
Abstract
Background
Type 2 diabetes mellitus (T2D) and Alzheimer's disease (AD) are two prevalent chronic diseases that pose significant global health challenges. Increasing evidence suggests a complex bidirectional relationship between these conditions, where T2D elevates the risk of AD, and AD exacerbates glucose metabolism abnormalities in T2D.
Objective
This review explores the molecular mechanisms linking T2D and AD, focusing on the role of insulin signaling pathways and oxidative stress.
Methods
A comprehensive literature search from PubMed, Web of Science, and other relevant databases was conducted and analyzed.
Results
Insulin resistance in T2D leads to impaired insulin signaling in the brain, contributing to cognitive decline and the development of AD. Hyperglycemia-induced oxidative stress exacerbates neuronal damage, promoting the formation of amyloid-β plaques and neurofibrillary tangles characteristic of AD. Clinically antidiabetic drugs such as metformin show potential against AD in preclinical studies; Many natural products such as Dendrobium nobile Lindl. have anti-T2D efficacy and are also effective against AD in various in vivo and in vitro models.
Conclusions
Improving insulin resistance and reducing oxidative stress are important strategies in the treatment of T2D and AD. To understand the bridging role of insulin singling and oxidative stress in T2D and AD will provide insights and broader applications in alleviating T2D and AD.
Introduction
Type 2 diabetes (T2D) is a chronic metabolic disorder characterized by elevated blood sugar levels and insulin-related issues, such as insulin resistance and a relative lack of insulin. 1 T2D has increased prevalence among the elderly. 2 This condition poses a significant global health threat, as the prevalence of impaired glucose tolerance (IGT) and diabetes has quadrupled over the past three decades. 3 Currently, T2D affects over 463 million people worldwide, a number projected to rise to 700 million by 2045. 4 T2D accounts for more than 90% of all diabetes cases. 5
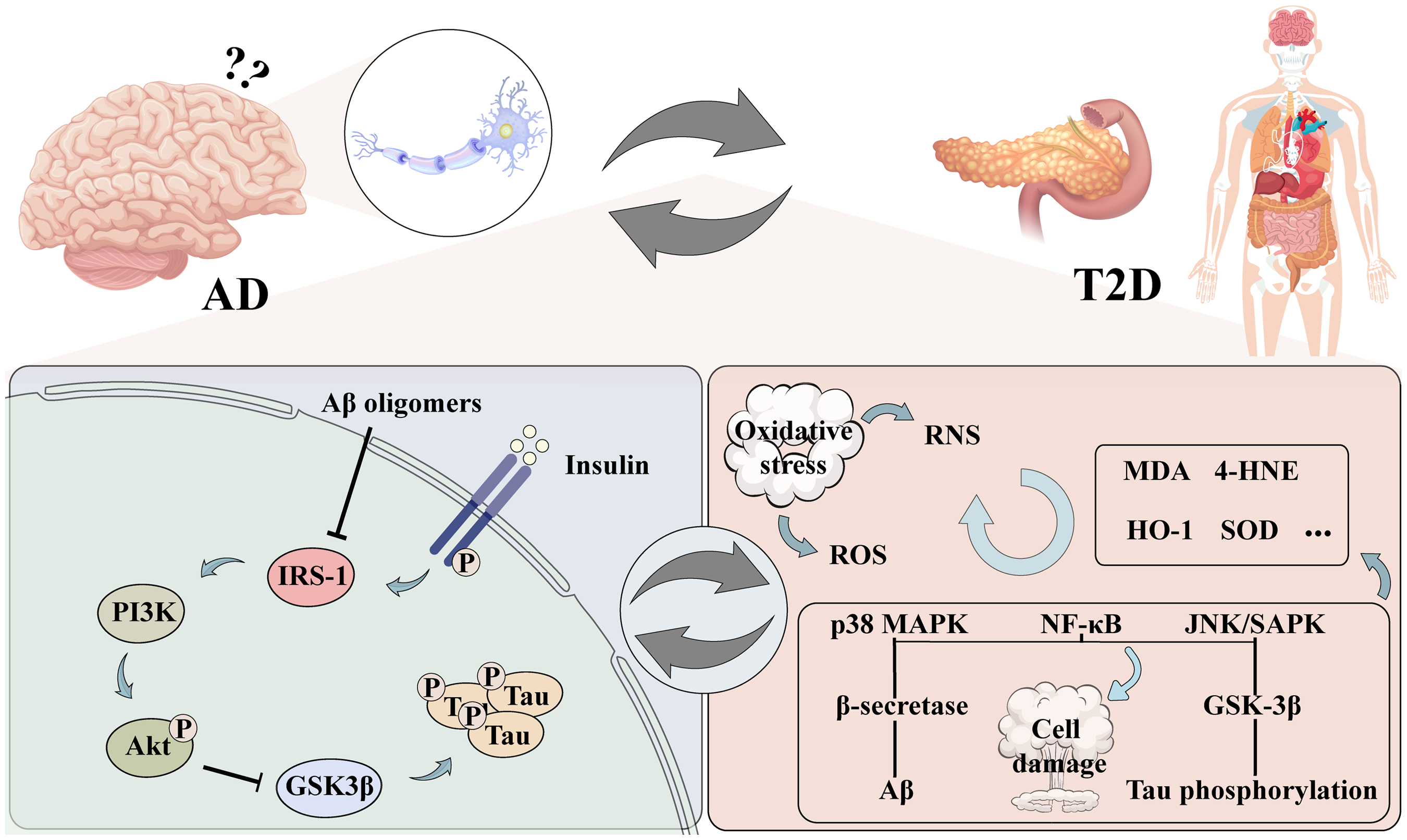
Impact and potential mechanisms of insulin signals and oxidative stress on AD and T2D.
