
Abstract
Autophagy is a fundamental cellular process critical for maintaining neuronal health, particularly in the context of neurodegenerative diseases such as Alzheimer's disease (AD). This review explores the intricate role of the SNARE complex in the fusion of autophagosomes with lysosomes, a crucial step in autophagic flux. Disruptions in this fusion process, often resulting from aberrant SNARE complex function or impaired lysosomal acidification, contribute to the pathological accumulation of autophagosomes and lysosomes observed in AD. We examine the composition, regulation, and interacting molecules of the SNARE complex, emphasizing its central role in autophagosome-lysosome fusion. Furthermore, we discuss the potential impact of specific SNARE protein mutations and the broader implications for neuronal health and disease progression. By elucidating the molecular mechanisms underlying SNARE-mediated autophagic fusion, we aim to highlight therapeutic targets that could restore autophagic function and mitigate the neurodegenerative processes characteristic of AD.
Introduction
Autophagy is a cellular process responsible for degrading and recycling damaged or unnecessary cellular components to maintain cellular homeostasis.1–3 This process involves the formation of double-membrane vesicles called autophagosomes, which engulf cellular cargo and subsequently fuse with lysosomes to form autolysosomes, where the enclosed material is degraded.4,5 The fusion of autophagosomes with lysosomes, known as autophagosome-lysosome fusion, is a critical step in autophagy, and its dysregulation is implicated in various neurodegenerative diseases, including Alzheimer's disease (AD).5–7
The 2021 World Health Organization's “Global Status Report on the Public Health Response to Dementia” highlights the significant societal and healthcare burden posed by neurodegenerative diseases. These conditions lead to cognitive and physical decline, resulting in loss of independence, reduced quality of life, and substantial strain on healthcare systems.8,9 AD, the most common cause of dementia, leads to severe cognitive decline and brain deterioration. According to 2023 Alzheimer's Disease Facts and Figures, nearly 6.7 million Americans aged 65 and older are affected by AD, making it the most prevalent form of dementia globally. 10
Recent studies have emphasized the importance of autophagy in the pathogenesis of AD.11–13 A prominent feature of AD pathology is the excessive accumulation of autophagosomes within affected neurons, indicating potential deficiencies in their efficient clearance. 14 This notable surplus strongly suggests that defects in the autophagosome-lysosome fusion process might play a crucial role in the disease progression. Emerging evidence suggests that dysregulation of this fusion process may contribute to AD pathogenesis.15,16 Therefore, understanding the molecular mechanisms governing autophagosome-lysosome fusion is crucial for developing therapeutic strategies to combat AD.
Central to the fusion of autophagosomes with lysosomes is the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex.17,18 This complex consists of specific proteins, including Syntaxin 17 (STX17), Vesicle-Associated Membrane Protein 8 (VAMP8), and Synaptosome-Associated Protein 29 (SNAP29), which mediate the membrane fusion process. Proper functioning of the SNARE complex is essential for maintaining efficient autophagic flux. However, in AD, the activity of the SNARE complex can be compromised, leading to impaired autophagosome-lysosome fusion and subsequent neuronal damage.
This review aims to unravel the role of the SNARE complex in lysosomal fusion within the context of AD. We will explore the mechanisms of autophagy in neuronal cells, highlighting the specificity of this process in maintaining neuronal health. The composition and function of the SNARE complex will be examined in detail, along with the regulation of its activity and interactions with other molecules involved in autophagosome-lysosome fusion. Additionally, we will discuss other key players in this fusion process, providing a comprehensive understanding of the molecular landscape governing autophagy in neurons.
Furthermore, the review will delve into the specific implications of SNARE complex-mediated autophagy in AD. We will discuss how the accumulation of autophagosomes and lysosomes in AD reflects disruptions in autophagic flux. The relationship between these abnormalities and disease progression will be analyzed, with a focus on how specific SNARE protein mutations or aberrant expression may contribute to the pathogenesis of AD. Impaired lysosomal acidification, another critical factor affecting autophagosome-lysosome fusion, will also be examined.
By elucidating these intricate molecular mechanisms, this review aims to highlight potential therapeutic targets that could restore autophagic function and mitigate the neurodegenerative processes characteristic of AD. Understanding the intersection of autophagy and the SNARE complex in lysosomal fusion provides a promising avenue for developing novel interventions to combat this debilitating disorder.
Autophagy: Mechanisms and specificity in neuronal cells
Overview of the autophagic process
Autophagy, often referred to as the cellular janitor, is a remarkable self-cleaning mechanism where cells degrade and recycle their damaged components, converting them into building blocks for renewal. 19 This process is vital for maintaining cellular homeostasis and responding to various stress conditions. Autophagy involves a series of interconnected processes and can be triggered by various intracellular and extracellular stimuli, such as nutrient starvation, hypoxia, and oxidative stress, which strongly induce a high level of autophagic activity. 20
The autophagic process begins with the nucleation of a phagophore, an initial membranous structure that elongates to enclose the cellular cargo. This step is critically regulated by the ULK1 (unc-51 like autophagy activating kinase 1) complex and the PI3 K (phosphatidylinositol-3-kinase) class III complex, which initiate the formation of the isolation membrane. Subsequently, a large number of double-membrane, cup-shaped isolation membrane, formed by structures such as the endoplasmic reticulum and mitochondria, emerge in the cytoplasm. 21 These membranes encircle the cargo destined for degradation, forming autophagosome precursors.
