing rituximab and ocrelizumab to be used in parallel based on access and affordability.
This study has limitations. Most importantly, despite our large sample size of 532 patients treated with anti-CD20 monoclonal antibodies, statistical comparison between treatment groups remained underpowered as the outcomes in question (relapses and new MRI activity) were very infrequent events. Consequently, we were unable to approach the analysis with a non-inferiority framework, which arguably is the most suitable approach for comparing two similar therapies. Other limitations include the retrospective observational nature of this study, which despite vigorous statistical methodology does not mitigate unknown and/or unaccounted confounders. In addition, no measures of baseline MRI activity, such as gadolinium-enhancing lesions, were included in baseline propensity score calculation, which is an important indicator of baseline disease severity. Since rituximab and its biosimilars were also pragmatically utilized without distinguishment in our cohort, only a group effect was determined, and variations in the treatment effects between the different medications cannot be ruled out. Data from this cohort also reflect the predominantly Caucasian RMS population and the practices of our local centres, and thus, our findings may not necessarily be generalizable to other patient populations or centres. Finally, this study was focused on the comparative effectiveness of ocrelizumab and rituximab, and we did not present significant data on disability worsening, adverse effects or treatment adherence of these agents. EDSS progression was not included as an outcome measure, given the sparsity of EDSS measurements (stemming from a large proportion of the study period occurring during the COVID-19 pandemic when in-person visits were limited). As our study excluded those who switched between anti-CD20 therapies and required completion of at least one post-induction maintenance dose with a minimum of 1-year clinical follow-up, we may have failed to capture individuals who discontinued or switched due to early breakthrough disease.
In conclusion, rituximab and ocrelizumab demonstrated high efficacy in this observational study, with very low relapse rates in both groups. Although there was a trend towards relatively greater reduction in relapses and longer time to first relapse with ocrelizumab, our study was underpowered for statistical comparisons due to the limited number of observed relapses. Whether rituximab is not non-inferior to ocrelizumab remains uncertain. Fortunately, ongoing randomized controlled trials examining this question may allow for definitive comparison.10–13
Supplemental Material
sj-docx-1-msj-10.1177_13524585261453817 – Supplemental material for Comparative effectiveness of rituximab versus ocrelizumab in relapsing multiple sclerosis on clinical relapses and radiological outcomes in British Columbia, Canada
Supplemental material, sj-docx-1-msj-10.1177_13524585261453817 for Comparative effectiveness of rituximab versus ocrelizumab in relapsing multiple sclerosis on clinical relapses and radiological outcomes in British Columbia, Canada by Nathan Y Chu, Jodie I Roberts, David J Hunt, Donna Kuipers, Jomana Morkous, Kyra West, Christopher E Uy, Mohammad Alhalabi, Ana-Luiza Sayao, Robert L Carruthers, Anthony Traboulsee, Virginia Devonshire, Izanne Roos, Tomas Kalincik and Alice J Schabas in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors would like to thank all the clinicians and patients involved in this study. They would also like to thank Brian Steele for insight and advice regarding the Postal Code Conversion File Plus.
Author Contributions
Nathan Y Chu: major role in the acquisition of data, design of the study, analysis/interpretation of the data and drafting/revision of the manuscript for intellectual content.
Jodie I Roberts: analysis/interpretation of the data and drafting/revision of the manuscript for intellectual content.
David J Hunt: major role in the acquisition of data and revision of the manuscript for intellectual content.
Donna Kuipers: major role in the acquisition of data and revision of the manuscript for intellectual content.
Jomana Morkous: major role in the acquisition of data and revision of the manuscript for intellectual content.
Kyra West: revision of the manuscript for intellectual content.
Christopher E Uy: major role in the acquisition of data and revision of the manuscript for intellectual content.
Mohammad Alhalabi: major role in the acquisition of data and revision of the manuscript for intellectual content.
Ana-Luiza Sayao: major role in the acquisition of data and revision of the manuscript for intellectual content.
Robert L Carruthers: major role in the acquisition of data and revision of the manuscript for intellectual content.