Alzheimer's disease (AD) is the most common age-related neurodegenerative illness, affecting approximately two-thirds of the over 50 million dementia sufferers worldwide. 6 Clinically, AD is marked by progressive memory loss, behavioral and personality changes, and a continual decline in cognitive abilities. Its primary pathological features include the accumulation of amyloid-β (Aβ) peptides, tau protein deposition, and significant neuronal loss in the brain. 7 Despite over a century of research, effective disease-modifying therapies remain elusive, largely due to the complex and multifactorial nature of the disease.
T2D and AD are interconnected, with individuals having diabetes more likely to develop AD. 8 T2D induces neurodegeneration through abnormal glucose metabolism and impaired insulin signaling, affecting normal cellular processes, cerebral blood flow, and neuronal structure, leading to hyperphosphorylation of tau protein and the formation of neurofibrillary tangles (NFTs).9,10 Concurrently, diabetes promotes the accumulation of Aβ peptides and the formation of Aβ plaques, further contributing to neurodegeneration and increasing the risk of AD. 11 Due to the association between disrupted glucose metabolism, insulin dysfunction and AD, some researchers refer AD as “type 3 diabetes”. 12
Oxidative stress is a key factor in the development of both T2D and AD.13,14 Excessive production of reactive oxygen species (ROS) damages mitochondrial function, alters cellular metabolism, and weakens antioxidant defenses, all of which negatively impact cognition function. 15 These changes can also directly affect neurotransmission and synaptic plasticity in the brain. In diabetes, oxidative stress contributes to cellular damage even before many diabetic complications arise. 16 Elevated glucose levels lead to increased ROS production, which reduces insulin sensitivity and induces the apoptosis of insulin-producing pancreatic cells, thereby exacerbating the progression of diabetes mellitus. 17
There are several hypotheses to link T2D and AD, for example, In T2D, AMPK can regulate glucose transporter-4 to improve insulin resistance. In AD, AMPK can improve abnormal brain energy metabolism by reducing Aβ deposition and tau hyperphosphorylation. 18 GSK-3β is a common kinase in insulin signaling transduction and tau protein phosphorylation in T2D and in AD. 19 Chronic TLR4 activation may contribute to insulin resistance in T2D, but also contribute to Aβ deposition and oxidative stress in AD. 20 Therefore, insulin signaling and oxidative stress are two major factors connecting T2D with AD. The aim of this review is to discuss the bridging roles of oxidative stress and insulin signaling in the disease process of T2D combined with AD and related therapeutic strategies (Figure 1).
