
Abstract
Alzheimer's disease (AD) is a neurodegenerative disease with a long preclinical and prodromal stage near 20 years. The neuropathological hallmarks of AD include amyloid plaques, neurofibrillary tangles, and neuroinflammation, those lead to neuronal and synaptic loss. Important fact, oxidative stress participates in the AD development by promoting amyloid-β deposition, tau hyperphosphorylation. However, the inflammatory response and pyroptotic death are mediated by the aberrant expression of NLRP inflammasome activated caspase-1, which leads to cleavage pro-inflammatory cytokines such as pro-interleukin-1β and pro-IL-18. IL-1β, TNF-α, and IL-6 which amplify the neuroinflammation loop, are produce by activated microglia and astrocytes, that can serve as early diagnostic markers or therapeutic targets in AD. In this review, we summarize our current understanding of the role of inflammasome in the pathogenesis of AD, highlighting key issues that need to be addressed to improve the development of new therapies.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder with an insidious onset. It is the most common form of dementia globally, affecting about 36 million individuals out of the total 55 million people worldwide who live with all forms of dementia. 1 Originally defined as a clinicopathological entity, the disease usually affects people over 65 age year and leads to disorders in the language, memory, comprehension, attention, judgement, and reasoning. 2
On November 3, 1906, at the 37th meeting of psychiatrists from southwestern Germany in Tubingen, psychiatrist and neuropathologist Alois Alzheimer reported “a particular severe pathological process of the cerebral cortex” in a 55-year-old woman with severe dementia. He was seeing a brand-new damage known as a neurofibrillary tangles (NFTs), with distinctive plaques. 3 AD was initially only referred to as affecting those with early dementia (pre-senile). Later, the definition of the word was broadened to encompass all dementia patients in whom plaques and NFTs could be seen. Histologically, extracellular senile plaques of amyloid-β (Aβ) and intracellular neurofibrillary tangles (iNFT) of hyperphosphorylated tau characterize AD.4–6
According to evidence, senile plaque deposits are not directly linked to the progression of cognitive decline, while the accumulation of tau appears to be more closely linked to it.7,8 Several studies have indicated a positive correlation between the quantity of tau aggregates, severity of cognitive impairment and symptoms in AD.9,10
Aβ and tau proteins are the specific biomarkers used for AD diagnosis, and they are becoming increasingly important in refining the differential diagnosis by enabling in vivo detection and quantification. 11 Biomarker analysis through MRI fails to differentiate between different types of dementia and cannot maintain the level of accuracy achieved by amyloid biomarkers in AD patients. 12 Although to different extents, individuals with tau-related dementia in the aging brain serve as notable instances of a distinct primary age-related tauopathy, which is commonly observed, there is substantial evidence to support the hypothesis that this condition emerges from a mechanism unrelated to Aβ, possibly involving a mechanical lesion. 13
Histological postmortem analyses is the diagnostic gold standard of AD patients currently confirm a diagnosis in favor of AD only in 83% of cases. 14 Brain inflammation is a neuropathological process associated with AD, evidence suggest that neuroinflammation plays a key role in AD pathogenesis.15,16 The activation of major glial inflammatory cells, such as microglia and astrocytes, can be caused by increased levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in an oxidative stress state. The neuroinflammatory response is positively influenced by the release of proinflammatory factors by these glial cells. 17
Alternatively, the blood-brain barrier (BBB) can be tampered with by factors released from the central nervous system (CNS) and peripheral cytokines, enabling lymphocytes like T cells to enter the CNS and contribute to the positive feedback of neuroinflammation. 18 In AD, the resident microglia population of the CNS primarily drives neuroinflammation. while it is acknowledged that microglia can have beneficial and restorative roles in AD by eliminating Aβ deposits through phagocytosis, the buildup of Aβ can also stimulate microglial cells, leading to their activation and the production of inflammatory mediators. 19
Additionally, as Aβ accumulates, microglial cells may experience a gradual decline in their capacity to phagocytose Aβ. 20 Furthermore, a state of microglial inflammation could also have repercussions on CNS resident cells, leading to synaptic dysfunction and neuronal damage. 21 The cellular participants in this neuroinflammatory response include microglia, astrocytes, and neurons, which, when activated, can generate high levels of inflammatory mediators, including pro-inflammatory cytokines and chemokines.22,23 Moreover, these cells have the capacity to produce prostaglandins, leukotrienes, thromboxanes, coagulation factors, free radicals like ROS and nitric oxide (NO), complement factors, proteases, protease inhibitors, and C-reactive protein, all of which can exacerbate the inflammatory cascade. 24 Aβ itself has the ability to induce the expression of various pro-inflammatory cytokines, such as interleukin IL-1β, IL-6, tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) within glial cells. This process can initiate a self-perpetuating cycle of inflammation. 25 The inflammatory events that occur contribute to both synaptic and neuronal damage, thereby playing a role in the neurodegenerative process. 22 Therefore, the neuroinflammatory initiated by Aβ and perpetuated as a self-reinforcing cycle, can actively contribute to the AD progression. 26
One potential mechanism underlying the chronic neuroinflammatory response in the early stages of AD could be the excessive inflammasomes activation, inflammasomes are linked with inflammatory caspases 1, 4, 5, 11, and 12, and they orchestrating certain programmed cell death pathways. 27 Nucleotide binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing 1 (NLRP1) is recognized for its capability to assemble into an inflammasome complex and trigger caspase-1 following the degradation of its N-terminal segment by the proteasome in neurons. In contrast, NLRP3 serves as the primary member of the NLRP family within the brain, with a predominant expression pattern observed in microglia. 28 In brief, the evidence mentioned earlier suggests that Aβ activates the NLRP3 inflammasome and then contributes to the development of AD through IL-1β, IL-18, and other inflammatory cytokines. 29 Furthermore, there is evidence that Aβ42 can also lead to gasdermine D lysis by activating the NLRP3-caspase-1 pathway and causing neuronal cell pyroptosis. 30
In this review, we summarize our understanding of the neuroinflammation mechanisms in AD pathogenesis through inflammasome and oxidative stress involvement and highlight key issues that need to be addressed to improve the successful development of new therapies.
