
Abstract
The accumulation of amyloid-β (Aβ) peptides is a hallmark of Alzheimer's disease (AD). Central to AD pathology is the production of Aβ peptides through the amyloidogenic processing of amyloid-β protein precursor (AβPP) by β-secretase (BACE-1) and γ-secretase. Recent studies have shifted focus from Aβ plaque deposits to the more toxic soluble Aβ oligomers. One significant way in which Aβ peptides impair neuronal information processing is by influencing neurotransmitter receptor function. These receptors, including adrenergic, acetylcholine, dopamine, 5-HT, glutamate, and gamma-aminobutyric acid (GABA) receptors, play a crucial role in regulating synaptic transmission, which underlies perceptual and cognitive functions. This review explores how Aβ interacts with these key neurotransmitter receptors and how these interactions contribute to neural dysfunction in AD. Moreover, we examine how agonists and antagonists of these receptors influence Aβ pathology, offering new perspectives on potential therapeutic strategies to curb AD progression effectively and improve patients’ quality of life.
Keywords
Introduction
Alzheimer's disease (AD) is one of the leading causes of dementia in the world. The pathology of AD includes the aggregation of amyloid-β (Aβ) proteins, the formation of insoluble plaques, and the production of neurofibrillary tangles resulting from hyperphosphorylated Tau protein aggregation. Most amyloid-β protein precursor (AβPP) is processed through the non-amyloidogenic pathway, where α-secretase cleaves AβPP to produce soluble AβPPα (sAβPPα), followed by γ-secretase cleavage, preventing the formation of intact Aβ. In amyloidogenic pathway, sequential proteolytic cleavage of the AβPP by β-secretase (β-site AβPP-cleaving enzyme 1, BACE-1) and γ-secretase, generates Aβ peptides ranging in length from 38 to 43 residues.1–3 Among the various isoforms of Aβ, Aβ42 and Aβ40 are the most commonly observed in human AD patients. Aβ42 is more prone to aggregation, while Aβ40 is less likely to form aggregates. 4 Therefore, the Aβ42/Aβ40 ratio serves as a marker for early AD progression. Familial AD is often linked to specific mutations in genes encoding AβPP and the catalytic subunit of γ-secretase, known as presenilin.5–7 These mutations tend to facilitate the amyloidogenic processing of AβPP, thereby increasing Aβ production. While amyloid plaque was traditionally regarded as neurotoxic, recent studies have underscored the toxicity of Aβ oligomers,8,9 including their association with long-term potentiation (LTP) impairment, 10 synapse loss, 11 and cognitive dysfunction. 12
Within the complex context of brain function, neurotransmitter receptors serve as important mediators of neuronal network dynamics underlying neural information processing. These receptors, including adrenergic receptors (ARs), acetylcholine receptors (AChRs), dopamine receptors (DRs), 5-HT receptors (5-HTRs), glutamate receptors (GluRs,) and gamma-aminobutyric acid receptors (GABARs) are the key receptors in regulating synaptic transmission and further perceptual and cognitive processes.13–17 Aβ has been shown to alter neuronal signaling by interacting with various neurotransmitter receptors (Figure 1).18,19 Dysregulation or impairment of these receptors in the context of AD can influence neural circuit dynamics and thus exacerbate perceptual and cognitive dysfunction. Despite the continuous effort towards the treatment of AD, little therapeutic progress has been made over the past two decades. Exploring the interplay between Aβ pathology and the activity of these common neurotransmitter receptors would be critical in uncovering the mechanisms underlying AD pathogenesis and identifying new therapeutic strategies.