The formation of autophagosomes is further driven by ATGs (autophagy-related genes), which orchestrate the conjugation of ATG12-ATG5 and the lipidation of LC3 to PE (phosphatidylethanolamine), essential for membrane expansion and closure.4,22,23 As the isolation membranes gradually extend, the targeted substances are completely enclosed within the autophagosome, which has a double-membrane structure. The autophagosome then fuses with lysosomes, resulting in the formation of an autolysosome where the cargo is degraded by lysosomal enzymes (Figure 1).

A simple diagram of the autophagy process. This diagram delineates the key stages of autophagy, commencing with the nucleation and elongation of the phagophore. The phagophore subsequently engulfs damaged organelles, misfolded proteins, and other cellular debris, forming a double-membraned autophagosome. The autophagosome then fuses with a lysosome, a process mediated by the SNARE complex. Essential SNARE proteins, including STX17, SNAP29, and VAMP8, facilitate this fusion, enabling lysosomal enzymes to degrade the autophagic cargo within the resulting autolysosome. STX17: syntaxin 17; YKT6: YKT6 v-SNARE homolog; VAMP8: vesicle associated membrane protein 8; STX7: Syntaxin 7; SNAP29: synaptosomal associated protein 29; SNAP47: synaptosomal associated protein 47; Rabs: Rab GTPases; EPG5: ectopic P-granules 5 autophagy tethering factor; HOPS: homotypic fusion and protein sorting; PLEKHM1: pleckstrin homology domain-containing protein family member 1; RILP: rab-interacting lysosomal protein; FYCO1: FYVE and coiled-coil domain-containing protein 1; ORP1L: oxysterol-binding protein-related protein 1L; Rab7: a small GTPase. This picture was drawn by Figdraw.
This intricate process not only ensures the removal of damaged or unnecessary cellular components but also contributes to the cellular response to stress, thereby playing a crucial role in various physiological and pathological conditions.24,25 Understanding the molecular mechanisms of autophagy is essential for developing therapeutic strategies for diseases associated with autophagic dysfunction.
Specificity of autophagy in neuronal cells
While the fundamental mechanisms of autophagy are conserved across various cell types, neuronal cells exhibit unique characteristics that underscore the specificity of this process. Neurons are highly polarized and typically do not divide in the adult brain, necessitating efficient autophagy to maintain cellular homeostasis. 26 Given their lack of regenerative capacity, neurons are particularly reliant on autophagy to clear long-lived proteins and damaged cellular structures, such as synapses, which is critical for preventing neurodegenerative diseases.27–29
Neurons contain numerous long-lived proteins and complex cellular structures. Autophagy plays a crucial role in degrading these proteins and damaged organelles, thereby preventing the accumulation of toxic materials that can lead to neurodegenerative conditions. 30 In neurodegenerative diseases such as AD and Parkinson's disease, dysregulation of autophagy is closely linked to disease progression. Reduced autophagic function leads to the accumulation of pathological proteins like tau and α-synuclein, which exacerbate disease pathology by forming toxic aggregates. 31
Additionally, neurons express specialized isoforms of autophagy-related proteins, including Beclin-1 and Atg4, which contribute to the unique autophagic processes observed in these cells. Beclin-1, an essential regulator of autophagy, interacts with the anti-apoptotic protein Bcl-2. This interaction not only inhibits cell death but also regulates autophagy, particularly within the nervous system.32,33 Additionally, Ambra1 (autophagy/beclin-1 regulator-1), which is crucial in neural development, can enhance Beclin 1-dependent autophagy. 34
Understanding the normal mechanisms of autophagosome-lysosome fusion is crucial for elucidating the pathogenesis of AD. 35 Dysregulation of this fusion process has been implicated in the accumulation of protein aggregates and the development of AD. Therefore, further investigation into the molecular mechanisms and signaling pathways involved in autophagosome-lysosome fusion holds great promise for the development of therapeutic strategies targeting neurodegenerative diseases.
SNARE complex: The central mediator of lysosomal fusion
Composition and function of the SNARE complex
The SNARE complex consists of proteins characterized by a coiled-coil SNARE motif, which facilitates membrane fusion events. These proteins are categorized as either target membranes SNAREs (t-SNAREs) or transport vesicles SNAREs (v-SNAREs), regulating membrane fusion in various physiological processes and across diverse species.36–38 In mammalian cells, the fusion of autophagosomes with lysosomes involves key SNARE proteins including STX17 (Qa), SNAP29 (Qbc), and VAMP8. 17 STX17 resides on the autophagosome membrane, VAMP8 is located on the lysosome membrane, and SNAP29 serves as a bridging molecule that facilitates the interaction between STX17 and VAMP8. The assembly of this complex brings the membranes of autophagosomes and lysosomes into close proximity, enabling their fusion and subsequent degradation of autophagic cargo.39–41
In addition to the STX17-SNAP29-VAMP8 complex, the YKT6-SNAP29-STX7 complex also plays a role in autophagosome-lysosome fusion, particularly during starvation-induced autophagy in mammals. 42 Recent research has overturned the traditional view that STX17-SNAP29-VAMP8 and YKT6-SNAP29-STX7 act independently in this fusion process. Instead, it indicates that the pre-assembly of YKT6-SNAP29-STX17 on the autophagosome membrane can significantly enhance the subsequent fusion process driven by STX17-SNAP29-VAMP8 between autophagosomes and lysosomes. 43
Recent studies have highlighted the existence of distinct sets of SNARE complexes that facilitate the integration of autophagosomes with late endosomes or lysosomes, indicating a complex and nuanced regulation of membrane fusion in autophagy (Figure 2(A)). For example, SNAP47, primarily localized to mitochondrial autophagosomes, functions in non-selective autophagy by forming a functional trimeric SNARE complex with STX17 and VAMP7/VAMP8 to mediate mitophagosome-lysosome fusion and is also implicated in hypoxia-induced mitophagy. 44 Additionally, ATG14 possesses tethering capabilities that enhance lipid mixing in protein-free liposome membranes and promote lipid and content mixing in proteoliposomes containing STX17, SNAP29, and VAMP8. 45 These findings indicate that under different induction conditions—whether selective or non-selective autophagy—the types and proportions of SNARE complexes involved in autophagosome-lysosome fusion vary, necessitating tailored research approaches.