Alzheimer's disease pathophysiology
The fundamental pathological processes in AD encompass brain β-amyloidosis characterized by Aβ plaques, and neurofibrillary degeneration that leads to the accumulation of hyperphosphorylated tau fibrils within iNFT. Multiple forms of Aβ are derived by proteolytic cleavage from the type I cell-surface protein amyloid-β protein precursor (AβPP). The amyloid hypothesis broadly posits that excessive amounts of Aβ peptide in the brain (particularly Aβ42) are responsible for AD-related pathology, including amyloid plaques, NFTs, synapse loss, and eventual neuronal cell death. 31
In patients with AD, hyperphosphorylation of certain amino acids in tau proteins causes the proteins to dissociate from the microtubules, disturbing the transport structure and resulting in starvation of neurons and, ultimately, cell death. Hyperphosphorylated tau thus has an important role in intracellular neurofibrillary changes and the pathogenesis of AD and related tauopathies. The majority of the studies have focused on the entorhinal cortex, particularly the second layer of stellate neurons, which is susceptible to early neurofibrillary degeneration and experiences a more rapid loss of neurons compared to other brain regions.32,33
Additionally, the hippocampal formation, specifically sector 1 of the cornu Ammonis, is consistently identified as one of the areas most affected in these investigations. 34 The neuronal loss within the entorhinal cortex leads to the disruption of the perforant pathway, resulting in functional impairment of entorhinal-hippocampal connectivity. 10 This, in turn, contributes to the structural isolation of the hippocampus, particularly in the advanced stages of AD.35,36 If the damage is confined to synapses near the plaque and does not propagate along axons, it is possible that the harm will be limited to those specific synapses. However, if the affected synapses remain in their original positions, further spread of damage along the axon could occur due to continuous calcium (Ca2+) influx and subsequent dysfunction of mitochondria, which are highly mobile within neurons. 37
The hypothesis proposed here suggests that an efficient microglial response may help prevent this by removing damaged synapses. It is probable that not only the loss of synapses but also other residual effects, such as localized Aβ induced phosphorylation of tau, may persist in the affected areas of the AD brain. 38 Over time, as more plaques affect multiple points along an axon, such damage would build up, causing tau to dissociate, ultimately resulting in the destruction of the axon. Consequently, this would render all of its connected synapses non functional. 38 Ultimately, this represents unidirectional process, and as more axons are lost, network dysfunction becomes an inevitable consequence. Within this framework, the threshold of “plaque load” that an individual can tolerate without experiencing cognitive decline would be contingent on their genetic predisposition, particularly related to the efficiency of their microglial cells in phagocytosing damaged synapses.39,40
Neuroinflammation as a pathological event in Alzheimer's disease
The initial indications of a connection between inflammation and the development of AD emerged from several epidemiological studies. These studies reported a potential reduction of up to 50% in the risk of developing AD and lower rates of cognitive decline among individuals who regularly used chronic non-steroidal anti-inflammatory drugs (NSAIDs).41–44 Likewise, in mouse models of AD, the use of ibuprofen has been shown to inhibit neuroinflammation, modify the processing and accumulation of Aβ, and mitigate cognitive impairments. 45 Neuroinflammation begins many years prior to the clinical AD onset and stands as one of the earliest pathomechanistic changes along the entire AD continuum. Proteomic investigations reveal that a complex network of abnormal molecular pathways, initiated and sustained by molecules such as TNF-α, IL-1β, transforming growth factor-beta (TGF-β), and the triggering receptor expressed on myeloid cells 2 (TREM2), play a role in the process of neuroinflammation. 46 The significant involvement of inflammation in the pathophysiology of AD was hypothesized over two decades ago.47–49
Recent research has confirmed that this early, disease exacerbating inflammation within the CNS begins many years before the onset of severe cognitive decline or the AD manifestations.50,51 In line with this, various longitudinal studies have demonstrated that inflammation and microglial activation take place several years before the onset of AD.52,53 Additionally, there exists a robust connection between neuroinflammation and the accumulation of Aβ and tau proteins in the human brain.53,54 The activation of the peripheral immune system triggers a coordinated reaction from the CNS, central to this communication between the immune system and the brain is the role of glial cells, including microglia and astrocytes. These cells interpret and transmit inflammatory signals within the brain, ultimately influencing physiological and behavioral responses. 55 Activated microglia are a typical pathophysiological feature of AD. 19 There are two types of microglia cells present in the brain: “resting” or “quiescent” microglia and “active” microglia as shown in Figure 1.17,49,56 Two opposing activation phenotypes for microglia exist: the M1 or ‘proinflammatory’ phenotype (classically activated), which produces proinflammatory cytokines and NO and inhibits the release of neurotrophic factors, leading to an exacerbated inflammation and cytotoxicity. 56 The M2 microglia type, which is considered anti-inflammatory, demonstrates the production of anti-inflammatory cytokines, an increased level of neurotrophic factors, and other signals associated with downregulation. The M1/M2 dichotomy (or polarization scheme) remains controversial as new evidence suggests that there is a continuum between M1 and M2 phenotypes for microglia activation. Furthermore, the disturbance of microglial activity, particularly dystrophic microglia, could either initiate, worsen, or both the formation of aberrant protein aggregates in the brain.37,57

The role of microglia in AD progression. The clearance of Aβ mediated by microglia contributes to the homeostasis maintenance of CNS. However, with the AD progression, excessive activation of microglia would release excessive pro-inflammatory factors to compromise neurons and their synapses. From microglia in the neuroinflammatory pathogenesis of Alzheimer's disease and related therapeutic targets. Copyright © 2022. Cai et al. 19