Anthony Traboulsee: major role in the acquisition of data and revision of the manuscript for intellectual content.
Virginia Devonshire: major role in the acquisition of data and revision of the manuscript for intellectual content.
Izanne Roos: analysis/interpretation of the data and revision of the manuscript for intellectual content.
Tomas Kalincik: analysis/interpretation of the data and revision of the manuscript for intellectual content.
Alice J Schabas: major role in the acquisition of data, conception/design of the study, analysis/interpretation of the data and revision of the manuscript for intellectual content.
All authors approved the final manuscript for publication.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: N.Y. Chu has received fellowship support from Canadian Network of MS Clinics, and speaker fees and honoraria from Novartis; J.I. Roberts has received compensation serving as an Editor, Associate Editor or Editorial Advisory Board Member for American Academy of Neurology, research support from Canadian Network of MS Clinics, Rebecca Hotchkiss International Exchange Program and the University of Calgary Medical Group Helios Award, and speaker fees and honoraria with Novartis and EMD Serono; D.J. Hunt reports no disclosures relevant to the manuscript; D. Kuipers has received travel funding and speaker fees from Biogen, Hoffman-La Roche, Sentrex, Novartis, EMD Serono and Apotec; J. Morkous reports no disclosures relevant to the manuscript; K. West reports no disclosures relevant to the manuscript; C.E. Uy reports no disclosures relevant to the manuscript; M. S. Alhalabi reports no disclosures relevant to the manuscript; A.L. Sayao received consulting fees from Pfizer Canada, Novartis, EMD Serono and Hoffman-La Roche, payment or honoraria from Pfizer, Novartis, EMD Serono, Hoffman-La Roche and Biogen, payment for expert testimony from Harper Grey LLP, and support for travel from Apotec and British Columbia Section of Neurologists Executive Member; R. Carruthers is Site Investigator for studies funded by Roche, Genentech, Novartis and EMD Serono and receives research support from Teva Innovation Canada, Roche Canada and Vancouver Coastal Health Research Institute, and has received honoraria from Teva, Roche, EMD Serono, Sanofi, Biogen, Novartis and Alexion; A. Traboulsee receives grants from Hoffman-La Roche, Biogen, Clene, Sanofi Genzyme, Abbvie, the National Institute for Health, MS Canada and the Consortium of MS centres, consulting fees from Hoffman-La Roche and Sanofi Genzyme, honoraria from Hoffman-La Roche, EMD Serono and Biogen, travel support from Sanofi Genzyme and EMD Serono, and is on a drug and safety monitoring board committee member at Sanofi Genzyme; V. Devonshire has received consulting fees, honoraria and support for travel from Hoffman-La Roche; I. Roos has received honoraria and speaker fees from Novartis, Merck and Roche, research support from MSIF-ARSEP and Melbourne Research Scholarship, and travel support from Merck and Roche; T. Kalincik has received honoraria from Roche, Sanofi Genzyme, Novartis, Merck, Biogen, National Health and Medical Research Council, MS Research Australia, Biogen, Janssen, Bristol Myers Squibb, WebMD Global, Teva, BioCSL and Eisai; research support from Biogen, Novartis, Genzyme, Roche, Celgene, Merck, MSBase Foundation, ARSEP and EDMUS Foundations, MS Research Australia, National Health and Medical Research Council, Australian Research Council, MS Society and Trish Foundation and has a non-compensated relationship as a scientific leadership group vice-chair with MSBase Foundation that is relevant to AAN interests or activities; A.J. Schabas has received honoraria from Biogen, Novartis, EMD Serono, Sanofi, Alexion, Hoffman-La Roche and Abbvie.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Ethics Approvals and Trial Registration
Ethics approval was obtained by the University of British Columbia Clinical Research Ethics Board (study ID H24-02237) and the University of Northern British Columbia Research Ethics Board (study ID 2025-008).
Consent for Publication
Not applicable.
ORCID iDs
Data Availability Statement
Anonymized data can be made available on a case-by-case basis by request from a qualified investigator.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.