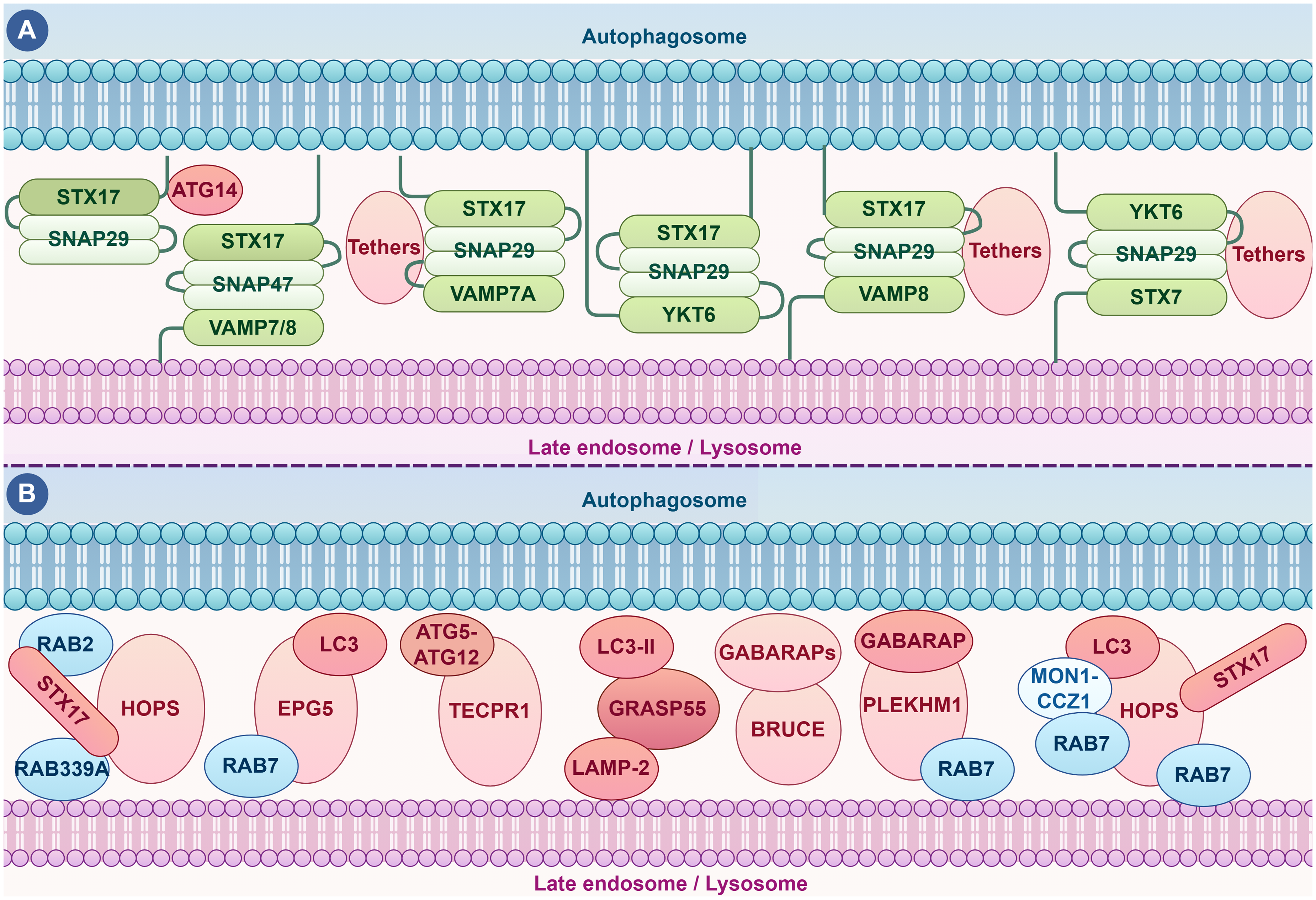
The integration of autophagosomes with late endosomes or lysosomes. (A) SNARE Complexes in Autophagosome-Lysosome Fusion: Recent research has identified specific SNARE complexes as essential mediators of the fusion between autophagosomes and lysosomes. These SNARE proteins assemble into a complex that brings the autophagosomal and lysosomal membranes into close proximity, facilitating their fusion and the subsequent degradation of autophagic cargo. (B) Other Regulatory Proteins Involved in Autophagosome-Lysosome Fusion: Key regulatory proteins include Rab proteins, such as Rab7 and Rab339A, which orchestrate the positioning and coordinated movement of autophagosomes and lysosomes. Additionally, molecules like the HOPS complex component PLEKHM1 function as tethering factors, ensuring the necessary proximity for efficient SNARE-mediated fusion. Together, these components provide a comprehensive overview of the molecular mechanisms and regulatory pathways involved in autophagosome-lysosome fusion, highlighting the complexity and specificity of this critical cellular process. Understanding these interactions is essential for developing targeted therapeutic strategies aimed at enhancing autophagic flux and mitigating the progression of neurodegenerative diseases, such as AD. ATG14: autophagy-related 14; ATG5: autophagy-related protein 5; ATG12: autophagy-related protein 5; VAMP7/8: vesicle associated membrane protein 7/8; Rab2: a Rab small GTPase; Rab39A: a Rab small GTPase; TECPR1: tectonin beta-propeller repeat containing 1; LC3-II: microtubule-associated protein 1A/1B-light chain 3 II, an important marker protein in the process of autophagy; LAPM2: lysosome-associated membrane protein-2; GRASP55: the Golgi reassembly stacking protein-55; BRUCE: an anti-apoptotic factor; MON1-CCZ1: an important protein complex involved in intracellular membrane trafficking and autophagy processes. This picture was drawn by Figdraw.