The resting to active functional state of microglia in AD may be caused by inflammation, which could be influenced by factors like stress or depressive-like behavior. 58 The activation profile that has been observed aligns with an M1 profile characteristic of microglia and macrophages. Microglia induce the production of cytokines, including IL-1β, TNF-α, and IL-6, which play a role in propagating this immune-derived signal within the brain and mediating both physiological and behavioral responses.59,60 Induction of chemokines by activated microglia serves as another means of transmitting neuroinflammatory signals and could potentially represent a mechanism through which resident microglia communicate with peripheral immune cells.61–63
In AD brain inflammation is primarily associated with activated microglia, which has a dual role. On one hand, they help clear Aβ deposits, but on the other hand, they release neurotoxic molecules that can contribute to neurodegeneration.61,63 IL-1β, a key player in brain neuroinflammation, is elevated in the AD brains and may contribute to AD pathology by enhancing the expression of the APP gene, promoting tau hyperphosphorylation, and impairing memory (Figure 2). 64 Despite efforts, anti-inflammatory treatments have not yielded the anticipated benefits in AD patients. 64 This suggests that microglial inflammation might be a consequence of AD rather than its cause. Notably, deteriorating neurons are well known triggers of inflammatory responses in the brain, and the loss of synapses remains the most significant correlated marker of dementia in AD. 65 In the 21st century, astrocytes are increasingly recognized as playing a central role in neuroinflammation and are considered crucial contributors to CNS pathologies. 66
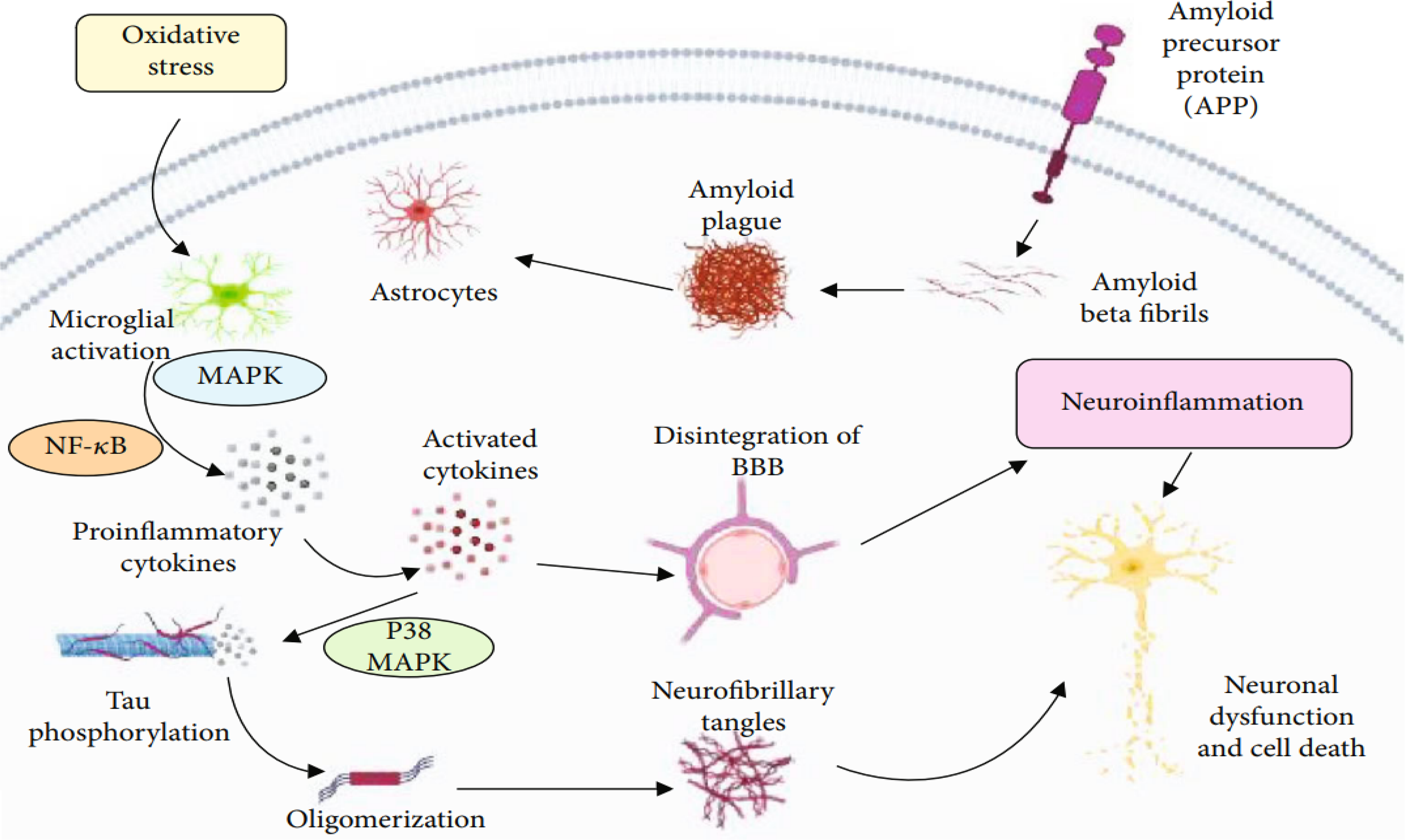
Aβ fibrils lead to neuronal death, which include reactive oxygen species generation, neurotoxicity, release of inflammatory cytokines, and activation of the complement system. Due to the accumulation of Aβ oligomers, neuronal degeneration may stimulate the microglial activation, which will initiate the liberation of proinflammatory mediators, neurotoxins, and free radicals but also play a pivotal role in the elimination of Aβ peptides. These peptides trigger oxidative stress and promote inflammatory processes in neurons, which enhance the production of Aβ peptides via increased AβPP expression. Activated a mitogen-activated protein kinase and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) lead to the production of proinflammatory cytokines, and their increased expansion promotes AβPP processing and disintegration of blood-brain barrier and aggravates the phosphorylation of tau protein and eventually leads to the formation of neurofibrillary tangles via the activation of p38-MAPK which leads to neuronal degeneration. From “Inflammation and Alzheimer's disease: mechanisms and therapeutic implications by natural products”. Copyright © 2021. Rather et al. 223
Neuroinflammation and Aβ neurotoxicity
The production and degradation of the Aβ peptide are ongoing under normal physiological conditions, and it is transported to the cerebrospinal fluid (CSF) and nearby blood vessels. In a pathological condition, Aβ accumulates abnormally in the brain and aggregates later. Chronic activation of the immune system in the CNS can be caused by these aggregates.67,68 The neurotoxic role of Aβ in AD is supported by undisputed evidence; however, the neuroinflammatory process and ongoing activation of microglia and astrocytes lead to alteration in the communication between glial cells and neurons. The process is combined with the prolonged exposure of neurons to pro-inflammatory mediators such as IL-1β, IL-18, IL-6, TNF-α and can result in neuronal dysfunction and contribute to cell death.69,70 The buildup of Aβ is a defining pathological feature of AD. The Aβ peptide is generated from a larger precursor known as the AβPP. 64 Notably, the 42-amino acid-long form of Aβ has a pronounced propensity to create the previously mentioned soluble oligomers and fibrils.71,72 When Aβ binds to CD36, toll-like receptors 4 (TLR4), and TLR6, it triggers the activation of microglia, which subsequently begin producing proinflammatory cytokines and chemokines such as CCL2, CXCL10, CCL3, and CX3CL1. 64 In sporadic cases of AD, the inefficient clearance of Aβ has been recognized as a significant pathogenic mechanism. 73 It has been proposed that elevated cytokine levels play a role in reducing the phagocytic capacity of microglia by downregulating Aβ phagocytosis receptors. 64 The extensive information offered by single-cell technologies offers an exceptional chance to explore the molecular pathways and cellular processes associated with Aβ pathophysiology in a cell specific manner. 39 This is particularly beneficial for examining the systematic changes in the inflammatory response of microglia and astrocytes, providing insights into the complex neuroimmune interactions that play a role in the AD pathophysiology. 74 Neuroinflammatory pathways and the activation of microglial cells are linked to the activation of the neuronal ectopic cell cycle. 75 Specifically, the activation of microglia by Aβ oligomers encourages neuronal ectopic cell cycle events through the tumor TNF-α and the c-Jun kinase (JNK) signaling pathways. 46 Upon activation by pathological stimuli such as neuronal death or protein aggregates, microglia extend their processes towards the site of injury. Subsequently, they begin migrating to the lesion and initiate an innate immune response. 76 The recognition of these pathological triggers is mediated by receptors in AD, microglia are capable of binding to soluble Aβ oligomers and Aβ fibrils through specific receptors. 77 These receptors include CD36, CD14, α6β1 integrin, CD47, and TLR2, TLR4, TLR6, and TLR9. This binding is considered a component of the inflammatory response in AD. Those were initially designed to detect danger or pathogen-associated molecular patterns (DAMPs/PAMPs). Rats that receive lipopolysaccharide (LPS) injections demonstrate a pro-inflammatory response, an increase in the expression of APP and β-secretase, a decrease in Aβ clearance, and cognitive impairment, these findings suggest that chronic inflammation may play a role in promoting AD pathology. 78 AD appears to involve the influence of several other factors, which raises questions about the simplicity of the amyloid cascade hypothesis. 79 It underscores the complexity of AD, which is characterized by multiple interconnected events that cannot be easily explained by a single hypothesis. 80 Among these factors, we can include lysosomal dysfunction, disturbances in Ca2+ homeostasis, neuroinflammation and ongoing oxidative damage.7,81,82
Neuroinflammation and NFT formation
The presence of extracellular tau is involved in the transition from quiescent to active microglia, In the quiescent state, the protein fractalkine CX3CL1, released by healthy neurons, binds to the cell receptor CX3CR1 found on microglia, which helps keep the microglia in their resting state. 83 Tau pathology is linked to neuroinflammatory processes, and microglia may play a role in the propagation of tau in tauopathies, and contribute to propagation of Aβ. In a study by d’Errico et al., 84 using transplantation of wild-type neurons, they show that Aβ enters wild-type grafts, and that this is accompanied by microglia infiltration. They showed that manipulation of microglia function reduced Aβ deposition within grafts. Furthermore, in vivo imaging identified microglia as carriers of Aβ pathology in previously unaffected tissue. 84
