
Abstract
Background:
Smokers with multiple sclerosis (MS) experience worse disease, yet underlying mechanisms remain unknown. Smoking disrupts bile acid and tryptophan metabolism in non-MS populations; both pathways involve host–microbiome co-metabolism and have been linked to MS.
Objective:
Determine whether smoking perturbs these metabolic pathways in MS and whether such alterations statistically mediate smoking’s effect on MS severity.
Methods:
We analyzed serum bile acid, tryptophan, and tobacco-related metabolites across four independent MS cohorts (N = 266) using discovery-replication analyses. Mixed-effects regression assessed replicating associations with current smoking and nicotine exposure. Mediation analyses tested if replicating metabolites were potential mediators between smoking and MS severity. Hypothesis-generating metagenomic analyses explored smoking-associated gut–microbial shifts and metabolite correlations.
Results:
Current smokers and nicotine-exposed MS subjects had reductions in bile acids and tryptophan metabolites, notably indolepropionate, a neuroprotective, anti-inflammatory gut–microbial metabolite. Lower indolepropionate statistically mediated ~20% of smoking’s adverse effect on MS severity. Metagenomic analyses identified potential smoking-enriched MS-linked taxa, and that indolepropionate broadly co-occurs with microbial networks (e.g. Lachnoclostridium appeared inversely associated with indolepropionate in smokers with MS).
Conclusion:
Tobacco exposure disrupts host–microbiome tryptophan and bile acid metabolism in persons with multiple sclerosis, with indolepropionate depletion partially mediating disease severity, highlighting a potential mechanistic pathway warranting further investigation in MS smokers.
Introduction
Tobacco smoke exposure accelerates disease progression and worsens clinical outcomes in persons with multiple sclerosis (PwMS).1,2 Unfortunately, the mechanisms that drive these relationships are unknown. As one of the few modifiable exposures that shape MS biology, defining any of these mechanisms may uncover pathways shared with other environmental insults and identify therapeutic targets that could benefit all PwMS.
Recent longitudinal studies demonstrated that gut–microbiome and related metabolomic shifts influence MS severity, including bile acid perturbations.3,4 These findings align with other metabolomic studies of MS outcomes, which highlight prominent roles for tryptophan and bile acid metabolism—two interconnected pathways that involve host–microbiome co-metabolism.5–7 Tryptophan gives rise to bioactive microbial metabolites, including indole derivatives such as indolepropionate (indole-3-propionic acid; IPA), that regulate immune responses and CD4+ T-cell metabolic programming through receptors such as the aryl hydrocarbon receptor (AHR). 8 Bile acids, beyond their digestive role, modulate immune and central nervous system signaling and have been implicated in MS pathobiology. 9 Interestingly, both tobacco smoke and nicotine disrupt these pathways and alter microbial composition and metabolic function, with downstream effects on immunity and neuroinflammation.10–13 Collectively, these findings support a potential mechanistic framework in which smoking perturbs gut–microbial metabolism, amplifying neuroinflammation and neurodegeneration in MS.
To date, no study has examined whether smoking alters microbiota-linked metabolites in PwMS or if such changes might mediate its impact on MS severity. We hypothesized that smoking and recent nicotine exposure would perturb tryptophan and bile acid metabolism (potentially reflecting disruption of host–microbiome co-metabolic pathways) and that smoking-associated metabolic alterations would be associated with greater MS severity in a biologically concordant direction. We further posited that smoking-related shifts in gut–microbial composition may contribute to these perturbations and explored two nonexclusive mechanisms: (1) a pathogenic strain expansion model, in which smoking would increase pro-inflammatory taxa and promote depletion of neuroprotective metabolites and (2) a loss-of-beneficial strain model, in which smoking would reduce protective commensal taxa, leading to diminished anti-inflammatory metabolites and potential expansion of pathogenic strains.
To evaluate this mechanistic framework, we leveraged serum metabolomic profiles of bile acid, tryptophan, and tobacco-related metabolites from four independent MS cohorts. Our objectives were to (1) identify tryptophan and bile acid metabolites associated with current smoking or recent nicotine exposure using a discovery-replication framework; (2) assess whether smoking-associated metabolites statistically mediate portions of the smoking-MS severity association; and (3) in a subset of samples, explore whether smoking is associated with altered gut–microbial strains and if these shifts align with smoking-perturbed metabolites in PwMS.
Materials and methods
Ethical approval
This study was approved by institutional review boards at the University of Miami (#20240098), Case Western Reserve University (#STUDY20210045), Mayo Clinic (#21-003944), and Brigham and Women’s Hospital (BWH; #2017P001169). Participants provided written informed consent.
Study design
This cross-sectional study included non-Hispanic White PwMS from three biorepositories divided into four cohorts based on clinical characteristics. PwMS were aged ⩾18 years at clinical onset and met the 2001, 2010, or 2017 McDonald diagnostic criteria.14–16 Inclusion/exclusion criteria, diagnostic definitions, and sample processing protocols are provided in eMethods. For Objective 1, the two early-stage (⩽5 years from onset and ⩽2 years from diagnosis), untreated (disease-modifying therapy naïve or untreated ⩾3 months) relapsing remitting MS (RRMS) cohorts (Accelerated Cure Project for MS (ACP) + Mayo Clinic Center for MS and Autoimmune Neurology (CMSAN1)) comprised the discovery sample. Replication was performed in two more heterogeneous cohorts (CMSAN2 + BWH), which included RRMS and progressive MS cases with variable disease duration. For Objective 2, all cohorts were included in mixed-effects mediation analyses. For Objective 3, exploratory metagenomic analyses were conducted in the BWH cohort with gut–microbiome data.
Metabolomic profiling
Bile acid, tryptophan-pathway, and tobacco-related serum metabolites were quantified using an untargeted LC-MS metabolomics platform (eMethods). Metabolomic data underwent cohort-specific quality control and inverse rank-normalization (mean = 0, SD = 1). Metabolites with high missingness were excluded, and remaining missing values were imputed using a minimum-value approach (eMethods). Fifty-one of 54 bile acid and 35 tryptophan metabolites were retained in ACP; the same 51 bile acids and 34 tryptophan metabolites in CMSAN; and 42 bile acids and 34 tryptophan metabolites in BWH. Six tobacco/nicotine-related metabolites were retained in ACP, four in CMSAN, and three in BWH; cotinine and hydroxycotinine were present across cohorts (eFigures 1A–D).