The precise control of membrane fusion by the SNARE complexes is pivotal for the normal functioning of the nervous system. In neurotransmission, SNARE complexes are essential for the release of neurotransmitters at the synapse, allowing for the propagation of signals across neurons. 46 Similarly, during autophagy, SNARE complexes promote the fusion of autophagosomes with lysosomes, which is a critical step for the degradation of cellular components. Autophagosome-lysosome fusion, as the rate-limiting step in the autophagy process, is essential for the degradation of pathological proteins associated with AD. Efficient clearance of these pathological proteins prevents their accumulation, thereby avoiding neuronal dysfunction and death.47,48 In summary, the SNARE complexes play a multifaceted role in cellular processes, with their involvement in lysosomal fusion being particularly critical for maintaining cellular and neurological health.
Regulation of SNARE complex activity
The activity of the SNARE complex, crucial for lysosomal fusion, is tightly regulated through various mechanisms, including post-translational modifications, regulatory proteins, and lipid components.49,50 This intricate regulation ensures precise control over the fusion process, facilitating the efficient and accurate delivery of cargo to the lysosome.
Post-translational modifications such as phosphorylation, acetylation, and lipidation play critical roles in modulating SNARE protein interactions and functionality. For instance, the deacetylation of STX17 not only promotes its complex formation of with SNAP29 and VAMP8 but also enhances its binding to the HOPS (Homotypic fusion and vacuole protein sorting) complex, further facilitating the fusion between autophagosomes and lysosomes. 51 Conversely,O-GlcNAc glycosylation inhibits the autophagosomal localization of SNAP29, the assembly of the STX17-SNAP29-VAMP8 complex, and subsequent autophagosome-lysosome fusion by potentially hindering the interaction between STX17 and VAMP8, thus reducing autophagic flux. 52 Phosphorylation can alter the conformation of SNARE proteins, affecting their ability to form the core complex necessary for membrane fusion. For example, phosphorylation of YKT6 disrupts its binding with SNAP29, thereby inhibiting autophagosome-lysosome fusion. 53 Similarly, phosphorylation of VAMP8 disrupts SCFD1 (sec1 family domain containing 1) recruitment, inhibiting the formation of the STX17-SNAP29-VAMP8 complex and autophagosome-lysosome fusion. 54 Additionally, the Sec1/Munc18 (SM) protein family plays a key regulatory role, stabilizing SNARE proteins in their prefusion state and facilitating their proper assembly, making it an essential component of the intracellular membrane fusion machinery alongside SNARE proteins. 55
Rab GTPases are crucial for the spatial and temporal regulation of SNARE-mediated fusion by recruiting SNARE proteins to specific membrane sites. 56 The lipid composition of membranes also affects SNARE complex activity; certain lipids can either promote or inhibit SNARE-mediated fusion. For example, STX17 interacts with phosphatidylinositol 4-phosphate (PtdIns(4)P) through its C-terminal positively charged amino acids and colocalizes with PtdIns(4)P on autophagosomes. Knocking down the gene PI4KIIα or using a PI4KIIα inhibitor hinders the recruitment of STX17 to autophagosomes and blocks autophagosome-lysosome fusion. 57
Collectively, these regulatory mechanisms ensure precise control over SNARE complex activity, enabling efficient lysosomal fusion and cellular trafficking processes. Understanding the intricate regulation of SNARE complex activity is essential for developing therapeutic strategies aimed at restoring normal autophagic processes.
Molecules interacting with SNARE proteins
The developmental progression of autophagosomes depends critically on their successful integration with the endolysosomal system, which includes early endosomes, late endosomes/multivesicular bodies, and lysosomes. 39 This fusion is crucial for autophagosomes to acquire the necessary enzymatic content and membrane properties required for maturation and effective function. Autophagosomes may either initially fuse with endosomes to form amphisomes, which subsequently merge with lysosomes for material degradation, or directly fuse with lysosomes to facilitate degradation. In both pathways, the fusion with lysosomes represents a critical step, with the SNARE complex playing an indispensable role in mediating these fusion events. Given the crucial role of the SNARE complex in autophagosome-lysosome fusion, factors interacting with SNARE complex proteins and influencing this fusion must be carefully considered. Existing research has identified several proteins that directly interact with SNARE proteins to impact autophagosome-lysosome fusion.45,58,59 These factors can either promote or hinder the fusion process, as summarized in Tables 1 and 2, respectively.
Factors interacting with SNARE members to promote the fusion of autophagosomes with lysosomes.

Factors interacting with SNARE members to hinder the fusion of autophagosomes with lysosomes.

Beyond their core role in mediating membrane fusion, SNARE proteins interact with various other proteins and lipids, contributing to the regulation and specificity of membrane fusion events. One notable interaction involves the association of SNARE proteins with tethering factors such as the HOPS complex, essential for facilitating autophagosome-lysosome fusion. 68 Technological advancements have revealed the existence of different SNARE subtypes that may compete with each other, exerting opposing effects during autophagosome-lysosome fusion. For example, the VAMP7 subtype VAMP7B, which lacks the long protein structure domain of the main VAMP7A, inhibits this fusion process by interfering with the interaction between VAMP7A and STX17. 60 VAMP7B competes with VAMP7A for binding to the SNARE domain of STX17 through its C-terminal domain. Additionally, the lysosome-localized protein DIPK2A disrupts this interaction, thereby promoting autophagosome-lysosome fusion by enhancing the interaction between STX17 and VAMP7A. 60 This enhancement is crucial for the efficient assembly of the SNARE complex and subsequent membrane fusion.