In this context, microglial CX3CR1 acts as a receptor for extracellular tau (Figure 3). The absence of CX3CR1 can disrupt the internalization of tau by microglia. 85 Therefore, extracellular tau can compete with CX3CL1 for the same receptor. Microglial cells that lack CX3CR1 are unable to properly respond to neuronal CX3CL1 signaling, and as a result, they do not remain in the resting state. 86 The lack of microglial CX3CR1 can disrupt the synaptic integration of adult-born hippocampal granule neurons. 87 NFT have been found to be physically associated with inflammatory markers, including CRP and TGF-β. 88 Similar to the situation with ROS/RNS, NFT can be seen as both a toxic outcome of neuroinflammation and a factor that contributes to the process itself. 89 As an example, tau accumulates in neurites of cultured neurons when exposed to TNF-α or when co-cultured with activated microglia. This process appears to be linked to cytokine-stimulated ROS. 90
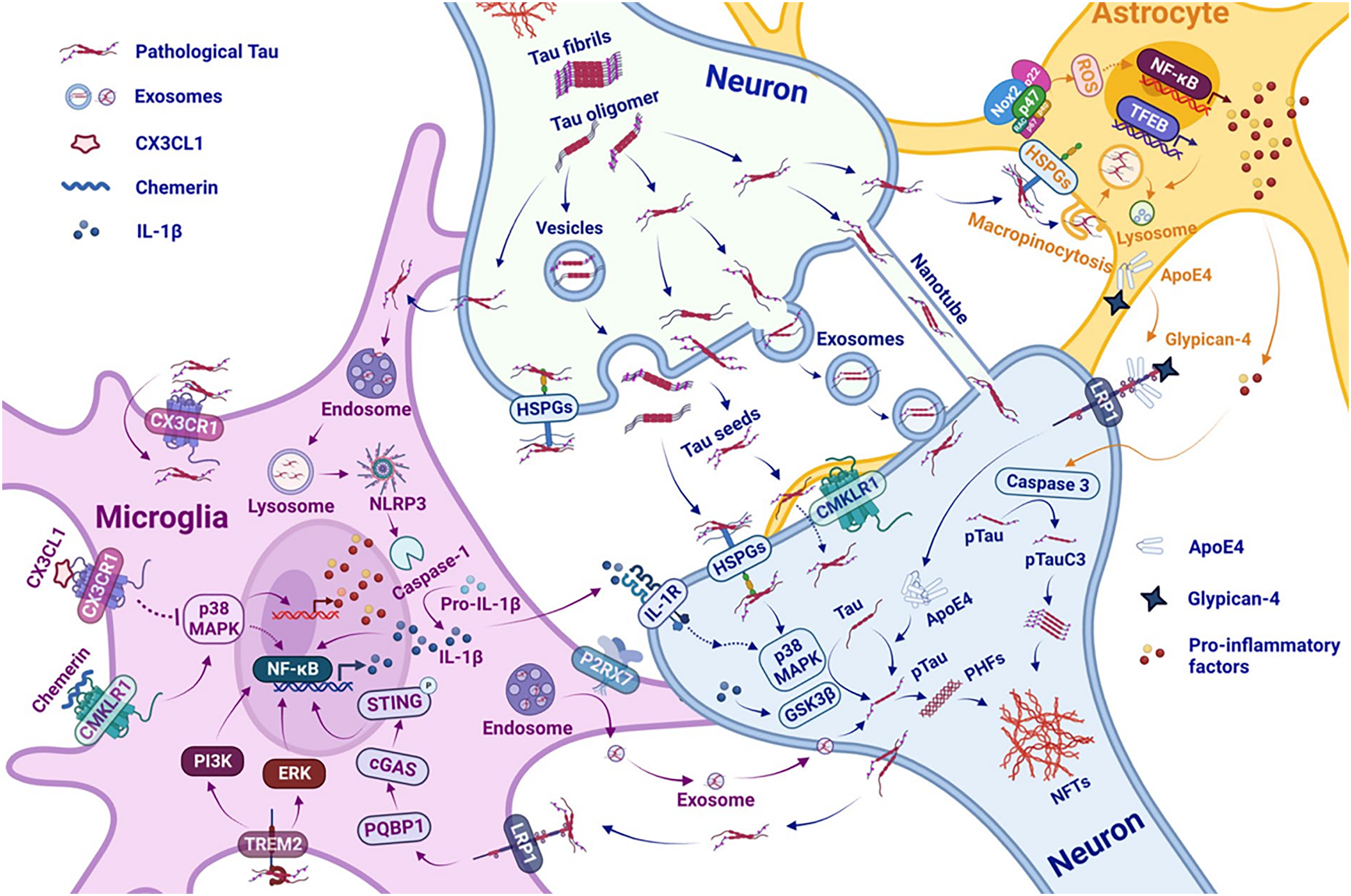
Schematic diagram showing the interaction between neuroinfammation and tau pathology contributing to the progress of AD pathogenesis. (ApoE4) apolipoprotein E4, (cGAS) cyclic GMP-AMP synthase, (CMKLR1) chemerin chemokine-like receptor 1, (CX3CL1) chemokine C-X3-C motif ligand 1. (CX3CR1) CX3C motif chemokine receptor 1, (GSK3β) glycogen synthase kinase-3 beta, (IL-1β) interleukin-1β, (IL-1R) interleukin-1 receptor, (HSPGs) heparan sulfate proteoglycans, (LRP1) low-density lipoprotein receptor-related protein 1, (MAPK) mitogen-activated protein kinase, (NF-κB) nuclear factor kappa B, (NFTs) neurofbrillary tangles, (NLRP3) NLR family pyrin domain-containing protein 3, (Nox2) NADPH oxidase 2, (P2RX7) P2X purinoceptor 7, (PHFs) paired helical flaments, (PQBP1) polyglutamine-binding protein1, (pTau) phosphorylated tau, (STAT1) signal transducer and activator of transcription 1, (STING) stimulator of interferon genes, (TFEB) transcription factor EB, (TNF- tumor necrosis factor (TREM2) triggering receptor expressed on myeloid cells2. From Tau and neuroinflammation in Alzheimer's disease: interplay mechanisms and clinical translation. Copyright © 2023. Yijun C and Yang Y. 224
Dysfunctional cytoskeletal tangles would likely impair various aspects of neuron function, potentially reaching neurotoxic levels. Debris released during cell death and the presence of residual NFT “ghosts” could feasibly trigger additional immune activation, thereby intensifying neuroinflammatory cycles. Microglial cells have a vital role in immune surveillance of the presynaptic microenvironment and are involved in synaptic remodeling through processes like axonal and dendritic terminal pruning, which involves reshaping proteolytic and phagocytic mechanisms. Additionally, microglial cells can recruit astroglia, or they can be recruited by astroglia themselves.91–93 Microglial cells are believed to contribute to the widely recognized age related regional synaptic vulnerability, as has been recently documented. 94 This is supported by the observation that an age related change in the ultrastructure and function of microglia cells is linked to heightened synaptic susceptibility and neurodegeneration. 93
Neuroinflammation and cytokines release
The role of interleukins in AD pathogenesis is notably intricate, as some interleukines have proinflammatory effects while others have anti-inflammatory actions. In this context, it is noteworthy to mention IL-12 and IL-23, which show increased levels in the CSF in both AD and mild cognitive impairment (MCI).95,96 Remarkably, the genetic removal of IL-12 and IL-23 or therapeutic strategies aimed at inhibiting IL-12 and IL-23 signaling have been shown to decrease AD-like pathology, which includes histopathological and behavioral alterations. These findings make IL-12 and IL-23 promising targets for potential treatments of AD. 97 During the AD brain autopsy, a substantial number of activated microglia were found in the vicinity of the lesions, and the expression of immune cytokines, including TNF-α, IL-6, and IL-10 could be detected (Figure 4).98,99
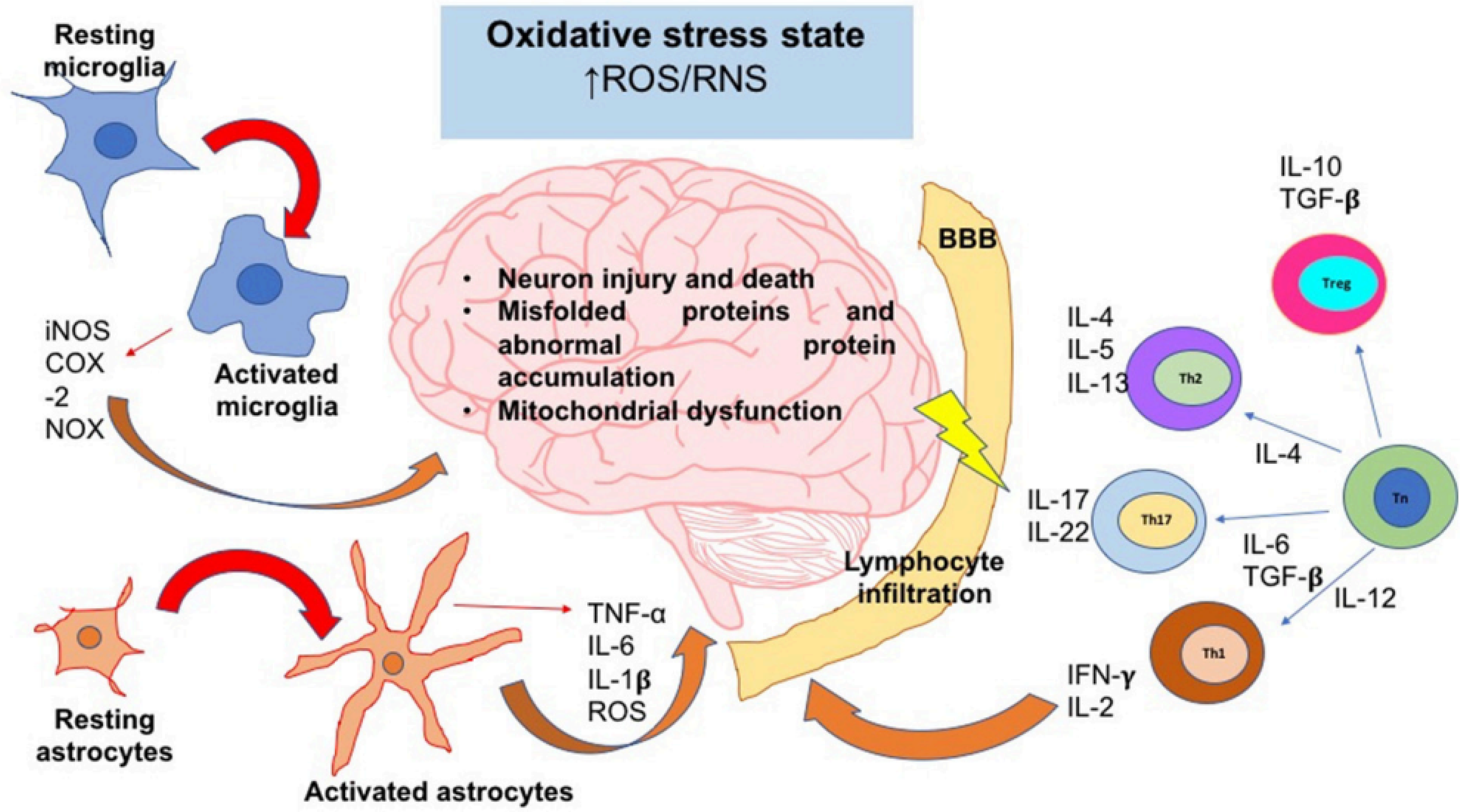
The oxidative stress state induces neuroinflammation and neurodegeneration. In an oxidative stress state, ROS and RNS levels are augmented; these reactive species can activate signaling pathways that lead to the activation of the major glial inflammatory characters: microglia and astrocytes. These glial cells secrete proinflammatory factors which positively feedback the neuroinflammatory response. On the other hand, SNC-secreted factors and peripheral cytokines are able to disrupt the blood-brain barrier (BBB) integrity; thereby, leukocytes such as T cells are able to infiltrate into SNC and take turn in the positive feedback of neuroinflammation. Inflammatory cells and secreted factors lead to neurodegeneration, in which the most characteristic feature is the neuron injury and death. iNOS, inducible nitric oxide synthase; COX-2, cyclooxigenase-2; NOX, NADPH oxidase; IL, interleukin; Th, T helper cell; Tn, T naive cell; Treg, T regulatory cell; ROS, reactive oxygen species; RNS, reactive nitrogen species; TNF-α, tumor necrosis factor alpha; TGF-β, transforming growth factor beta. From effect of chronic oxidative stress on neuroinflammatory response mediated by CD4 + t cells in neurodegenerative diseases. From “Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4 + T cells in neurodegenerative diseases”. Copyright © 2018. Solleiro-Villavicencio H and Rivas-Arancibia S. 15
IL-1β is a crucial molecule involved in inflammatory responses, as well as processes such as cell proliferation, differentiation, and apoptosis. The plasma concentrations of IL-6 and IL-1β are notably elevated in individuals with AD compared to those with normal cognitive function, as indicated by separate meta-analyses.100,101 The inflammatory cytokine IL-1β is responsible for the harmful effects on synapses caused by the Aβ peptide. 102 Interestingly, the interleukin-1 receptor antagonist (IL-1Ra) has the ability to reverse the changes in synaptic plasticity induced by the administration of the Aβ. There is evidence to suggest that microglia cells produce and secrete IL-1β in response to Aβ deposition, leading to chronic neuroinflammation and, ultimately, neuronal disruption, dysfunction, and neurodegeneration.103,104 Additionally, a negative correlation has been observed between CSF concentrations of IL-1β and cognitive scores in AD. 105
Furthermore, IL-6 levels are associated with the severity of cognitive decline. 106 It is worth noting that peripheral IL-6 concentrations positively correlate with cerebral ventricular volumes and matched CSF samples in AD. 107 The peripheral changes in IL-6 levels may start during the prodromal phase of AD. A recent meta-analysis indicates higher IL-6 concentrations in subjects with MCI compared to controls. 106