Insulin signaling in type 2 diabetes and Alzheimer's disease
Insulin signaling and T2D
T2D is known as non-insulin-dependent diabetes and is impacted by a number of hereditary and environmental variables. It is also associated with obesity and islet beta cell degeneration.21,22 Insulin resistance represents a hallmark in the pathogenesis of T2D. At physiological state, insulin binds to the insulin receptor, leading to the phosphorylation of insulin receptor substrate 1 (IRS1). This activation causes PI3 kinases (PI3 e) to phosphorylate PIP2, triggering the Akt pathway. GLUT4 is consequently translocated to the plasma membrane in insulin-sensitive tissues such as skeletal muscle and adipocytes. This translocation lowers blood glucose levels by promoting glucose uptake in the cells.23,24 Additionally, elevated insulin levels alter PI3 K function, leading to the phosphorylation of Rac instead of PIP2, activating NADPH oxidase 4, and generating substantial amounts of ROS. 25 Research indicates that prolonged exposure to glucose adversely affects insulin gene expression in beta cell lines. Experiments conducted with the HIT-T15 beta cell line demonstrated that extended exposure to high doses of glucose resulted in decreased levels of insulin mRNA, reduced insulin content, and impaired glucose-induced insulin release. 26 Thus, regulating insulin signaling, reducing insulin resistance, and lowering blood sugar may help treat T2D, and is a key target for improving T2D related complications.
Insulin signaling and AD
Insulin is a peptide hormone that is primarily secreted by pancreatic β-cells, which is known to influence glucose metabolism, maintain brain function, promote neuronal growth, neurite growth and repair.27,28 Insulin signaling pathway involves binding to insulin and insulin-like growth factor receptors, activating PI3 K, and subsequently promoting Akt phosphorylation, which ensures glucose homeostasis.29,30 Insulin resistance (IR) was originally defined as the need for substantial insulin amounts in T2D patients. Currently, it refers to reduced responsiveness to insulin's metabolic actions, such as impaired gluconeogenesis in the liver and decreased glucose uptake in muscle and adipose tissue. 31 Clinically, IR is characterized by insulin-target cells being less sensitive to elevated insulin levels. 32 It underlies many modern disorders, including metabolic syndrome, such as T2D and AD. 33
It is worthy of note that insulin is an essential element of the nervous system and affects the number of neurological illnesses that arise from insulin abnormality, it regulates behavioral and cognitive processes as well as central nervous system functions.34,35 Under normal physiological conditions, insulin readily traverses the blood-brain barrier (BBB) via receptor-mediated transport mechanisms. However, conditions like inflammation and obesity can modulate this process. 36 In addition, insulin can reach parts of the brain, like the hypothalamus, that are not protected by the BBB. 37 With aging, there is a noted decline in insulin transport into the cerebrospinal fluid (CSF).38,39 This decline may contribute to compromised brain insulin action, predisposing individuals to age-related cognitive dysfunction and neurodegenerative diseases. This assertion is substantiated by findings of decreased CSF insulin levels observed in individuals with AD in several studies. 40
Furthermore, in elderly individuals without dementia, a reduction in cortical insulin levels and receptor binding correlates with advancing age. 41 In AD, disruptions in insulin signaling are apparent in brain tissue. These alterations involve the reduced mRNA and protein expression of insulin, insulin receptor, and IRS1. 28 Moreover, a positive association between AD severity and elevated phosphorylation levels of IRS1 and other insulin signaling pathways is found. 42 In addition, people with AD exhibit decreased sensitivity to peripheral insulin and hyperinsulinemia during fasting and in reaction to an oral glucose tolerance test. 43 Compared to controls, AD patients have fewer or mis-localized insulin receptors—that is, receptors that are not on the membrane surface—and a lower receptor affinity for insulin in their brains. 44 In animal models of AD, both transgenic and non-transgenic mice frequently show compromised insulin signaling. Specifically, insulin resistance signals have been observed in the hypothalamus of APP/PS1 mice. 45 Moreover, central administration of amyloid-β oligomers (AβOs) could induce peripheral insulin resistance, similarly affecting models like APP/PS1 and 3xTg-AD mice. 46
It is reported that the main component of amyloid plaques in the brains of AD patients is Aβ, a peptide derived from amyloid-β protein precursor (AβPP), which AβPP is cleaved by the enzyme α-secretase within its extracellular domain. Proteolysis of AβPP by beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1), β-secretase, and γ-secretase results in the production of Aβ, a neurotoxic peptide. Disruptions in the production, clearance, and aggregation of Aβ lead to its accumulation, which may be a primary etiological factor in AD.47,48 Insulin is reported to influence AβPP metabolism, thereby altering the balance between Aβ synthesis and degradation, while reduced insulin levels are linked to increased Aβ production and amyloid plaque formation in the brain. 49 Additionally, insulin has been demonstrated to reduce extracellular levels of Aβ1–40 and Aβ1–42 in primary cultures of rat cortical neurons and neuroblastoma SH-SY5Y cells. 50 In addition, small oligomers of Aβ are involved in downstream events, including synaptotoxicity, that trigger neurodegenerative processes in AD. it has been discovered that Aβ oligomers may suppress of IRS-1 in both in vivo and in vitro models.51,52 In additional research on the impact of Aβ oligomers in animals, the Aβ42 peptide was discovered to cause APP/PS1E9 mice to develop hepatic insulin resistance by activating Janus Kinase 2 (JAK2).