In addition to protein interactions, SNARE proteins also interact with lipids, influencing their localization and function. Phosphoinositides, for example, regulate the localization and activity of SNARE proteins during membrane fusion events. The successful fusion of autophagosomes and lysosomes is to some extent dependent on the presence of PtdIns4P on autophagosomes, as well as the precise balance and levels of PtdIns(3,5)P2 and PtdIns3P on lysosomes.69,70 PI4KIIα converts PtdIns to PtdIns(4)P, and its gene knockdown inhibits autophagosome-lysosome fusion, leading to the accumulation of autophagosomes. Both PI4KIIα and PtdIns(4)P promote autophagosome-lysosome fusion, underscoring the importance of lipid composition in SNARE-mediated membrane fusion. 69 The specific lipid composition of cellular membranes can impact the recruitment and function of SNARE proteins, thereby influencing the efficiency and specificity of membrane fusion.
In summary, the interactions of SNARE proteins with other proteins and lipids play crucial roles in regulating membrane fusion and vesicle trafficking processes. These interactions contribute to the specificity, efficiency, and regulation of membrane fusion events, highlighting the intricate and multifaceted nature of SNARE protein function in cellular physiology.
Other key players in autophagosome-lysosome fusion
Autophagy progresses through the orderly development of three distinct membrane structures: the phagophore, autophagosome, and autolysosome. Central to the final step of this process are SNARE proteins, including STX17, VAMP8, and SNAP29, which mediate the fusion of autophagosomes with lysosomes.40,71,72 Beyond the SNARE complex, numerous other proteins are essential for successful autophagosome-lysosome fusion (Figure 2(B)). Over 40 ATGs are crucial for the dynamic membrane processes involved in autophagy. 22 The fusion of autophagosomes with lysosomes is a critical step that requires the coordinated action of various key molecules and signaling pathways. RAB proteins, such as Rab7 and Rab24, regulate the positioning and approach of autophagosomes and lysosomes, playing a regulatory role in the fusion process.73–75
Specifically, the GTPase activity of Rab7 is essential for the formation of late-stage autophagic vacuoles, while its effectors RILP and ORP1L facilitate the membrane association of the dynein–dynactin motor machinery, enabling the retrograde transport of autophagic vacuoles and late endosomes/lysosomes. Additionally, PLEKHM1 (pleckstrin homology and RUN domain containing M1), as an effector of RAB7A, recruits HOPS complex to lysosomal membranes and thereby controlling the fusion step in autophagy. 76 Consequently, current research increasingly focuses on enhancing Rab7 GTPase activity and recruiting Rab7 effectors to drive the convergence of autophagosomes and lysosomes. For instance, UVRAG and Rab24 both recruit the Rab7 effector RILP, promoting the mutual approach of autophagosomes and lysosomes.77,78
Furthermore, LAMP2A serves as a receptor for autophagosomal SNAREs, facilitating their interaction with lysosomal SNAREs. 79 The HOPS complex plays a vital role as a tethering factor, bringing autophagosomes and lysosomes into close proximity. Specifically, VPS39 and VPS41 within the HOPS complex act as tethering factors, creating the proximity necessary for SNARE function. Understanding these key molecules and pathways is vital for developing therapies to enhance autophagic flux and mitigate disease progression.
The role of SNARE complex-mediated autophagy in Alzheimer's disease
Accumulation of autophagosomes and lysosomes in Alzheimer's disease
The accumulation of autophagosomes and lysosomes is a hallmark of disrupted autophagic flux, a process crucial for cellular homeostasis and survival.28,80 Autophagosomes are double-membraned vesicles that sequester damaged organelles, misfolded proteins, and other cellular debris, delivering them to lysosomes for degradation and recycling.5,81 When the fusion between autophagosomes and lysosomes is impaired, these vesicles accumulate within the cell. This can result from genetic mutations, environmental stressors, and inhibitory molecules that interfere with the SNARE complex.1,82–84 Understanding the mechanisms underlying this accumulation is essential for developing therapeutic strategies aimed at restoring normal autophagic flux and maintaining cellular health.
A 2005 study found that neurons in APP/PS1 double transgenic AD mouse models exhibited a 23-fold increase in autophagic vacuoles compared to age-matched controls, with numerous immature autophagic vacuoles in dystrophic neurites, while AD patient brains showed significant accumulation of autophagosomes in cortical and hippocampal neurons and elevated levels of LC3-II compared to healthy individuals. 85 Studies examining postmortem brains of AD patients have identified immature autophagosomes within dystrophic axons. 86 This accumulation is more likely attributable to a clearance blockade rather than an increased induction of autophagy. The presence of these immature autophagosomes suggests that impaired autophagic flux, rather than enhanced autophagic initiation, plays a significant role in the pathogenesis of AD. 14 These findings highlight the importance of autophagic clearance mechanisms in maintaining neuronal health and the potential impact of their dysfunction in neurodegenerative diseases.