TNF-α also plays a crucial role in the early proinflammatory process observed in preclinical AD, as supported by studies in animal models of AD and human longitudinal studies.52,108–111 TNF-α is continuously released throughout the course of AD pathology, possibly by activated microglia, neurons, and astrocytes stimulated by elevated levels of extracellular Aβ. 111 Oligomeric forms of Aβ activate microglia through abnormal TNF-α mediated pathways in mouse models. 112 This atypical stimulation of cerebral innate immunity contributes to reduced serotonergic tone, a primary event in depression associated with Aβ and a prodromal symptom of AD. 113 Additionally, TNF-α can stimulate γ-secretase activity, leading to increased synthesis of Aβ peptides and further elevation in TNF-α release.111,114 This auto amplified loop in the AD brain is believed to contribute to the persistence of elevated TNF-α levels, which can further stimulate Aβ synthesis, neuronal loss, and inhibit microglial phagocytosis of Aβ.111,115 TNF-α plays a significant role in promoting insulin resistance and subsequent cognitive decline in AD.116,117 Elevated TNF-α levels have been detected in both MCI and AD. Interestingly, individuals with Down syndrome who have preclinical AD exhibit significant associations between elevated levels of plasma TNF-α, Aβ accumulation, and subsequent cognitive decline in the years that follow. Furthermore, both TNF-α and TNF-RI concentrations are increased in the post-mortem brains of early stage AD patients. 113 Subjects with MCI present somewhat conflicting data. Longitudinal studies have reported associations between TNF-R concentrations and the risk of conversion from MCI to AD.52,118
Indeed, the TNF-α receptor complex and its functional proteins are believed to play a critical role, as they connect neuroinflammatory pathways to the process of amyloid deposition in a chronically damaging and self-perpetuating manner.53,119 Multiple studies suggest that inhibiting the TNF-α pathway in AD models leads to various positive outcomes, including improved memory and cognitive function as assessed by different behavioral tests, decreased immunohistochemical and histopathological markers such as Aβ plaques and NFTs, and reduced numbers of microglial cells in the AD brain. 120 When microglia is exposed to pre aggregated Aβ, it can lead to an increase in the production of various pro-inflammatory cytokines, including pro-IL-1β, IL-6, TNF-α, as well as macrophage colony-stimulating factor (M-CSF). This heightened inflammatory response is a characteristic feature of AD-associated neuroinflammation. 121
Additionally, studies have shown that levels of M-CSF in the plasma and CNS of AD patients are significantly higher compared to age-matched healthy controls or individuals with MCI.122,123 There is also evidence of elevated caspase-1 activation, which is necessary for maturing IL-1β, in the brains of individuals with MCI and AD. 124 In microglial cells surrounding Aβ plaques in AD brains and CSF, primary pro-IL-1β levels are observed to be elevated.125,126 IL-1β can, in specific conditions, contribute to Aβ deposition by influencing the expression and proteolysis of AβPP. 127 For example, individuals with elevated TNF-α levels and reduced TGF-β levels in CSF are at a higher risk of progressing from MCI to AD. 128 The secretion of anti-inflammatory cytokines such as IL-4, IL-10, IL-13, and TGF-β characterizes the M2 state microglial activation. In this state, microglia exhibit an increased phagocytic capacity without producing toxic NO. 115 There is a strong neurobiological link in the AD brain between the deficiency of anti-inflammatory cytokines, such as TGF-β1, and the early proinflammatory processes observed in preclinical AD. 129
TGF-β1 is also a neurotrophic factor whose deficiency plays a significant role in AD. There is a selective impairment of the TGF-β1 pathway in early AD, which is evident in both the AD brain and AD animal models.130–133 The deficit of TGF-β1 in AD appears to play a critical role in promoting neuroinflammation in the AD brain. TGF-β1 has anti-inflammatory and neuroprotective properties, and it also stimulates the clearance of Aβ by microglia.134–136 Moreover, TGF-β1 is involved in synaptic plasticity and memory formation processes, contributing to the transition from early to late long term potentiation. 137 It is important to reevaluate the significance of TGF-β1 in neuroinflammation associated with microglial activation, as it appears to play a role in reactivating the neuronal cell cycle. 138 In this context, it is proposed that the reactivation of the neuronal cell cycle could be facilitated by the disruption of Smad dependent TGF-β1 pathways. These studies indicate that the deficiency of Smad-dependent on TGF-β1 pathways may contribute to neuroinflammation and cognitive impairment in AD. 113 On the contrary, IL-10 appears to have a protective role. 139 When IL-10 is delivered via adeno-associated virus, it leads to a significant reduction in microgliosis and astrogliosis and can reverse cognitive impairment in transgenic AD mice. 140
Neuroinflammation is cause and/or a consequence of oxidative/nitrosative stress
Oxidative stress and neuroinflammation are believed to have a significant impact on both typical aging and age-related neurological conditions such as AD. 141 Oxidative stress occurs when there is an imbalance between the production of ROS and RNS and the body's ability to counteract them with antioxidants. 142 ROS and RNS are highly reactive molecules that can potentially damage various cellular components, including DNA, proteins, and lipids. This oxidative damage is associated with various diseases and aging processes.143–145 ROS and RNS are molecules that contain oxygen or nitrogen and have high reactivity due to unpaired electrons. 146 In controlled amounts, ROS play roles in cell signaling, immune defense, and other physiological processes, they can serve as important signaling molecules in cellular processes, but their excess or uncontrolled production can lead to oxidative stress and cellular damage. ROS include molecules like O2, hydroxyl radicals (OH−), peroxyl radicals (RO2), hydrogen peroxide (H2O2), organic peroxides (ROOH), and peroxynitrite (ONOO−), they are produced during processes such as mitochondrial respiration, enzymatic reactions, and immune responses. 147
RNS, on the other hand, are primarily nitrogen containing molecules like NO, nitrogen dioxide (NO2), nitrous acid (HNO2), and peroxynitrite (ONOO−). 147 NO, in particular, is a well-known signaling molecule involved in vasodilation and neurotransmission. 148 However, excessive production of RNS can contribute to oxidative stress and inflammation. 149 When this balance is disrupted, it can lead to oxidative stress, which is implicated in various diseases, including neurodegenerative disorders.143,150 Today, there is broad consensus that oxidative stress is closely linked to the inflammation seen AD. 151