Smoking status, nicotine exposure, MS severity, and covariates
Smoking status was harmonized as never, former, or current smoker (eMethods). We defined participants as recently nicotine-exposed or unexposed based on detectable cotinine levels, evident in a few non-active smokers (eFigures 1A–D; eFigure 2). Age, sex, body mass index (BMI), disease duration (from clinical onset), and disease course (RRMS, secondary progressive (SP), primary progressive (PP)) were available. Expanded Disability Status Scale (EDSS) was available except for 42 ACP and 4 CMSAN2 participants; EDSS and age were used to construct the global age-related MS severity score (gARMSS) (eMethods). 17
Gut–microbiome metagenomics
Stool samples for the BWH cohort were processed as previously described. 3 DNA was extracted from 200 mg of stool using Qiagen DNAeasy PowerLyzer kits. The V4V5 region of 16S rRNA was amplified using Earth Microbiome Project primers and sequenced on an Illumina MiSeq platform. Bioinformatics processing used QIIME2, DADA2 denoising, and SILVA taxonomic classification (v138-99-515-806). Species in <10% of samples were excluded.
Statistical analysis
Smoking-associated tryptophan and bile acid metabolites (Objective 1)
Discovery analyses (ACP + CMSAN1) used multivariable linear regression: each metabolite was the dependent variable and smoking status (never smoker = reference) as the independent variable, adjusting for age, sex, BMI, and cohort. Parallel analyses were performed for nicotine exposure as the independent variable. Significant discovery associations were defined by two-sided p < 0.05 and false discovery rate (FDR; Benjamini–Hochberg method) q < 0.20 to balance FDR control with sensitivity to detect biologically meaningful signals. Discovery-significant metabolites were tested for replication in CMSAN2 + BWH cohorts using similar models, and statistical significance was a two-sided p < 0.05 and FDR q < 0.20. To obtain final effect estimates for replicating associations, a mixed-effects linear regression was conducted across cohorts, adjusting for age, sex, and BMI, with cohort as a random effect. Final models were repeated with a former smoker as the reference to contrast with the final associations, and sensitivity analyses including disease duration and disease course were conducted. Heterogeneity was assessed using I2 statistics.
Mediation of smoking-severity association (Objective 2)
Metabolites associated with current smoking or nicotine exposure (Objective 1) were evaluated as potential mediators of the relationship between smoking and MS severity, measured by normalized gARMSS (mean = 0, SD = 1; N = 220). A sequence of mixed-effects linear regression models adjusted for age, sex, BMI, disease duration, and disease course, with cohort as a random effect, was performed: Path A (smoking → metabolite), Path B (metabolite → MS severity—smoking), Path C (smoking → MS severity), and Path C’(smoking → MS severity—metabolite) (eMethods; eFigure 3). The average causal mediation effect (ACME) of smoking on MS severity via the metabolite was estimated using the product-of-coefficients approach with SEs derived via the delta method. Given the directional hypotheses, one-sided 95% confidence intervals and p values, and FDR q, were reported for ACME. Because gARMSS in younger or earlier-stage cases may partly reflect relapse-related disability rather than accrued severity, we performed sensitivity analyses restricted to ages ⩾40 or ⩾50 years and with disease duration ⩾10 years for metabolites demonstrating statistical mediation.
Exploring smoking, gut–microbial strains, and metabolite correlates (Objective 3)
Microbial taxa associated with smoking status (current vs non-smoker) were identified in the BWH cohort using Multivariable Association Discovery in Population-scale Meta-omics Studies (MaAsLin2), 18 a linear modeling framework that evaluated associations between microbial features and exposures, adjusting for sex, age, and BMI. MaAsLin2 used a generalized linear approach that accounts for zero inflation and relative abundance compositionality and applies FDR control.
Microbe–metabolite association modeling was performed using MMvec (microbe–metabolite vectors), a neural network framework for estimating microbe–metabolite interactions through their inferred conditional co-occurrence probabilities, following standard methods. 19 MMvec uses a multinomial neural embedding approach to model conditional co-occurrence probabilities rather than pairwise correlations. Because MMvec does not generate p values, standard errors, or FDR-corrected significance, associations were interpreted based on the highest-magnitude conditional probabilities (i.e. strongest inferred co-occurrence patterns) learned by the model, consistent with established practice.
Targeted microbe–metabolite pairwise correlations were then performed using Spearman’s rank correlation to explore relationships between smoking-associated taxa and mediating metabolites from Objective 2, in current smokers and non-smokers. The small number of current smokers precluded reliable MMvec modeling. An uncorrected two-sided α = 0.05 guided interpretation.
Results
Study population
The sample included 266 PwMS (Table 1). Objective 1 discovery cohorts (ACP + CMSAN1) included 146 early-stage, untreated RRMS cases with the mean age of 39 years (SDs: 10–11.5), 70%–80% female, and a median disease duration ⩽1.1 years. In ACP, 44% were ever smokers and 37% nicotine exposed, versus 27% and 18% in CMSAN1. Replication cohorts (CMSAN2 + BWH) included 140 older PwMS (mean: 55 years, SDs: 8–11.5) with longer disease duration (median: 8–19 years). CMSAN2 was composed of roughly one-third RRMS, SPMS, and PPMS, whereas BWH included 50% RRMS, 43% SPMS, and 7% PPMS. Females comprised 50%–73%. Ever smoking and nicotine exposure were present in 30% and 18% of CMSAN2 and 34% and 9% of BWH, respectively. Mean gARMSS ranged from 3.0 to 4.7 (SDs: 2.1–2.5) across cohorts (eFigure 4).
Study sample attributes (all cases were non-Hispanic White).