In conclusion, the accumulation of autophagosomes and lysosomes observed in AD primarily results from disruptions in the autophagic process. 2 Specifically, impediments in the maturation and conversion of autophagosomes, as well as lysosomal dysfunction, lead to the buildup of autophagic vesicles. 87 This accumulation contributes to neuronal damage and disease progression. The inability to effectively degrade and recycle cellular components through autophagy exacerbates neurodegenerative processes, highlighting the critical role of autophagic impairment in AD pathogenesis. As research continues to unravel the molecular intricacies of the SNARE complex, the potential for developing targeted therapies to restore or enhance autophagic flux in AD becomes increasingly promising. The SNARE complex, as the central mediator of autophagosome-lysosome fusion, stands at the forefront of this promising research frontier, offering hope for novel therapeutic strategies against neurodegeneration.
The relationship between autophagosome-lysosome fusion abnormalities and Alzheimer's disease
The relationship between autophagosome-lysosome fusion abnormalities and AD involves complex molecular mechanisms and pathophysiological changes. Autophagy, an intracellular clearance mechanism, degrades and recycles damaged proteins and organelles, maintaining cellular function and homeostasis. 88 In AD, abnormalities in autophagosome-lysosome fusion are key factors driving disease progression. The deposition of amyloid-β (Aβ) and tau proteins, hallmarks of AD pathology, is exacerbated by disruptions in this fusion process, impeding their effective clearance and resulting in pathological accumulation. 89
The pathological changes in AD induced by the continuous accumulation of Aβ lead to abnormal autophagy and a reduction in autophagosome-lysosome fusion, creating a vicious cycle that exacerbates the progression of AD. 90 Autophagy is activated in the early stages of AD, accelerating the degradation and recycling of APP-rich metabolic substrates through the autophagy-lysosome pathway. 86 However, in the later stages of AD, the persistent accumulation of Aβ impairs autophagic function. The toxicity of Aβ interferes with the anchoring effect between the endoplasmic reticulum and microtubules, reducing microtubule stability.91–93
In late-stage AD model mice and the cortical and hippocampal neurons of AD patients, abnormal accumulation of autophagosomes and immature autophagic vesicles indicates disrupted fusion processes.16,80 This disruption hampers the effective clearance of Aβ and contributes to neurodegeneration. Dysfunction in the autophagy-lysosome system results in the formation of tau protein oligomers, directly linking autophagic impairment to tau aggregation. The failure of this pathway to degrade and recycle cellular components leads to tau oligomer accumulation, precursors to the neurofibrillary tangles characteristic of AD. 94
Understanding the mechanisms behind impaired autophagy and autophagosome-lysosome fusion in AD is crucial for developing effective therapeutic strategies.95,96 By investigating these molecular mechanisms and pathophysiological changes, novel therapeutic approaches can be developed. Future research should focus on the specific molecular mechanisms of these fusion abnormalities and how modulating this process might delay or prevent AD progression.
The potential role of specific SNARE protein mutations or aberrant expression in Alzheimer's disease
The terminal phase of membrane reconfiguration within the autophagic pathway is characterized by the genesis of the autolysosome, a process that entails the coalescence of fully-formed autophagosomes with lysosomes, orchestrated by the concerted action of specialized SNARE proteins.97,98 The SNARE complex, a critical component of cellular machinery, consists of conserved proteins that facilitate membrane fusion events. This complex operates through the formation of a four-helix bundle, which overcomes barriers like electrostatic repulsion and hydration layers, effectively pulling the membranes of two organelles together. 99
During autophagy, STX17 can form a SNARE complex with VAMP8 and SNAP29 to mediate the fusion of autophagosomes with lysosomes. Notably, after treatment with Aβ31–35, STX17 expression in mouse hippocampal neural tissue and cells is significantly downregulated, while autophagy marker proteins LC3II and P62 are increased, indicating a pivotal role for STX17 in AD progression. 100 Methamphetamine exposure, which induces AD-like pathological changes, significantly upregulates proteins such as BACE-1, CTFs, pS396-tau, and pS404-tau, while markedly decreasing STX17 expression. 101 This suggests membrane fusion defects between autophagosomes and mature endosomes/lysosomes, as evidenced by increased LAMP1 expression and upregulated proteases cathepsin L (CTSL) and cathepsin D (CTSD). Furthermore, overexpression of STX17 notably reduces the accumulation of Aβ42 and BACE-1 within autophagosomes, while enhancing their delivery to lysosomes. 101 BRUCE interacts with STX17, and that silencing BRUCE can reduce autophagosome-lysosome fusion in AD. 63 This interaction was also confirmed in previous research, which showed that BRUCE modulates STX17-mediated autophagosome-lysosome fusion. 102 This interaction highlights the critical role of SNARE protein alterations in AD pathology and suggests potential therapeutic targets for restoring autophagic function and alleviating disease progression (Figure 3).