Neuroinflammation can be both a trigger and a result of chronic oxidative stress. Microglia activated by cytokines produce significant quantities of ROS and RNS, which can place stress on neighboring neurons. 152 Conversely, oxidants can trigger the transcription of pro-inflammatory genes in glial cells, giving rise to various inflammatory responses. 153 Neuroinflammatory processes can indeed serve as both a cause and an effect of chronic oxidative stress. 154 In this context, microglia play a pivotal role, pro-inflammatory microglial activities in AD may be harmful due to the generation of ROS and RNS, leading to oxidative stress induced neuronal death. This neuronal damage could be further worsened by chronic stress. 155 There is accumulating evidence suggesting that in AD, microglial inflammation induced oxidative stress is increased. 156
However, microglial mediated clearance mechanisms appear to be dysfunctional in this context.17,156 Aβ peptides can directly promote the formation of ROS through interactions with transition metal ions via reductive processes. 157 Additionally, the production of H2O2 by Aβ peptides may play a significant role in initiating the activation of glial cells, as H2O2 itself can activate cultured rat astrocytes to produce cytokines, including TNF-α. 158 Activated astrocytes and microglia in the AD brain produce substantial amounts of NO. Additionally, elevated levels of nitrotyrosine-modified proteins, have been observed in this case. 159 Elevated mRNA levels for inducible nitric oxide synthase (iNOS) and argininosuccinate synthetase have been detected within the cortex of individuals with AD, indicating the potential involvement of NO in AD pathophysiology. 160
The activation of microglia and astrocytes by oxidative stress, along with the subsequent release of cytokines, may indeed contribute to the progression of AD, this neuroinflammatory response can exacerbate the neurodegenerative processes seen in AD. There is compelling evidence implicating oxidative stress in the pathophysiology of AD. Oxidative stress promotes tau hyperphosphorylation and neurofibrillary pathology by inhibiting phosphatase 2A, which in turn activates glycogen synthase kinase 3β.161,162 Additionally, it contributes to the accumulation of Aβ through ROS induced inhibition of the proteasomal system, which occurs via impaired mammalian target of rapamycin (mTOR) signaling. 163 Meda and colleagues demonstrated that Aβ and IFN-γ work synergistically to activate microglia, leading to the production of reactive nitrogen intermediates and TNF-α. Their research suggests that the production of reactive nitrogen intermediates is in part, mediated by the induced release of TNF-α. 164 TNF-α, which induce the expression of COX-2 through the NF-κB dependent pathway and generate ROS via a cytosolic phospholipase A2-linked cascade.165,166 The pattern recognition receptors (PRRs) pathway, TREM2 signaling, and ROS-mediated pathway are among the molecular pathways that activate and maintain the chronic inflammation state in the CNS. 167 Various processes such as PRR signaling, ROS production, and inflammasome assembly may be linked by mitochondrial cells to neurodegenerative and neuroinflammatory pathologies in the CNS.168,169
ROS play a significant role in the activation of microglia, leading to the secretion of proinflammatory cytokines and the production of additional ROS in a harmful cascade. These released cytokines such as IL-1β, TNF-α, IL-6 which activate glial cells and trigger ROS induced apoptosis of pericytes, ultimately leading to the breakdown of the BBB have been observed in the brain, CSF, and serum of patients with AD.168–170
The NLRP inflammasome activation in Alzheimer's disease pathogenesis
Inflammasomes, discovered only in 2002, are now recognized as central regulators of innate immunity, playing a crucial role in the host's defense against stressful conditions and pathogens that could potentially harm the host. 171 Nonetheless, when inflammasomes become overly activated, leading to excessive or prolonged inflammation, they can also create a harmful and destructive environment, potentially contributing to the development of inflammatory diseases, including neurodegenerative diseases. 172 Inflammasomes are cytosolic multiprotein complexes that, once assembled, activate the pro-inflammatory caspase-1 enzyme. Caspase-1 is responsible for the maturation and secretion of the inflammatory cytokines IL-1β and IL-18. 173
In response to inflammasome inducing stimuli, PRR proteins oligomerize and recruit pre-existing procaspase-1 zymogens into the complex. This proximity-induced interaction leads to the autoactivation of procaspase-1, converting it into its active form, caspase-1. 174 Consequently, caspase-1 cuts the biologically inactive pro-peptides pro-IL-1β and pro-IL-18 into mature cytokines, which are then discharged by the cell. Besides its involvement in maturing pro-IL-1β and pro-IL-18, caspase-1 can also initiate a pro-inflammatory type of cell death known as pyroptosis. This process is marked by early rupture of the plasma membrane, releasing soluble intracellular contents that contribute to the inflammatory response.172,175 IL-1β and IL-18 have significant functions in the CNS, and various cell types in the brain express receptors for these cytokines. 176 Activation of these receptors initiates inflammatory signaling cascades that can potentially contribute to neuronal injury and cell death in the CNS.177,178 Therefore, elevated levels of IL-1β and IL-18 are frequently detected in cases of CNS infection, brain injury, and neurodegenerative diseases.179–181
Pyroptosis represents a recently identified form of programmed cell death intricately linked with inflammation. It plays a significant role in inflammasome-driven pathology by releasing various other inflammatory mediators and DAMPs.182–184 Initially attributed to caspase-1-mediated cell death, pyroptosis was subsequently redefined as a form of programmed necrosis, primarily facilitated by Gasdermin-D. 185
Although inflammasome signaling in the CNS is mainly attributed to microglia, the key innate immune cells of the brain, expression of inflammasome components has also been reported in other cell types of the CNS, including neurons, astrocytes, perivascular CNS macrophages, oligodendrocytes, and endothelial cells.186–188 Many recent studies have implicated inflammasomes in the development of AD, elevated expression of IL-1β has been reported in microglia that surround Aβ plaques in AD patients.188,189
Involvement of NLRP3 inflammasome with caspase-1 and IL-1β in Alzheimer's disease