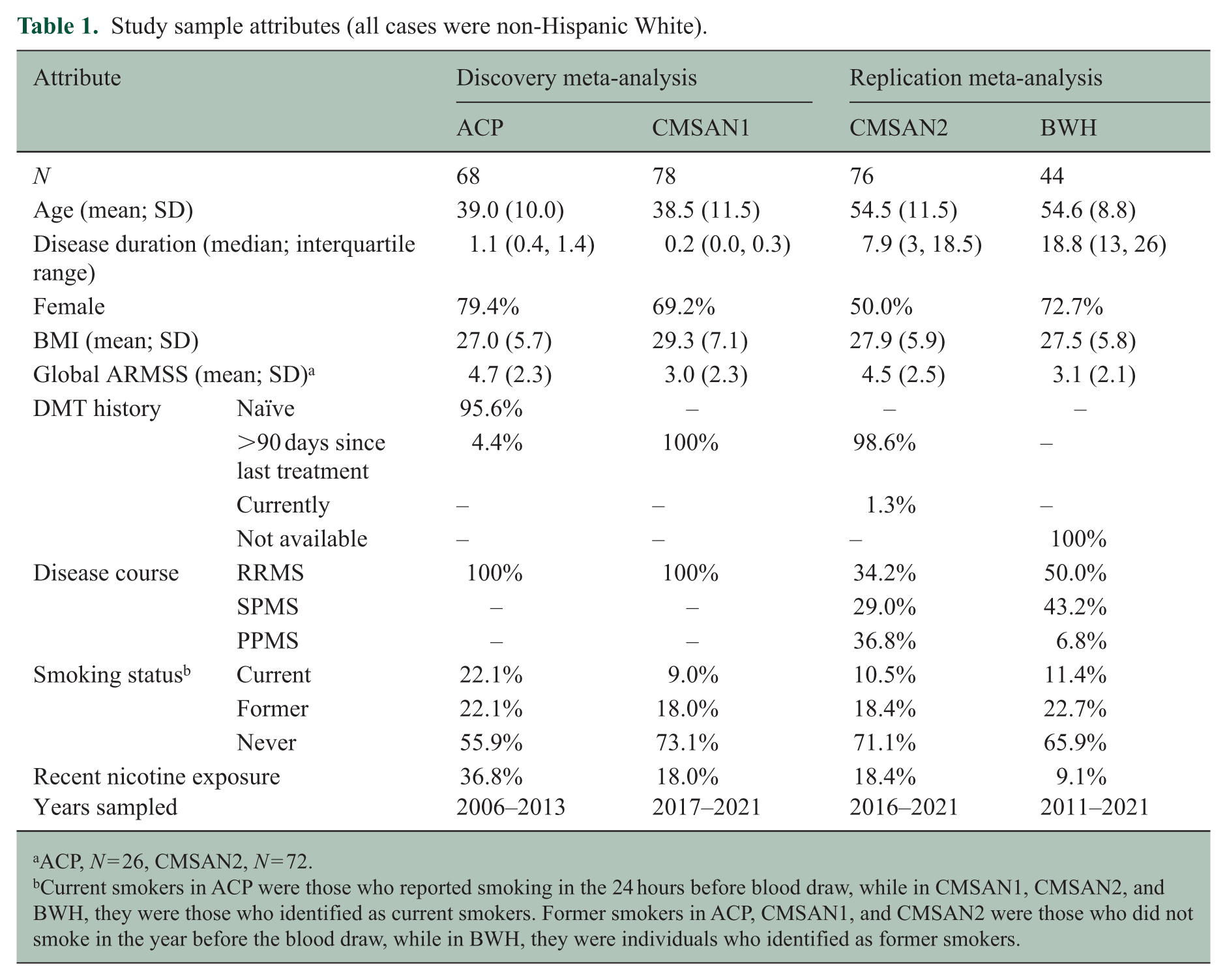
ACP, N = 26, CMSAN2, N = 72.
Current smokers in ACP were those who reported smoking in the 24 hours before blood draw, while in CMSAN1, CMSAN2, and BWH, they were those who identified as current smokers. Former smokers in ACP, CMSAN1, and CMSAN2 were those who did not smoke in the year before the blood draw, while in BWH, they were individuals who identified as former smokers.
Metabolites associated with current smoking
Of 86 metabolites tested in discovery analyses, 15 bile acid and 5 tryptophan metabolites were significantly (two-sided p < 0.05, FDR q < 0.2) altered in current versus never smokers, each with effect sizes ranging from 0.4 to 0.74 SDs (Table 2; eTable 1). None of these differed between former and never smokers (eTable 1). Nineteen were available for replication, and six replicated (two-sided p < 0.05, FDR q < 0.2) (Table 2). In the pooled analyses, current smokers had 0.5–0.7 SDs lower levels of one primary bile acid (tauro-β-muricholate spectral analog), two secondary bile acids (glycolithocholate, taurolithocholate), and two tryptophan metabolites (anthranilate, IPA). One bile acid intermediate (3beta-hydroxy-5-cholestenoate) was elevated by 0.7 SD in current versus never smokers. All replicating metabolites were lower in current versus former smokers (p < 0.05; eTable 2). Results were unchanged when adjusting for disease duration and disease course (eTable 3). Heterogeneity was low (I2 < 0.15). Boxplots of replicating metabolites are shown in Figures 1(a)–(d) and 2(a) and (b).
Significant discovery associations for current smokers versus never smokers for 86 metabolites, their associations in the replication analysis, and the combined meta-analytic associations for replicating metabolites. a
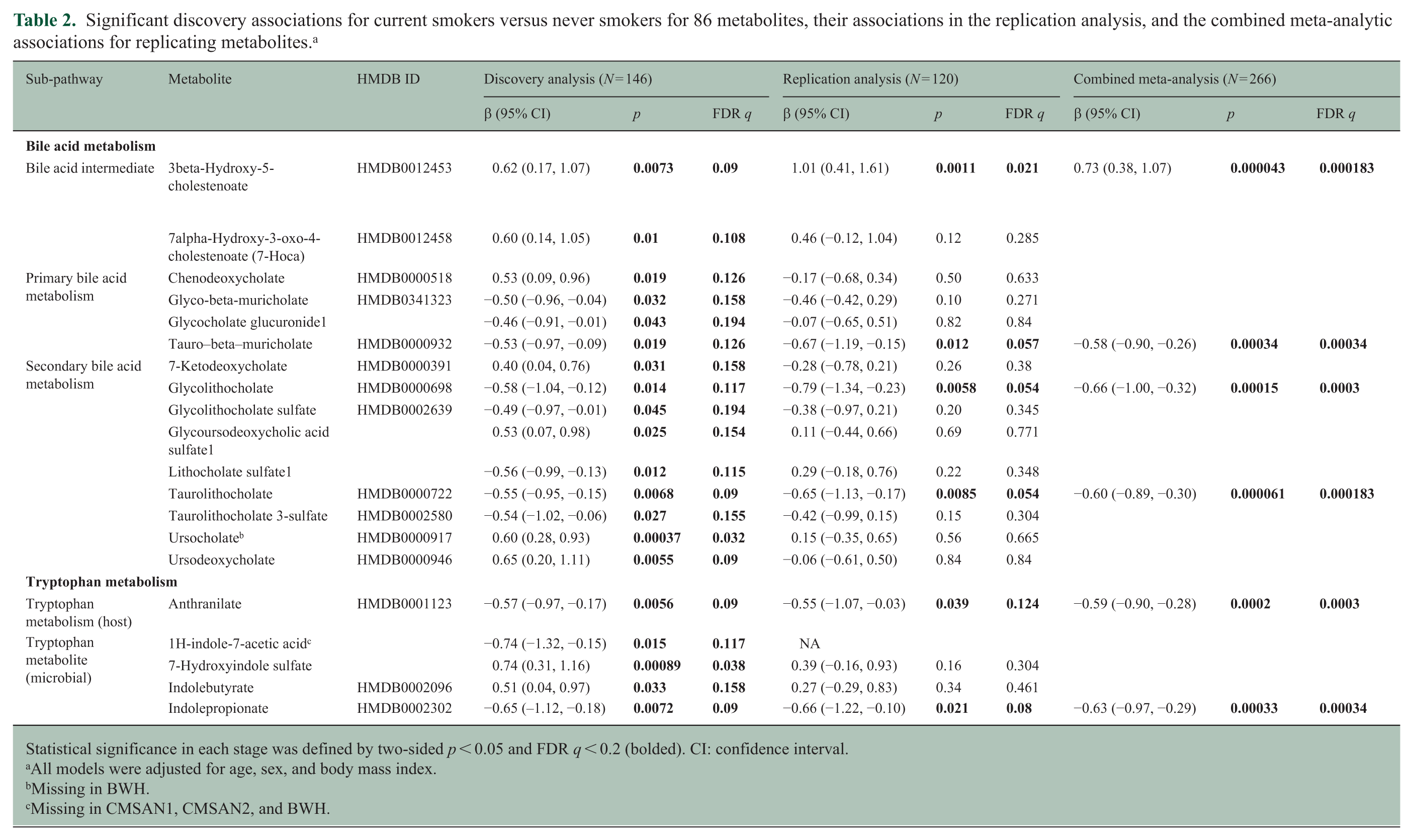
Statistical significance in each stage was defined by two-sided p < 0.05 and FDR q < 0.2 (bolded). CI: confidence interval.