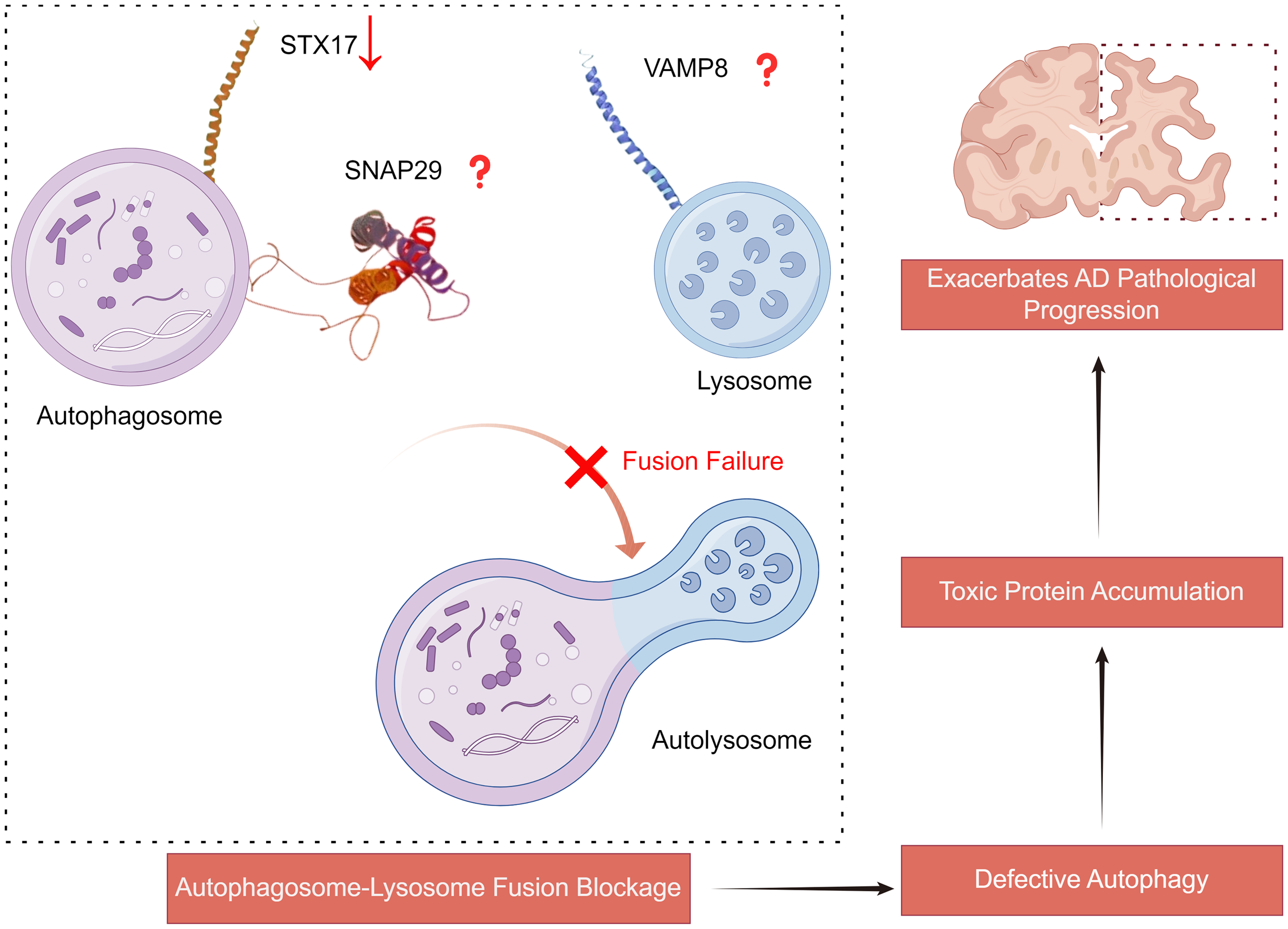
The role of SNARE complex mediated autophagy in AD. Reduced expression of STX17 or abnormalities in other SNARE proteins lead to impaired fusion of autophagosomes with lysosomes. This autophagic defect further results in the accumulation of toxic proteins, thereby exacerbating the pathological progression of AD. STX17: syntaxin 17; VAMP8: vesicle associated membrane protein 8; SNAP29: synaptosomal associated protein 29. This picture was drawn by Figdraw.
SNARE proteins are not only crucial for facilitating the fusion of autophagosomes and lysosomes but also indispensable in neurotransmitter release, promoting the fusion of vesicles with the cell membrane through specific protein complexes. 55 While current research predominantly focuses on SNARE protein mutations in the context of neurotransmitter release, studies on their involvement in autophagosome-lysosome fusion are progressively advancing.
Genetic variability is common in SNARE proteins and can affect their function and stability. In AD, single nucleotide polymorphisms (SNPs) in SNARE complex genes are associated with cognitive impairment. Specifically, certain SNPs in SNAP-25 and Syntaxin-1A (STX1A) are linked to cognitive decline in AD and mild cognitive impairment patients. 103 Furthermore, STX1A expression levels are significantly elevated in individuals with high-functioning autism, possibly related to abnormalities in early brain serotonin synthesis. 104 Additionally, mutations at the O-GlcNAc sites in SNAP-29 significantly enhance the formation of SNAP-29-containing SNARE complexes, which in turn promotes the fusion of autophagosomes with endosomes and lysosomes, thereby facilitating autophagic flux. 52
SNARE proteins as pivotal in both autophagy and neurotransmitter release, making them promising therapeutic targets for AD. The downregulation of STX17 and its interaction with BRUCE may impede autophagosome-lysosome fusion.63,100 Enhancing STX17 function could improve autophagic flux and reduce toxic protein accumulation in neurons. 101 Furthermore, genetic variations in SNARE proteins, such as SNPs in SNAP-25 and STX1A, highlight the potential for personalized medicine, where targeted interventions could enhance treatment outcomes. 103 Additionally, modulating post-translational modifications, such as those that enhance SNAP-29 complex formation, might further facilitate autophagic processes. 52 Overall, targeting SNARE proteins and their associated pathways offers innovative strategies to restore cellular homeostasis and mitigate cognitive decline in AD.