NLRP3 inflammasome activation promote neuroinflammation and the formation of senile plaques through various pathways. 190 In AD patients, NLRP3 inflammasomes often remain active and regulate the release of harmful inflammatory molecules in the AD brain, inhibiting the NLRP3 inflammasome activation could be a significant avenue for future treatments of AD patients (Figure 5). 191 Neuroinflammatory events mediated by glial cells and driven by Aβ are now recognized as a significant contributing factor in the AD pathogenesis.192,193
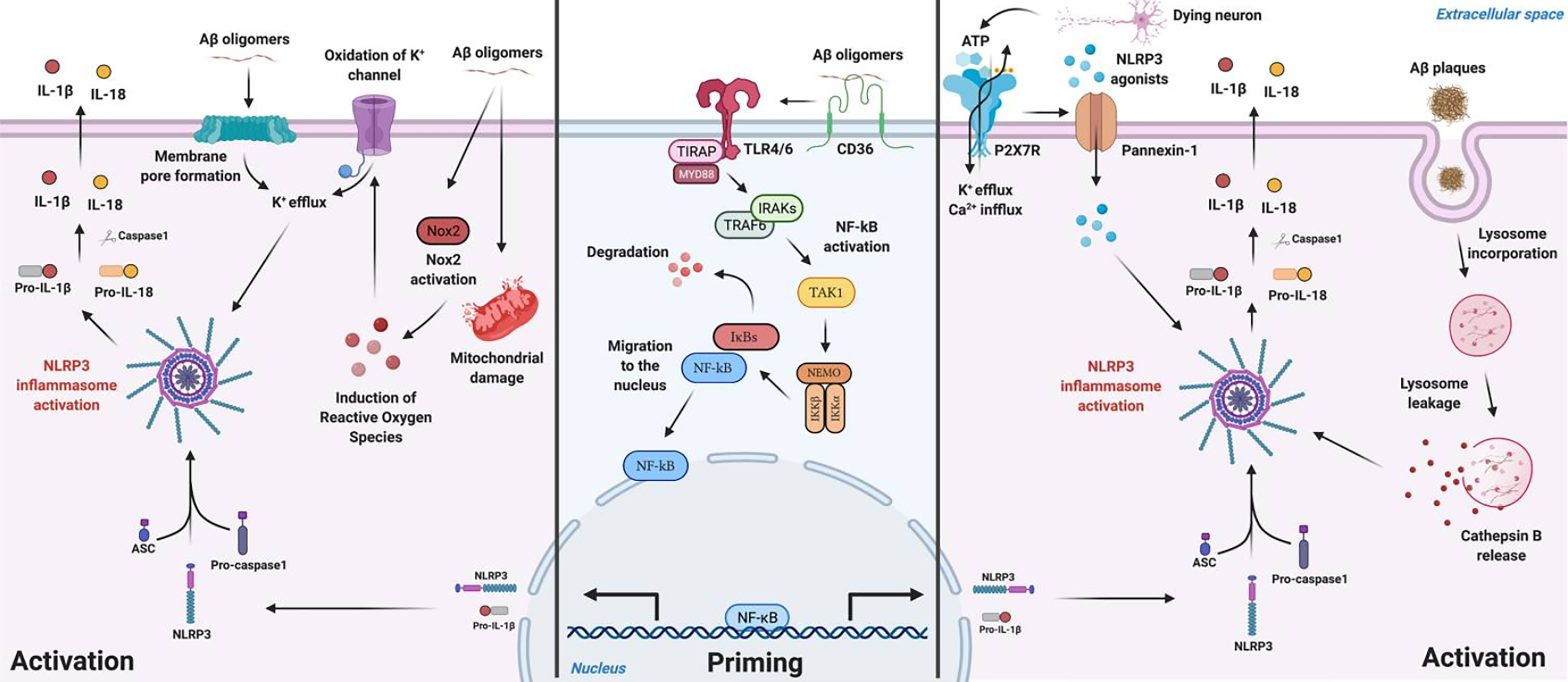
Schematic illustration for Aβ mediated NLRP3 inflammasome priming and activation mechanisms described in microglia. Aβ species can work either as a priming stimulus (middle panel) or as an activating stimulus (left and right panels). As a priming signal, Aβ oligomers bind to the CD36 surface receptor, triggering the formation of a TLR4-TLR6 heterodimer. This heterodimer activates a cascade of signaling molecules resulting in the activation and nucleus translocation of the transcription factor NF-κB that promotes the transcription of NLRP3 domain and of pro-IL-1β. Aβ plaques can act as an activating signal by two main mechanisms (right panel). Aβ is known to cause synaptic dysfunction and neuronal damage. Considering this, P2X7R is activated by ATP released from dying neurons and recruits the Pannexin-1 channel that allows the entrance of NLRP3 agonists to the cell. In addition, ATP binding to the purinergic receptor induces K+ efflux and Ca2+ influx, known to promote NLRP3 activation. On the other hand, Aβ plaques can also be phagocytized and incorporated into lysosomes, boosting lysosomal destabilization and consequent content release. Cathepsin B, a lysosomal proteolytic enzyme, promotes the assembly of the inflammasome by a still unknown mechanism. Aβ oligomers are also able to activate NLRP3 through a mechanism that does not involve phagocytosis (left panel). Soluble oligomeric Aβ species can induce pore formation in the cell membrane and ROS production, which then promotes oxidation of K+ channels. These events might culminate in K+ efflux promoting the activation of the inflammasome. From NLRP3 inflammasome: A starring role in amyloid-β- and tau-driven pathological events in Alzheimer's disease. Copyright © 2021. Zellera et al. 225
Moreover, the activation of the NLRP3 inflammasome, a cytosolic multiprotein complex involved in inflammation, is believed to play a prominent role in this process.124,194 When the NLRP3 inflammasome is activated, it can contribute to the development of AD through two mechanisms: first, it can regulate IL-1β, leading to the production of neurotoxic substances that contribute to neuronal degeneration. 195 Second, it can promote an increase in Aβ deposition, initiating an Aβ self-perpetuating positive feedback loop that ultimately contributes to the AD progression. 40
In a study, using postmortem brain tissue from individuals with AD and age-matched non-AD individuals as controls, the relationship between the NLRP3 inflammasome and specific markers like autophagy lysosome labeled A0205 protein, phosphorylated tau protein, and glial maturation factor was analyzed through immunohistochemistry. Ahmed and colleagues discovered that the NLRP3 inflammasome's neuroinflammatory impact can be intensified and controlled by GMF. Moreover, this interaction can hinder the clearance of protein aggregates through the autophagy signaling pathway. 196 Saresella et al. observed that peripheral blood mononuclear cell (PBMCs) in AD patients showed signs of activation in both the NLRP3 inflammasome and the NLRP1 inflammasome when stimulated with LPS or Aβ, they suggested that the migration of peripheral monocytes across the BBB could be a significant factor contributing to neuroinflammation in AD. 197
The loss of function of the NLRP3 inflammasome may reduce tau hyperphosphorylation and aggregation by regulating tau kinases and phosphorylases. 198 This study corroborated the involvement of microglial NLRP3 inflammasome activation in the pathogenesis of tau-related diseases. Additionally, it provided further support for the Aβ cascade hypothesis in AD pathogenesis. Moreover, this study illustrated how NFTs contribute to the subsequent development of microglial activation induced by Aβ. 198 This process exacerbates the harm to neighboring tissues and intensifies the neurotoxic effects of Aβ. 199 The accumulation of Aβ and the activation of the NLRP3 inflammasome may potentially have a bidirectional causal relationship. Activation of the intracellular NLRP3 inflammasome can trigger the M1 phenotype activation of microglia, consequently fostering the deposition of Aβ and exacerbating cognitive impairment in mouse models of AD. Conversely, microglia with a specific functional impairment of the NLRP3 inflammasome tend to adopt the M2 phenotype. This shift reduces extracellular Aβ deposition, protects nerve cells from synaptic dysfunction, and alleviates cognitive decline in the brain. 200 The theory proposing that neuroinflammation induced by the activation of the NLRP3 inflammasome suggests that congenital immune disorders and neuroinflammation play crucial roles in the pathogenesis of AD. 191
Gene expression analysis of PBMCs from AD patients revealed higher expression of NLRP3, caspase-1, caspase-5, as well as the cytokines IL-1β and IL-18.197,201 In vitro studies have demonstrated that when microglia phagocytose fibrillar Aβ, it can activate the NLRP3 inflammasome, resulting in the activation of caspase-1 and the release of IL-1β. 202 NLRP3 inflammasome activation has been observed in vivo in the transgenic AβPP/PS1 mouse model of AD. Interestingly, deficiency in NLRP3 has been shown to significantly improve spatial memory deficits and hyperactive behavior in these mice. This improvement was associated with reduced Aβ deposition in the hippocampus and cortex, smaller plaque volumes, decreased levels of pro-inflammatory cytokines, and enhanced microglial phagocytic ability. 124
Similarly, studies conducted in AβPP/PS1 mice that lacked caspase-1, a key enzyme in the inflammasome pathway, yielded similar results. 124 Indeed, studies in mice deficient in the IL-1 receptor antagonist (IL-1ra) have shown that IL-1β plays a pathogenic role in AD. These mice exhibited increased vulnerability when injected with human oligomeric Aβ42, suggesting that IL-1β may contribute to the development and progression of AD pathology. 203 It appears that injection of IL-1β into the cerebral hemisphere can lead to increased levels of AβPP in wild-type mice. 204 The sustained overexpression of IL-1β in the hippocampus of AβPP/PS1 mice seems to have a somewhat different effect. In this case, it was observed to reduce plaque pathology, possibly due to the increased phagocytic activity of microglia and macrophages. This suggests that the role of IL-1β in AD is complex and can have both pro-inflammatory and potentially beneficial effects depending on the context and timing of its expression. 205
NLRP1 inflammasome activation induce pyroptosis