All models were adjusted for age, sex, and body mass index.
Missing in BWH.
Missing in CMSAN1, CMSAN2, and BWH.
Significant discovery associations for recent nicotine exposure for 86 metabolites, their associations in the replication analysis, and the combined meta-analytic associations for replicating metabolites.
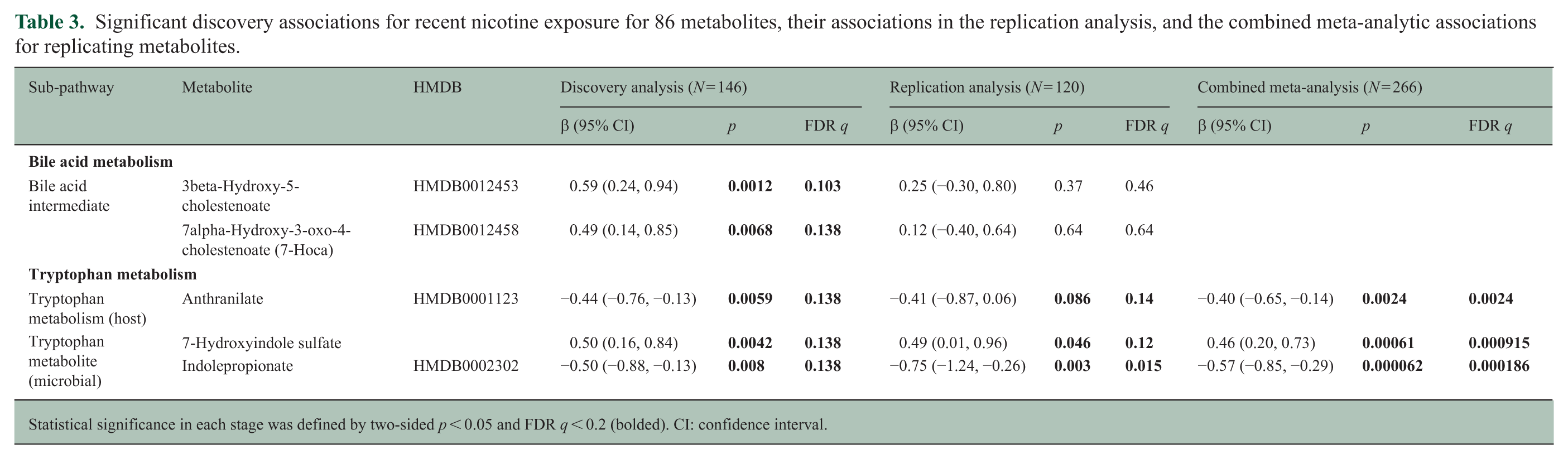
Statistical significance in each stage was defined by two-sided p < 0.05 and FDR q < 0.2 (bolded). CI: confidence interval.
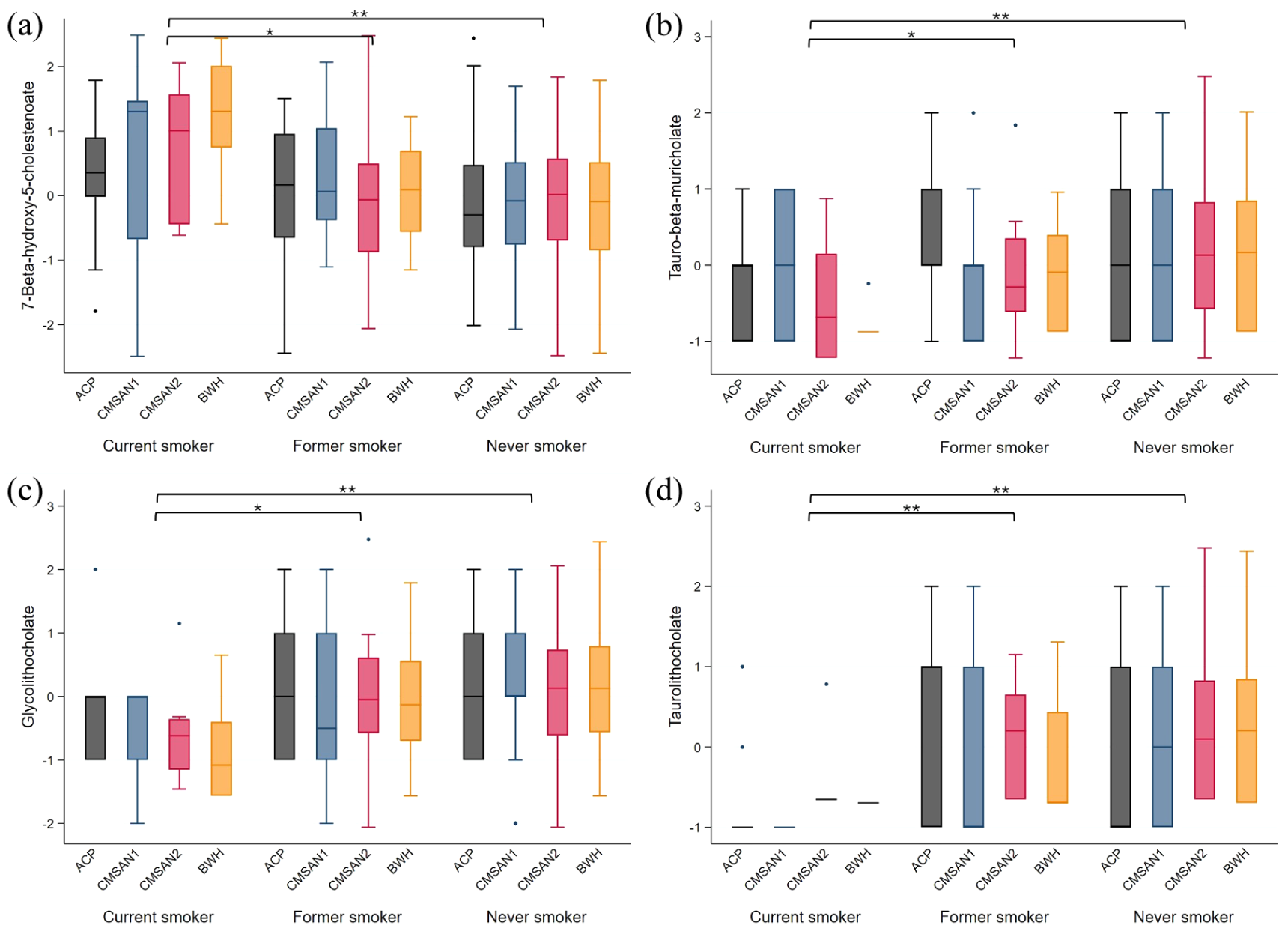
Distribution of bile acid metabolites with replicating associations (within each cohort mean = 0, standard deviation = 1).