Impaired lysosomal acidification may also affect the fusion of autophagosomes and lysosomes
Lysosomes are critical organelles responsible for degrading and recycling cellular waste, thus playing a vital role in maintaining cellular homeostasis. They contain a diverse array of hydrolytic enzymes capable of breaking down various macromolecules, including proteins, lipids, nucleic acids, and carbohydrates. 105 These degradation products are then recycled back into the cell for reuse in biosynthetic pathways. However, in the presence of defective lysosomal acidification, while fusion between lysosomes and aotophagosomes remains possible, the resulting autolysosomes exhibit impaired degradation capabilities. 106 Mutations in presenilin 1, a major contributor to familial AD, lead to impaired lysosomal acidification. 107
Lysosomal membrane proteins, such as LAMP1 and LAMP2, contribute to the protection of the lysosomal membrane from enzymatic degradation and help maintain the acidic environment required for optimal enzyme activity. A database-based study found that the expression of autophagy-related LAMP1 and LAMP2 is upregulated in AD. 108 In neurons, efficient lysosomal function is crucial due to the high metabolic demands and the necessity for long-term maintenance and function. Impaired lysosomal acidification may affect the close apposition of the SNARE complex, critical for autophagosome-lysosome fusion.
In AD, accumulating evidence indicates significant lysosomes dysfunction.109,110 Studies have shown that lysosomes in AD are often enlarged and exhibit altered enzyme activities. For instance, the level of CTSD, a critical lysosomal protease associated with the dysfunction of the autophagy-lysosomal system, is significantly elevated in blood exosomes, potentially serving as a biomarker for both preclinical and clinical stages of AD. 111 Additionally, the intraluminal acidification of lysosomes to a pH of 4.5–5.0 is critical for their function, and failure in this acidification process is a potential unifying pathogenic mechanism in AD. 112 Recent studies further suggests that the onset of AD is initiated by dysfunction in the acidification of the autophagic-lysosomal system. 113
The acidic environment of lysosomes is crucial for the formation and function of the SNARE complex.114,115 Acidification defects can affect the localization and interaction of SNARE proteins, such as STX17, SNAP29, and VAMP8, thereby hindering the fusion of autophagosomes with lysosomes. Another study arrived at an opposite conclusion regarding methamphetamine-induced AD-like changes. Following methamphetamine exposure, the expression level of LAMP1 increased, and there was a significant upregulation of proteases CTSL and CTSD. These results suggest that lysosomal proteolytic activity may not directly affect the upregulation of AD-like pathological proteins induced by methamphetamine. 101
Discussion
The lysosomal system, particularly the autophagic pathway, serves as the primary mechanism for degrading proteins with long half-lives, as well as for the turnover of organelles and the clearance of large protein aggregates or inclusions within cells. 116 Autophagy plays a pivotal role in maintaining cellular homeostasis by clearing misfolded proteins like Aβ and hyperphosphorylated tau, and by removing damaged organelles, such as mitochondria, which are essential for normal neuronal function and energy metabolism.95,117–119
Central to the regulation of autophagic processes is the SNARE complex, which facilitates the fusion of autophagosomes with lysosomes, thereby ensuring the effective degradation of autophagic cargo. In the context of AD, the interplay between the SNARE complex and autophagy is crucial for neuronal health. The accumulation of autophagosomes and lysosomes in AD neurons indicates significant disruptions in autophagosome-lysosome fusion, highlighting the pivotal role of SNARE proteins in this process. 95 Specifically, the SNARE proteins, including STX17, SNAP29, and VAMP8, orchestrate the docking and merging of autophagosomes with lysosomes, facilitating the degradation of autophagic cargo.40,45
Dysregulation or altered expression of SNARE proteins can severely impede the fusion process, exacerbating the pathological accumulation of autophagosomes and lysosomes observed in AD. In both AD patients and mouse models, there is an abnormal buildup of autophagosomes within cortical and hippocampal neurons, suggesting impaired fusion between autophagosomes and lysosomes. 120 During autophagy, STX17 can form a SNARE complex with VAMP8 and SNAP29 to mediate the fusion of autophagosomes with lysosomes. 121 Furthermore, reduced levels of the SNARE complex in postmortem brain samples from AD patients suggest a correlation between SNARE dysfunction and the onset or progression of AD. 122 Additionally, specific mutations or dysregulation of SNARE proteins may directly contribute to the pathological landscape of AD, underscoring their potential as therapeutic targets.52,63,100,103
Beyond SNARE proteins, impaired lysosomal acidification represents another critical barrier to effective autophagosome-lysosome fusion. 123 Proper lysosomal acidification is necessary for the enzymatic degradation of autophagic contents; disruptions in this process can result in the accumulation of undegraded materials, thereby exacerbating neurodegenerative processes.124,125
As AD progresses, the increase in autophagosomes coupled with restricted fusion to lysosomes leads to significant neuronal loss and death. 126 Therefore, enhancing the fusion process is essential for mitigating AD pathology. Future research should focus on elucidating the precise molecular mechanisms governing SNARE-mediated autophagosome-lysosome fusion and the impact of lysosomal pH modulation. Therapeutic strategies that enhance SNARE complex functionality and ensure proper lysosomal acidification hold promise for addressing the autophagic deficits observed in AD. By restoring autophagic flux, it may be possible to alleviate neuronal damage and slow disease progression. A comprehensive understanding of SNARE complex function is thus vital, as it plays an indispensable role in the fusion of autophagosomes with lysosomes—a critical step in the autophagic pathway. Insights into these mechanisms can inform the development of targeted therapeutic interventions for neurodegenerative diseases characterized by autophagy dysregulation.
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
Author contributions
Siyu Li (Conceptualization; Data curation; Formal analysis; Project administration; Supervision; Validation; Writing – original draft; Writing – review & editing); Yangyang Wang (Conceptualization; Project administration; Supervision; Validation; Writing – review & editing); Xiao Liang (Visualization; Writing – review & editing); Yu Li (Conceptualization; Formal analysis; Funding acquisition; Project administration; Supervision).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Natural Science Foundation of China (NSFC: 82371440) and the Nature Science Foundation of Chongqing (CSTB2023NSCQ-MSX0797).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available within this article.