Moreover, a mild elevation in insulin levels will lead to an increase in insulin-degrading enzyme (IDE) activity. 53 Under normal conditions, Aβ can be broken down by IDE; therefore, if IDE activity is high enough (not IDE levels), it will help remove Aβ from the brain. However, during insulin resistance, insulin levels rise significantly and affect other pathological processes. Under this condition, IDE primarily functions to degrade insulin, which in turn reduces the clearance of Aβ, ultimately leading to Aβ accumulation.54,55
Tau is a protein predominantly located in neuronal axons, where it plays a crucial role in stabilizing microtubules. In AD, one of the primary pathological characteristics is the excessive phosphorylation of tau protein, which leads to the formation of NFTs. These NFTs are believed to impair neuronal function and contribute to the progressive degeneration of neurons. Additionally, smaller aggregates of hyperphosphorylated tau are implicated in further disrupting cellular processes and exacerbating neurodegeneration in AD.56,57 Tau hyperphosphorylation is increased in an insulin-deficient AD mice model. 58 Impaired insulin signaling leads to decreased activity of the PI3K-Akt pathways and activation of GSK-3β, which in turn promotes tau phosphorylation. 59 Insulin inhibits GSK-3β, which prevents tau from being phosphorylated. While insulin resistance is present, GSK-3β activity is elevated, leading to tau phosphorylation and the formation of NFTs. 60 Additionally, peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Studies have demonstrated a significant increase in tau phosphorylation upon deletion of the IGF-1 and IRS-2 genes. And tau phosphorylation is specially enhanced at two distinct GSK-3β targeted sites following deletion of the IGF-1 gene.61,62 According to an analysis, people with T2D had higher CSF tau concentrations than normal people. 63 Increased peripheral insulin resistance has been associated with lower cognitive scores and higher levels of both phosphorylated and total tau protein in patients. Studies using tau knockout mice have demonstrated that reduced brain insulin sensitivity is associated with abnormal synaptic plasticity and deficits in short-term memory. 64
Interaction of insulin signaling in T2D and AD
Impaired insulin signaling in T2D leads to hyperglycemia and hyperinsulinemia, which may affect brain insulin levels and function through the blood-brain barrier. Disrupted brain insulin signaling in AD reduces the expression and functionality of insulin receptors and IRS1. These disruptions in insulin signaling may promote the production and deposition of Aβ and pathological changes in tau protein, exacerbating neuroinflammation and neuronal death. Earlier research revealed that reduced brain insulin receptors (IR) sensitivity, hypo-phosphorylation of the IR, and reduced downstream signaling including IRS1 are all observed in AD patients. 65 Impaired insulin signaling leads to defective glucose uptake and utilization, resulting in decreased energy production, a phenomenon consistent with that observed in the diabetic Zucker rat model.65–67 This energy deficit is accompanied by increased levels of ROS and reactive nitrogen species, which not only cause oxidative and nitrosative damage to mitochondria but also disrupt the pathways involved in synaptic plasticity, leading to neuronal loss, cognitive decline, and cerebral degeneration.68–70 Furthermore, this imbalance results in an excess of ROS within mitochondria, promoting the accumulation of Aβ peptides and causing oxidative damage to proteins, lipids, and nucleic acids in the brain, ultimately resulting in neuronal death.71,72
Oxidative stress in type 2 diabetes and Alzheimer's disease
Oxidative stress and T2D
Pancreatic beta-cells, similar to neurons, exhibit limited expression of antioxidant enzymes, rendering them more vulnerable to oxidative stress. 73 ROS modify various signaling pathways within cells, playing a significant role in influencing glucose tolerance and insulin sensitivity. 74 The generation of ROS activates stress-sensitive pathways, including NF-κB, JNK/SAPK, and p38 MAPK, leading to cellular damage and complications in T2D.26,75 Studies examining islets isolated from the pancreases of individuals with T2D have revealed increased markers of oxidative stress, including the lipid peroxidation products malondialdehyde and 4-hydroxynonenal,76,77 and these markers are closely associated with the degree of impairment in glucose-stimulated insulin secretion.