Reducing the activity of the NLRP1 inflammasome resulted in significantly reduced neuronal pyroptosis and led to the recovery of cognitive impairments in these mice. A pathway involving NLRP1, Caspase-1, and Caspase-6 has been identified in human neurons.188,206 This pathway connects neuron-specific NLRP1 inflammasome activation with IL-1β mediated neuroinflammation and the activation of Caspase-6, which is associated with axonal degeneration. 207 In addition, NLRP1 is elevated in neurons affected by AD, where it co-localizes with Caspase-6 activity. 208 The NLRP1 inflammasome found in human neurons is responsible for activating Caspase-1 in human neuron cultures exposed to stressors (lipopolysacharide). 188
The use of antibodies against NLRP1 in cell-free extracts, NLRP1 siRNAs in primary neuron cultures, and Nlrp1 knockout mice all prevent stress induced Caspase-1 activity and IL-1β production. 188 Additionally, there is a significant increase in NLRP1 levels in hippocampal and cortical neurons of both sporadic and familial AD cases, indicating the importance of NLRP1 in AD neurons. The recent discovery of genetic polymorphisms in the NLRP1 gene associated with AD further underscores the potential role of NLRP1 in the pathogenesis of AD. 209 Recent research has linked Nlrp1 to behavioral deficits, cell death, Caspase-1 activation, and IL-1β production in mutant PSEN1 ΔE9/AβPP Swedish (PS1ΔE9/AβPPSw) transgenic mice. This suggests a potential role for Nlrp1 in the pathogenesis of AD in this mouse model. 210 Whether sporadic or familial, age related neuronal stress factors like reduced growth factors, oxidative stress, or environmental influences, or the expression of mutant forms of AβPP, PSEN1, or PSEN2, can trigger the activation of NLRP1. This activation may contribute to the neuroinflammatory processes observed in the disease. Upon activation, NLRP1 recruits and activates Caspase-1, which in turn converts pro-IL-1β into the secreted form of IL-1β. This initiates a second phase of inflammation involving astrocytes and microglia. 170
Additionally, Caspase-1 activation leads to Caspase-6 mediated axonal degeneration and an increased production of Aβ42.211–213 These processes contribute to the complex neuroinflammatory and neurodegenerative cascades observed in AD. 214 Additionally, very high concentrations of Aβ42 can activate NLRP1 in rodent neurons, as demonstrated by tan and colleagues. This further underscores the associated role of Aβ42 and NLRP1 in the pathogenesis of AD. 210 This neuronal NLRP1-Caspase-1-Caspase-6 pathway, activated in response to non-pathogenic insults, suggests that inflammasome activation in neurons may not always require the presence of microbial pathogens. The discovery of the NLRP1-Caspase-1-Caspase-6 pathway in AD neurons may provide an explanation for the limited effectiveness of NSAIDs in treating AD. NSAIDs primarily target cyclooxygenase prostaglandins and do not inhibit inflammasome activity. 188
Therefore, more promising therapeutic targets for AD could be NLRP1 or Caspase-1. 188 There are a number of potential therapeutic targets including caspase-1 activation and signaling at the IL-1R1 receptor. Neither caspase-1 or IL-1R1 have been pharmacologically targeted in animal models of AD. However, genetic deletion of caspase-1 has been shown to increase amyloid phagocytosis in isolated microglia and reduce neuroinflammation following striatal amyloid injections in mice. 202 Fenamate tolfenamic acid, which is structurally very similar to mefenamic acid, has been found to be therapeutic in the AβPPSwe R1.40 mouse model of AD, lowering plaque burden, tau pathology, and cognitive deficits.215–217 However, similar therapeutic effects in similar animal models of AD were seen solely from the genetic deletion of the NLRP3 inflammasome and the inflammasome adapter molecule ASC, suggesting that inhibitory activity of fenamates on NLRP3 activation could exclusively explain their efficacy.202,218 Collectively, this evidence demonstrates that through NLRP3 inhibition and other potential mechanisms, fenamates have been found to be therapeutic in several animal models of AD and are therefore a promising potential therapeutic in AD. 124
In summary, pyroptosis represents a recently identified form of controlled cell demise that has been implicated in the development of various neurological disorders.219,220 It is marked by the creation of membrane pores, a process that ensues following activation of both canonical and non-canonical inflammasome pathways. 220 Throughout pyroptosis, the formation of pores in the cell membrane hinges on the cleavage of the GSDMD protein by inflammatory caspases. The cleavage of GSDMD triggers augmented membrane permeability, causing water influx, cellular swelling, and eventual osmotic rupture, leading to the release of proinflammatory cytokines and cytosolic contents into the extracellular environment. During pyroptosis, similar to apoptosis, cells exhibit chromatin condensation and DNA fragmentation.221,222
Conclusion
Neuroinflammation in AD begins many years prior to the clinical onset and is now clear that inflammation plays a fundamental role in the pathophysiology of AD. Inefficient clearance of Aβ and tau pathologies leads to increased cytokine levels, which reduce microglial phagocytose. These results in harmful effects on synapses through inflammatory cytokines IL-1β and TNF-α, which can further stimulate Aβ synthesis, neuronal loss, and inhibit microglial phagocytosis of Aβ. This self-amplifying loop in the AD brain contributes to the persistence of neuroinflammation. Cytokine-activated microglia produce significant quantities of ROS and RNS, which stress neighboring neurons. Oxidative stress promotes tau hyperphosphorylation and contributes to Aβ accumulation, with involvement of NLRP3 inflammasome regulating IL-1β, leading to production of neurotoxic substances contributing to neuronal degeneration. Furthermore, NLRP1 is associated with axonal degeneration through Caspase-1 and Caspase-6, ultimately leading to pyroptosis. All these data can contribute to opening of new therapeutic pathways and future directions because inflammasome have multiple points in the activation pathway which can be inhibited as well as inhibiting pro-inflammatory cytokines, caspases and inflammasome components, specially the NLRP3 inflammasome which, is an attractive pharmacological target, as inhibition would specifically abate pathological inflammation without altering basal microglia function or leaving the patient overly susceptible to infection.
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
Author contributions
Mourad Belkhelfa (Conceptualization; Data curation; Methodology; Resources; Validation; Writing – original draft; Writing – review & editing); Narimene Beder (Conceptualization; Investigation; Resources; Writing – review & editing); Hakim Leklou (Conceptualization; Writing – review & editing).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
Mourad Belkhelfa is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.