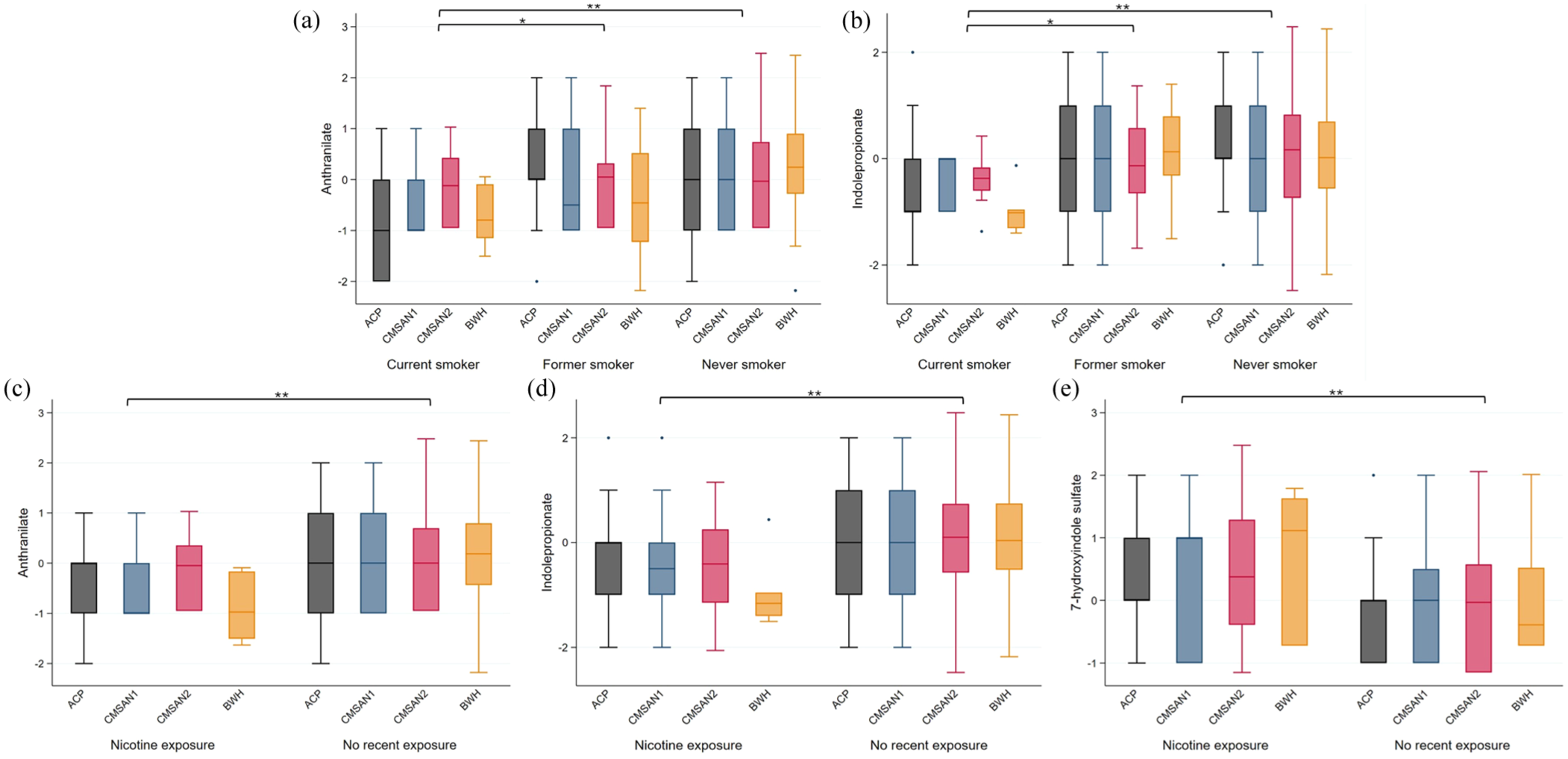
Distribution of tryptophan metabolites with replicating associations (within each cohort mean = 0, standard deviation = 1).
Metabolites associated with recent nicotine exposure
In total, 5 of 86 metabolites were significantly associated with nicotine exposure in discovery analyses (Table 3; eTable 1), all overlapping with current smoker discovery findings (Table 2). Overall, recent nicotine exposure results aligned with those for current smokers (eTable 1; eFigure 5). Three tryptophan metabolite associations replicated. In pooled analyses, nicotine-exposed PwMS had 0.4 and 0.57 SD lower levels of anthranilate and IPA and 0.46 SD higher levels of 7-hydroxyindole sulfate compared with nonexposed PwMS (Table 3; Figure 2C–E). Results were unchanged when adjusting for disease duration and disease course (eTable 3). Heterogeneity in final models was low (I2 < 0.15).
Mediation results of smoking-MS severity association
Replicated smoking- and nicotine–metabolite associations persisted in the gARMSS subset (N = 220; Table 4: Path A). IPA was inversely associated with gARMSS when adjusting for smoking or nicotine exposure (p < 0.05, FDR q < 0.2; Path B), and both exposures were associated with greater MS severity (p < 0.05; Path C). ACME for IPA was significant (p < 0.05, FDR q < 0.2) in both smoking models, suggesting IPA depletion statistically mediated ~20% of the smoking-MS severity association, although the proportion-mediated estimates were imprecise (SDs: 11%–15%). Sensitivity analyses were directionally consistent (eTable 4).
Causal mediation models examining indirect effects of current smoking or nicotine exposure on MS severity (gARMSS) via tryptophan and bile acid metabolites (N = 220).