Additionally, prolonged exposure to fatty acids in pancreatic beta cells has been shown to suppress insulin gene expression.78,79 A cross-sectional study involving elderly individuals with impaired fasting glucose or T2D revealed a negative association between serum 25-hydroxyvitamin D levels and circulating oxidative stress biomarkers.80,81 These biomarkers included advanced glycation end products, advanced oxidation protein products, susceptibility of low-density lipoprotein to oxidation, and nitric oxide metabolites. 82 Moreover, multiple studies indicate that lipotoxicity induces oxidative stress in beta-cells, thereby compromising their function and viability,83,84 and contributing to the initiation and progression of T2D. It can be seen that while ROS is a cellular signal that is necessary for certain physiological processes, its accumulation caused by oxidative stress, high blood glucose and progressive insulin resistance interact, exacerbate the process of T2D disease. Therefore, blocking the vicious cycle of oxidative stress-T2D circles is important in the treatment of disease.
Oxidative stress and AD
Oxidative stress occurs when there is an imbalance between harmful free radicals and the body's antioxidant defenses, leading to an excess of free radicals that can damage lipids, proteins, and nucleic acids. 85 The extent and nature of the damage caused by free radicals can vary depending on their site of generation, the specific type of free radicals involved and their half-lives, their ability to move to different parts of the body from their point of origin. 86
There are two categories of ROS: radicals and non-radicals. Radicals include superoxide (O2•⁻), alkoxyl (RO•), peroxyl (ROO•), hydroxyl (OH•), hydroperoxyl (HO2•), and nitric oxide (NO•). Non-radicals consist of ozone (O3), singlet oxygen (1O2), hypochlorous acid (HOCl), hydrogen peroxide (H2O2), organic peroxides (ROOH), and aldehydes (RCHO). 87 There is mounting evidence that oxidative stress plays a critical role in the development of various human diseases, including both types of diabetes, the progression of neurodegenerative disorders, and cardiovascular disease.85,86
The brain is susceptible to the harmful effects of oxidative stress due to its structure, rapid metabolism, and high lipid levels. 88 Subjects with mild cognitive impairment exhibit significantly higher levels of oxidative changes in the biological macromolecules of hippocampal tissue. 89 The studies suggest that in elderly individuals who are cognitively intact but exhibit AD pathology, oxidative stress is elevated and synaptic protein levels are lowered. 90 This indicates that oxidative damage is already present in the initial preclinical phase of AD. Furthermore, oxidative stress markers such as F2-isoprostanes, malondialdehyde, and trans-4-hydroxy-2-nonenal are found to be upregulated in brain samples and various biological fluids (CSF, plasma, and urine samples) collected from patients with AD and moderate cognitive impairment.91,92 Oxidative stress has also been demonstrated to worsen the accumulation of Aβ and the deposition of NFTs in AD patients, and in models with PS1/PS2 and AβPP defects, as well as in transgenic Tg2576 mouse brains. 93 Pre-clinical research has shown an increase in oxidative stress in the brain's cortex and hippocampus tissues as AD progresses. In the case of model AD rats, the introduction of Aβ42 peptide into the forebrain can result in oxidative stress in the hippocampal tissue, 94 while oxidizing agents can raise local Aβ42 levels in the interstitial fluid in the hippocampal tissue of the mice. 95 On the one hand neurons exposed to Aβ1–42 demonstrated heightened vulnerability to oxidative stress, potentially due to elevated levels of the pro-apoptotic protein Bcl-2-Associated X-protein and downregulation of the anti-apoptotic Bcl-2 protein. 93 On the other hand, excessive ROS might affect the production of Aβ by increasing the amount of amyloid precursor protein and changing the activity of important enzymes like β-secretase and γ-secretase, which are involved in digesting Aβ. Furthermore, oxidative stress stimulates autophagy, which in turn causes Aβ to accumulate in lysosomes and lysosomal enzymes released in turn cause neuronal death. 96
Under high levels of oxidative stress, p38 MAPK activation increases β-secretase production and Aβ deposition, while GSK-3β activation leads to tau phosphorylation and NFT formation. 97 In addition, tau-containing NFTs and elevated levels of heme oxygenase-1 (HO-1) colocalize in the brains of AD patients. Significant HO-1 activity is observed in the hippocampus, cortex, and subcortical white matter. In AD, senile plaques and NFTs also show HO-1 immunoreactivity.98,99 HO-1 degrades the prooxidant heme into biliverdin, which is then reduced to bilirubin, releasing ferrous iron and carbon monoxide in the process. This reduction in oxidative stress is attributed to bilirubin's ability to scavenge radicals, while carbon monoxide offers neuroprotection through its anti-apoptotic and anti-inflammatory properties. 100 Research involving Cu/Zn superoxide dismutase (SOD) deficient animals revealed that increased oxidative stress, particularly in hippocampal tissue, results in cognitive decline similar to that seen in elderly mice.101,102 Furthermore, studies have indicated that both AD patients and animal models exhibit decreased levels of antioxidant enzymes. 103
Preclinical research has demonstrated that the progression of AD in mouse and rat models can be slowed down by correcting mitochondrial dysfunction through the targeted delivery of antioxidants. 104 Vitamin E, a lipophilic antioxidant found in membranes, has been associated with memory loss in aging and dementia. Supplementing with vitamin E increases its levels in AD and decreases the susceptibility of lipoproteins to oxidation. 105 Additionally, vitamin E has been shown to reduce Aβ and tau levels in Tg2576 mice. 106 In general, improving the body's antioxidant system's defense capabilities and improving oxidation restoration balance, can significantly improve the disease process of AD and is an indispensable part of AD treatment.
Interaction of oxidative stress in T2D and AD
Studies suggest that increased diabetic mortalities can be attributed to the rise in vascular diseases, potentially due to oxidative damage.107,108 Likewise, in diabetic rats and the streptozotocin (STZ) model of T2D, the prefrontal cortex, amygdala, and hippocampal regions exhibit decreased activity of antioxidant enzymes such as catalase and SOD, along with a simultaneous increase in lipid peroxidation products.100,109 An overproduction of ROS creates an oxidative stress environment, destabilizing redox signaling. 75 Impaired redox signaling promotes pro-inflammatory and pro-fibrotic pathways, which negatively impact insulin metabolic signaling. Excessive ROS production inhibits the expression of MaFA (mature, adult β-cells express transcription factors), a critical component of the insulin secretion pathway, and the transcription of the insulin gene is controlled by the basic leucine zipper transcription factor family. 110 Adult mice deficient in MafA displayed reduced expression of insulin secretion genes, fasting hyperglycemia, and impaired glucose-stimulated insulin secretion. 111 Inhibition of p38 MAPK-mediated MaFA degradation can enhance insulin production in db/db mice. 112 Oxidative stress can activate p38 MAPK, leading to MaFA degradation and affecting β-cell function and insulin production. 113 Another aspect is chronic oxidative stress-induced inflammation can damage β-cells and induce apoptosis by activating the NF-κB signaling, which promotes the expression of pro-apoptotic Bcl-2 family members.114,115 Moreover, the brain, known for its high cholesterol content, also experiences elevated levels of oxysterols such as 27-OHC, 7β-OHC, and 7-KC-in AD murine brains, 116 which further exacerbate cellular damage by sustaining free-radical chain reactions and worsening insulin resistance.
Additionally, a study revealed that Nrf2 depletion in mice on a high-fat diet can induce hepatic insulin resistance by activating the NF-κB signaling pathway.117,118 Nrf2-KO animals exhibited lower glutathione levels and higher malondialdehyde levels, indicating increased oxidative stress in T2D and AD.119,120 The results suggest that targeting oxidative stress and insulin resistance may enhance antioxidant benefits and thereby improve both T2D and AD.
New therapeutic strategies for type 2 diabetes and AD
Drugs for T2D may have potential against AD
Due to the overlap in the pathological processes of T2D and AD, medications developed for T2D are being investigated for their potential to conquer AD. Among these medications are metformin (MET), glucagon-like peptide-1 receptor agonists, peroxisome proliferator-activated receptor gamma agonists, DDP-4 inhibitors and SGLT2 inhibitors. 121 Research on these treatments demonstrates the promising avenues for combating AD in preclinical settings.