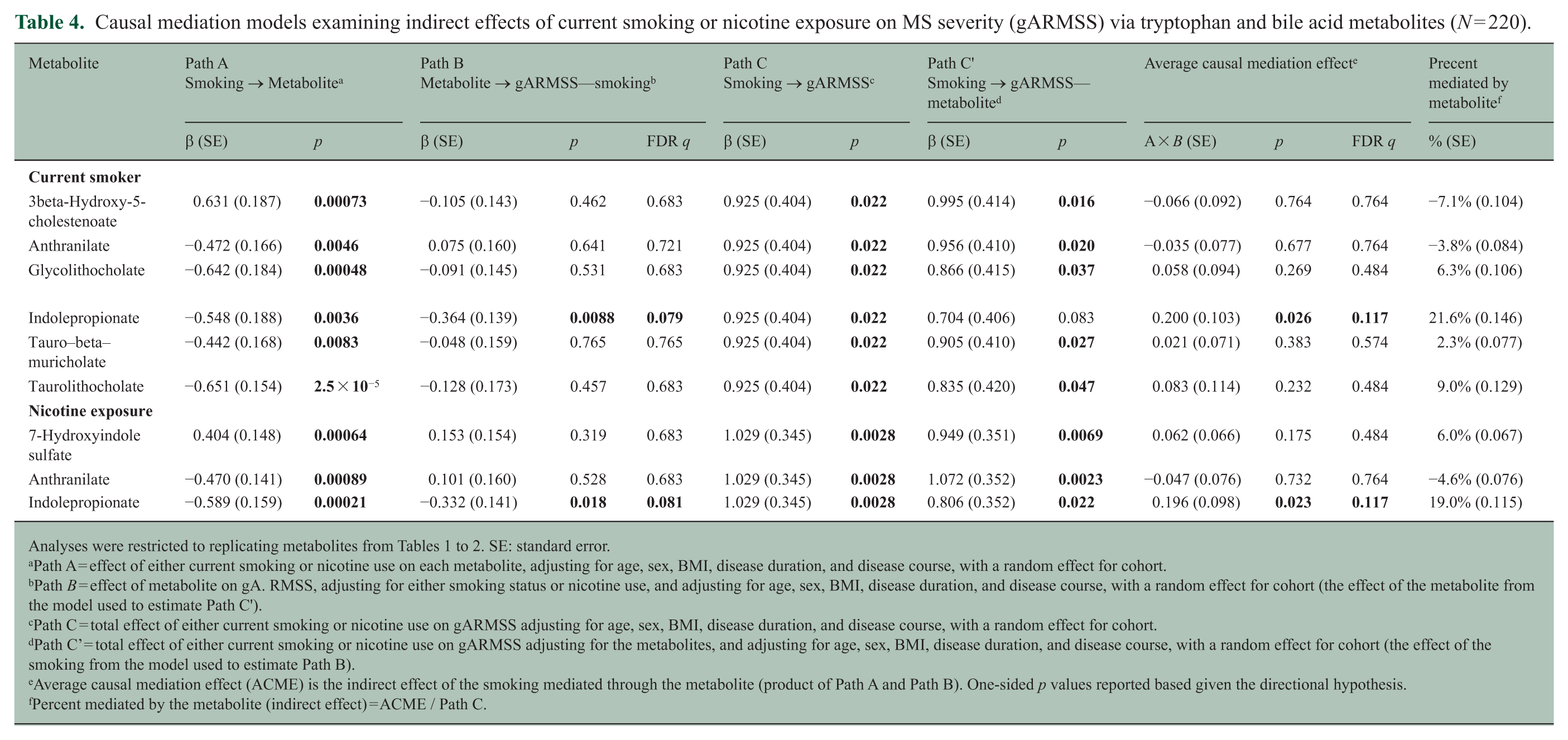
Path A = effect of either current smoking or nicotine use on each metabolite, adjusting for age, sex, BMI, disease duration, and disease course, with a random effect for cohort.
Path B = effect of metabolite on gA. RMSS, adjusting for either smoking status or nicotine use, and adjusting for age, sex, BMI, disease duration, and disease course, with a random effect for cohort (the effect of the metabolite from the model used to estimate Path C’).
Path C = total effect of either current smoking or nicotine use on gARMSS adjusting for age, sex, BMI, disease duration, and disease course, with a random effect for cohort.
Path C’ = total effect of either current smoking or nicotine use on gARMSS adjusting for the metabolites, and adjusting for age, sex, BMI, disease duration, and disease course, with a random effect for cohort (the effect of the smoking from the model used to estimate Path B).
Average causal mediation effect (ACME) is the indirect effect of the smoking mediated through the metabolite (product of Path A and Path B). One-sided p values reported based given the directional hypothesis.
Percent mediated by the metabolite (indirect effect) = ACME / Path C.
Exploring smoking, gut–microbial strains, and metabolite correlates
Among 60 BWH participants with stool metagenomic data, 10 of 440 taxa were associated with smoking status in the first exploratory analysis (p < 0.05; Figure 3, eTable5). Three notable strains were more abundant in smokers versus non-smokers: Lachnoclostridium, a genus that contains a pro-inflammatory strain recently implicated as a driver of MS risk (the strongest association), 20 Ruminococcaceae UBA1819, a taxon elevated in PwMS and correlated with MS severity,21,22 and Clostridia UCG-014, a strain observed to be expanded in MS oropharyngeal samples. 23 Conversely, Agathobacter species, a key butyrate-producing, anti-inflammatory genus, was significantly depleted in MS smokers. 24

Microbiota associations with current smoking versus non-smoking in MS. (a) Heatmap showing MaAsLin2 coefficients for significant associations between gut bacterial taxa and smoking status in MS adjusting for age and sex. Color scale: red indicating positive association and blue indicating negative association. Asterisks indicate statistical significance: ***p < 0.001, **p < 0.01, *p < 0.05.
In the second exploratory analysis, we observed coherent microbe–metabolite conditional co-occurrence patterns (Figure 4). SCFA-related metabolites had positive associations with Monoglobus, Colidextribacter, and Oscillospiraceae taxa. Indole derivatives such as IPA and tryptophan betaine displayed uniformly positive associations across many taxa, whereas kynurenine pathway metabolites (e.g. quinolinate, xanthurenate, and kynurenate) had predominantly negative associations, particularly with Barnesiella. Bile acid associations were divergent: glycocholate and taurocholate were positively associated with Oscillospiraceae and Monoglobus, while Barnesiella exhibited consistently negative associations with several tauro-conjugated bile acids and anthranilate.
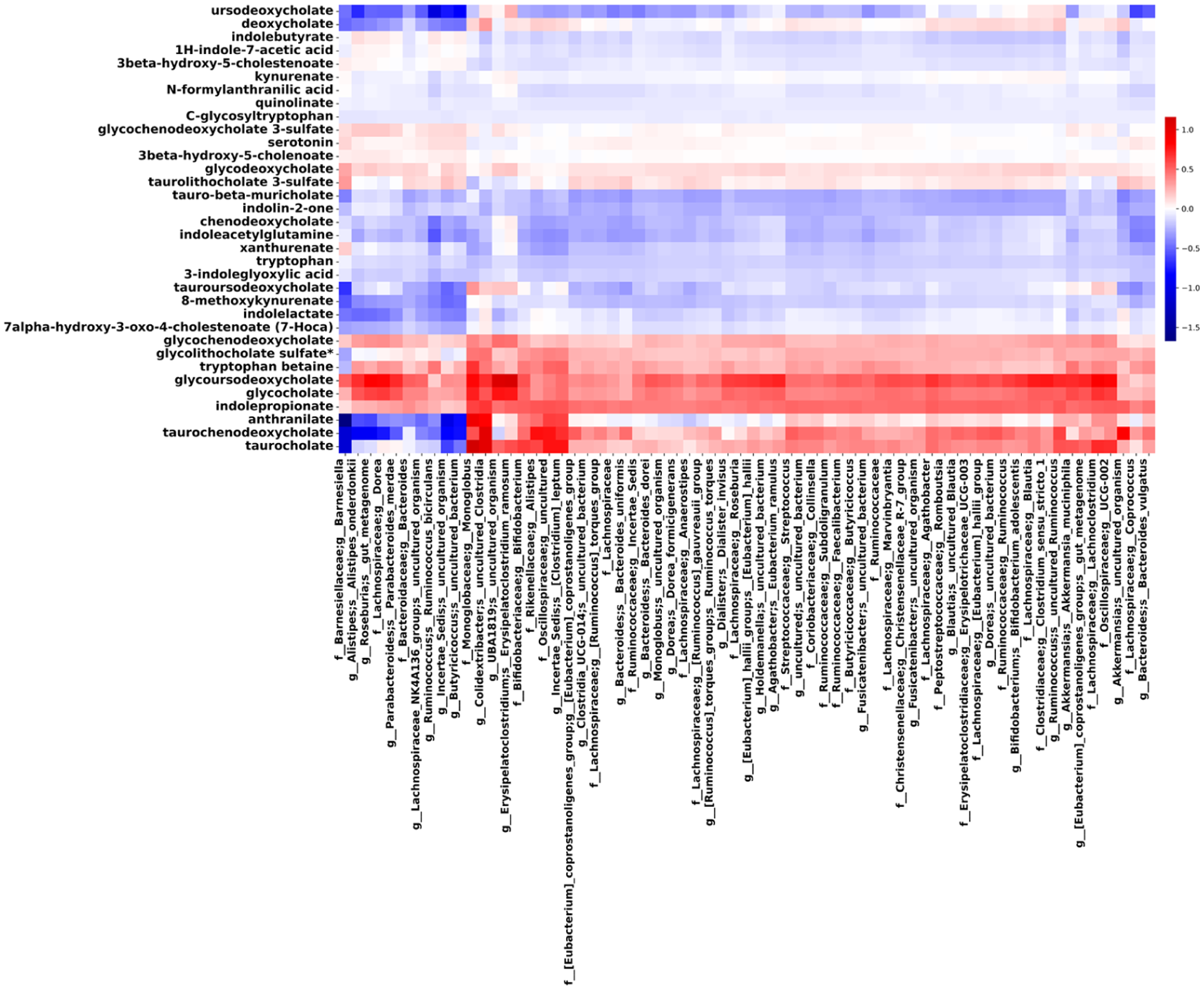
Microbe–metabolite co-occurrence patterns inferred by MMvec across the BWH cohort. Heatmap displays inferred conditional co-occurrence scores between gut–microbial taxa and serum metabolites associated with smoking status. Warmer colors indicate stronger positive co-occurrence, and cooler colors indicate weaker or negative co-occurrence patterns. MMvec estimates co-occurrence probabilities rather than statistical significance; values reflect relative association strengths across taxa–metabolite pairs.
In the final exploratory analysis, among the 10 smoking-associated taxa, Lachnoclostridium was inversely correlated with IPA in current smokers (N = 5; ρ = −0.89, p = 0.041), whereas no such relationship was observed in non-smokers (N = 56; ρ = 0.11, p = 0.40).
Discussion
In this multi-cohort study of PwMS, we demonstrate that tobacco smoke exposure (current smoking and nicotine exposure) disrupts two interconnected host–microbiome co-metabolic pathways: tryptophan and bile acid metabolism. The most biologically coherent finding was the depletion of IPA (indolepropionic acid; IPA), a gut–microbial tryptophan metabolite with neuroprotective and anti-inflammatory properties, which mediated a meaningful proportion of smoking’s adverse effect on MS severity. Additional reductions in other tryptophan and bile acid metabolites point to broader smoking-related perturbations in host and host–microbiome metabolic interactions—the latter supported by exploratory enrichment of MS-associated microbial taxa in smokers with MS
Tryptophan metabolism and IPA (primary discovery)
Tryptophan metabolism has been linked to multiple aspects of MS presentation through host and microbial mechanisms.5,25,26 Tryptophan gives rise to bioactive microbial metabolites, including IPA, which regulates immune responses and CD4+ T-cell metabolic programming through receptors such as AHR. 8 Our findings extend these observations, suggesting smoking disrupts tryptophan metabolism and reduces IPA levels. In a randomized trial of healthy older adults, probiotic supplementation with Bifidobacterium bifidum and B. longum increased circulating IPA, which correlated with brain-derived neurotrophic factor (BDNF); mechanistic analyses showed IPA reduced microglial inflammation and promoted BDNF and nerve growth factor expression via microglia–neuron interactions. 27 Beyond neuroprotection, IPA reprograms CD4+ T-cell metabolism to inhibit pro-inflammatory Th1/Th17 differentiation. 8 In pediatric MS, IPA was inversely associated with disease severity,5 consistent with our findings: IPA was depleted in smokers, inversely associated with MS severity, and statistically mediated 21.6% (SE 14.6%) of smoking’s association with gARMSS.
Exploratory metagenomic analyses further supported this framework. Smoking was associated with shifts in several taxa, and IPA broadly co-occurred within microbial–metabolite networks dominated by non-smokers. Among smoking-associated taxa, exploratory analyses suggested an inverse correlation between Lachnoclostridium, a genus containing a recently identified pro-inflammatory MS-risk strain, 20 and IPA in smokers but not non-smokers with MS. Although based on small numbers, these findings raise the possibility that smoking destabilizes microbial networks contributing to IPA production.
Nicotine exposure was also associated with higher circulating 7-hydroxyindole sulfate, a sulfated derivative of microbially produced indoles whose structural analogs exert pro-oxidative and pro-inflammatory effects. 28 Both current smokers and nicotine-exposed PwMS had reduced anthranilate, a kynurenine pathway metabolite linked to host–microbial metabolism. Because cigarette smoke contains AHR-activating ligands, chronic exposure may intersect with tryptophan-derived AHR signaling pathways. Together, these findings indicate that smoking perturbs multiple tryptophan-related host–microbial processes. Preclinical studies demonstrating that tryptophan supplementation reduces neuroinflammation in experimental MS and that IPA administration attenuates inflammation in murine colitis,8,29 provide context; however, clinical implications of modulating microbial-derived tryptophan metabolites in PwMS warrant rigorous longitudinal and interventional investigation.