For example, MET is a widely used oral anti-diabetic medication that lowers blood glucose levels through various mechanisms. Studies in STZ-induced Swiss mice and APP/PS1 mice indicate that MET may have therapeutic potential for AD. 121 Additionally, MET was found to markedly decrease BACE1 protein expression and activity in cell culture models, thereby reducing BACE1 cleavage products and the production of Aβ. 122 the pancreatic peptide amylin analogue pramlintide reduces amyloid accumulation in the brain and improves memory and learning in AD model mice. 123 Similarly, in the senescence-accelerated mouse model (SAMP8), which exhibits pathophysiological features similar to those of AD, MET is effective against AD symptoms. Pramlintide (an amylin analog used for the management of T2D) has been shown to promote synaptogenesis by reducing inflammation and oxidative stress and improve AD pathology and cognitive impairment. 124 However, despite its promising results in many preclinical studies, the outcomes in clinical trials still need further investigation. 121
Natural products in T2D and AD treatment
Natural products are a valuable source of lead compounds for T2D and AD treatment due to their unique structures, multi-target capabilities, and diverse activities. 12 These natural products include flavonoids (quercetin), polyphenols (resveratrol), polysaccharides (from Dendrobium nobile Lindl.), alkaloids (berberine), etc.12,125,126
Take Dendrobium nobile Lindl. alkaloids (DNLA) as an example, in animal models of T2D, such as in KK-Ay mice, DNLA lower blood sugar levels, enhance IR, improving insulin signaling, and protect the pancreatic β cells of the pancreatic islets. 127 In db/db mice model, the pancreas of db/db mice exhibits shorter telomeres, and DNLA has been shown to delay this shortening while increasing telomerase activity. 128 In high-fat diet-induced diabetic rat models, DNLA alleviates pancreatic damage. 129 It should be noted that in Aβ25–35-induced AD rat models, DNLA exerts neuroprotective effects by preventing neuronal and synaptic loss while improving cognitive dysfunction through the upregulation of synapses and postsynaptic density protein 95.130,131 In the brain of rats, DNLA can also lessen neuronal death and ameliorate the neuronal disturbance produced by LPS, oxygen-glucose deprivation and reperfusion. 12 DNLA is also effective in high methionine-diet causes AD-like symptoms mouse model, 132 in the accelerated aging SAMP8 mouse model133,134 and in APP/PS1 model mice, 135 DNLA demonstrates anti-aging effects by inhibiting cellular aging processes and decreasing neuronal degeneration, increasing Aβ clearance and alleviating neuronal damage. In in vitro experiment, DNLA not only activates autophagy through the mTOR/ULK1 signaling pathway, 136 but also reduces the level of cellular oxidative stress and thus inhibits the mitochondrial-mediated apoptosis pathway to protect PC12 cell from Aβ25–35 cytotoxicity. 137 Thus, DNLA is effective against both T2D and AD by reducing oxidative damage and improving insulin signaling pathways, thereby mitigating the harmful effects of both T2D and AD. Due to their dual action on these pathological mechanisms, DNLA holds promise as a natural therapeutic option for managing and potentially preventing the progression of T2D and AD.
Conclusions and perspectives
Insulin resistance (IR) and oxidative stress are key factors that link the pathologies of T2D and AD. IR is commonly associated with T2D, leads to impaired glucose metabolism and increased oxidative stress, which in turn damages neurons and contributes to the development of AD features, such as Aβ plaques and tau protein tangles. In the brain, IR disrupts signaling pathways essential for cognitive function, further accelerating neurodegeneration. Oxidative stress exacerbates these effects by promoting inflammation and neuronal damage. The bidirectional relationship between T2D and AD suggests that improving insulin signaling and targeting oxidative stress could offer promising therapeutic strategies for managing both conditions.
Current clinical treatments for T2D and AD can only alleviate symptoms to a limited extent, and halting the disease's progression remains challenging. By conducting a more in-depth investigation into the relationship between T2D and AD, therapeutic strategies for them could focus on addressing the shared pathological mechanisms, particularly focusing on the interactions between IR and oxidative stress. To understand the bridging role of insulin singling in T2D and AD will provide insights and broader applications in alleviating T2D and AD.
Footnotes
Acknowledgments
Thanks for the support of Guizhou Provincial Major Project (No: [2015] 5010).
Author contributions
Fengqing Xu (Investigation; Methodology; Validation; Visualization; Writing – original draft; Writing – review & editing); Jingshan Shi (Conceptualization; Funding acquisition; Supervision; Writing – review & editing).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data that support the findings of this study are available from the corresponding author, [J.S S], upon reasonable request.