Bile acid metabolism findings
Tobacco smoke and nicotine disrupt microbial communities and bile acid metabolism in non-MS populations.11–13 We extend these observations to MS, demonstrating current smokers have lower serum levels of several primary and secondary bile acids, including glycolithocholate, taurolithocholate, and a human spectral analog of tauro-β-muricholate. Exploratory metagenomic analyses identified shifts in taxa with biliary roles: Ruminococcus gauvreauii and Christensenellaceae species were depleted while MS-risk-associated Lachnoclostridium species were elevated in MS smokers.20,30,31 These findings align with a prior study that reported reduced secondary bile acids in pediatric and adult PwMS. 6 Notably, longitudinal work has demonstrated that bile acid metabolites predict MS progression, and tauroursodeoxycholate (a secondary bile acid) supplementation was associated with shifts in gut–microbial composition and decreases in central memory CD4+ and Th1/17 cells, while CD4+ naïve cells increased compared with placebo. 32 These recent findings underscore the relevance of bile acid–microbiome interactions in MS disease trajectory. Whereas prior studies have focused on progression broadly, our findings suggest tobacco exposure may represent one potentially modifiable factor influencing this bile acid axis.
Other findings
In general, the exploratory analyses suggest additional dysbiosis patterns in MS smokers. Agathobacter, a butyrate-producing genus with anti-inflammatory, neuroprotective, and microglia-modulating effects, was the most depleted. 24 Conversely, smoking-enriched Ruminococcus torques (associated with leptomeningeal enhancement and greater disability in MS), 3 Clostridia UCG-014 (implicated in immunotherapy toxicity 33 and increased in progressive MS oropharynx), 23 and Eubacterium coprostanoligenes (cholesterol-reducing anaerobe), 34 suggesting smoking may promote strains associated with mucosal disruption and neuroinflammation.
Strengths and limitations
Key strengths include our multi-cohort discovery-replication design spanning early RRMS to progressive cases, with adjustment for cohort heterogeneity, including higher smoking prevalence in ACP likely due to an earlier recruitment period and broader geographic recruitment, thereby enhancing external validity. Associations were consistent after adjustment for disease duration and MS course, supporting robustness across clinical phenotypes. We employed complementary exposure definitions (self-reported smoking and biochemical nicotine exposure), with strong concordance validating assessment (eFigure 1). Availability of gARMSS and metagenomic data enabled exploratory mediation and mechanistic analyses. Because gARMSS in younger or earlier-stage PwMS may partly capture relapse-related disability rather than accrued severity, sensitivity analyses in older participants and PwMS with longer disease duration were directionally consistent, particularly the inverse relationship between indolepropionate and gARMSS. Limitations include the following: cross-sectional design precluding causal inference, although mediation analyses were conducted within a statistical framework and should be interpreted cautiously; despite covariate adjustment, residual confounding cannot be excluded (i.e. diet); participants were non-Hispanic White, limiting generalizability; cotinine does not distinguish nicotine sources (cigarettes, e-cigarettes, nicotine replacement, secondhand smoke); and hypothesis-generating microbiome findings were based on small numbers (e.g. IPA microbiome correlation was in five current smokers), which are exploratory and require replication in independent cohorts.
Conclusion
This multi-cohort study demonstrates that tobacco smoking and nicotine exposure in PwMS disrupt bile acid and tryptophan metabolism, including depletion of IPA. Mediation analyses showed IPA depletion accounted for ~20% of smoking’s association with MS severity. Exploratory analyses suggest smoking promotes expansion of MS-associated taxa inversely correlated with IPA. Collectively, these findings provide mechanistic evidence linking tobacco exposure to adverse MS outcomes through host–microbiome co-metabolism.
Supplemental Material
sj-docx-1-msj-10.1177_13524585261454207 – Supplemental material for Tobacco smoking disrupts bile acid and tryptophan metabolism in multiple sclerosis
Supplemental material, sj-docx-1-msj-10.1177_13524585261454207 for Tobacco smoking disrupts bile acid and tryptophan metabolism in multiple sclerosis by Farren BS Briggs, Jevin Litwiler, Federico Montini, Mahboubeh Fereidan Esfahani, Jessica Sagen, Jacob L McCauley, Flavia Nelson, Simon Gregory, Roberta Brambilla, Erika S Trapl, Jessica N Cooke Bailey, Luke A Schwerdtfeger, Laura Cox, Howard Weiner and W Oliver Tobin in Multiple Sclerosis Journal
Supplemental Material
sj-docx-2-msj-10.1177_13524585261454207 – Supplemental material for Tobacco smoking disrupts bile acid and tryptophan metabolism in multiple sclerosis
Supplemental material, sj-docx-2-msj-10.1177_13524585261454207 for Tobacco smoking disrupts bile acid and tryptophan metabolism in multiple sclerosis by Farren BS Briggs, Jevin Litwiler, Federico Montini, Mahboubeh Fereidan Esfahani, Jessica Sagen, Jacob L McCauley, Flavia Nelson, Simon Gregory, Roberta Brambilla, Erika S Trapl, Jessica N Cooke Bailey, Luke A Schwerdtfeger, Laura Cox, Howard Weiner and W Oliver Tobin in Multiple Sclerosis Journal
Supplemental Material
sj-xlsx-1-msj-10.1177_13524585261454207 – Supplemental material for Tobacco smoking disrupts bile acid and tryptophan metabolism in multiple sclerosis
Supplemental material, sj-xlsx-1-msj-10.1177_13524585261454207 for Tobacco smoking disrupts bile acid and tryptophan metabolism in multiple sclerosis by Farren BS Briggs, Jevin Litwiler, Federico Montini, Mahboubeh Fereidan Esfahani, Jessica Sagen, Jacob L McCauley, Flavia Nelson, Simon Gregory, Roberta Brambilla, Erika S Trapl, Jessica N Cooke Bailey, Luke A Schwerdtfeger, Laura Cox, Howard Weiner and W Oliver Tobin in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The author would like to thank Dr Kristy Miskimen and Dr Jonathan Haines for their invaluable contributions to ACP and CMSAN sample processing and management. They also appreciate the support of the Accelerated Cure Project for Multiple Sclerosis and the contributions of their participants.
Author Contributions
Acquisition of data: F.B., J.S., F.M., M.F.E., H.L.W., L.M.C., and W.O.T. Conception and design of the study: F.B., L.M.C., F.M., S.G., J.M., F.N., R.B., E.T., J.C.B., and L.S. Statistical analyses: F.B., L.J., F.M. Drafting of text and preparing figures: F.B., J.L., and F.M. Manuscript editing and interpretation: all authors.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: F.B., J.S., S.G., and W.O.T, and metabolomic profiling of the ACP and CMSAN samples were supported by the NIH/NINDS R01NS121928. HW and BWH metabolomic profiling were supported by the Water Cove Charitable Foundation, the Clara E. and John H. Ware Jr. Foundation, and NIH/NINDS R01NS087226-01A1. LMC was supported by NIH/NINDS R21NS126866 and the Nancy Davis Race to Erase MS Young Investigator Award.
Data Availability Statement
Data are available to qualified investigators through the University of Miami, the Mayo Clinic, and Brigham and Women’s Hospital.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.