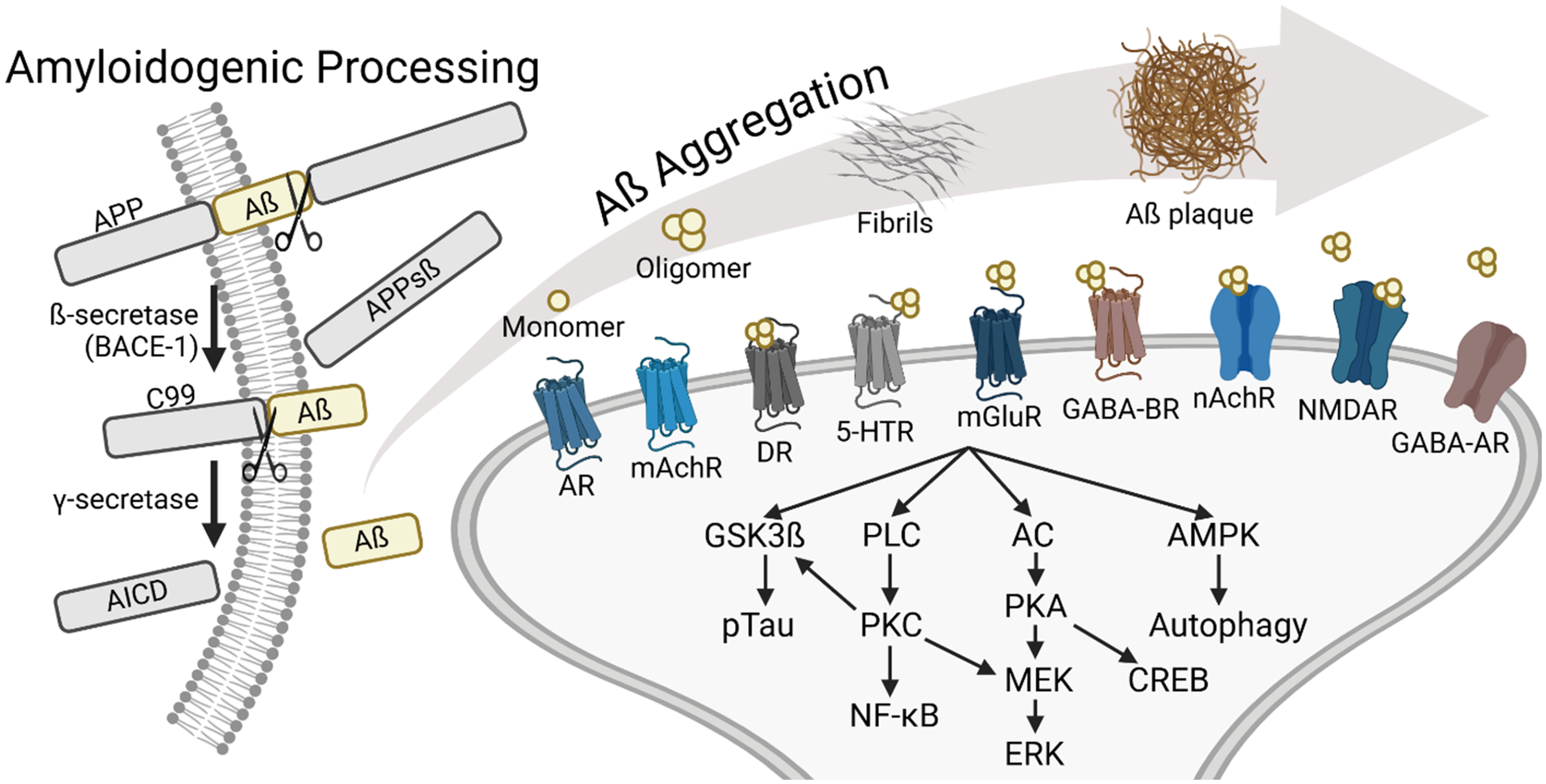
The production of amyloid-β (Aβ) peptides and their interaction with neurotransmitter receptor-mediated signaling pathways. Illustration was created in https://biorender.com.
In this review, we focus on elucidating the role of neurotransmitter receptors in shaping the outcomes of AD treatment. By exploring the evidence and mechanisms through which the activity of neurotransmitter receptors interacts with Aβ pathology, we aim to provide insights that could potentially inform the development of therapeutic interventions for mitigating the impact of AD on brain functions and quality of life.
Interaction between adrenergic receptor activity and Aβ pathology
Adrenergic receptors are a class of G protein-coupled receptors that are targets of norepinephrine. These receptors play essential roles in the regulation of a variety of brain functions.20–22 They are broadly classified into α1, α2, and β subtypes. In the CNS, α2 and β2 adrenergic receptors are the most common. The α2 receptors inhibit adenylyl cyclase activity through Gi proteins, while β receptors stimulate it via Gs proteins. Meanwhile, α1 receptors activate Phospholipase C (PLC) through Gq proteins. 23 Studies have demonstrated that AD is related to the change of locus coeruleus – norepinephrine (LC-NE) system.24–26 For instance, AD mouse models exhibited age-related loss of LC neurons, 27 and this loss coincided with increasing β adrenergic receptor activity. 28 Another example is by increasing β adrenergic activity, researchers preserved Aβ-induced LTP impairment in AD mouse models. 29 Thus, it is important to elucidate different types of adrenergic receptors and their interplay with Aβ.
β2 adrenergic receptors
The β2 adrenergic receptor has been the most studied among all the adrenergic receptor subtypes. Soluble Aβ can directly bind to the β2 adrenergic receptor and trigger a cascade of downstream reactions, including activation of Protein Kinase A (PKA) signaling for GluR1 phosphorylation and enhancement of AMPAR-mediated excitatory postsynaptic current (EPSCs).30,31 The majority of studies on enhancing β2 adrenergic receptor activity concluded that the activation of β2 adrenergic receptors plays a protective role against Aβ toxicity. Specifically, isoproterenol treatment and enriched environments, both of which stimulate β2 adrenergic receptors, have been shown to counteract Aβ-induced hippocampal impairments by activating cyclic AMP (cAMP)-PKA pathway. 32 Moreover, activation of the β2 adrenergic receptor by clenbuterol not only reduced Aβ plaque accumulation by modulating AβPP metabolism on molecular level, but also promoted hippocampal neurogenesis and memory function.33,34 These protective effects extend to the epigenetic level, with procaterol, another β2 adrenergic receptor agonist, inhibiting Aβ-induced synaptotoxicity through regulating histone acetylation. 35 From a translational perspective, human subjects receiving β2 adrenergic receptor agonists exhibited a reduced risk of developing AD, emphasizing the therapeutic potential of these compounds. 36 In contrast, some research suggested a potential adverse effect of β2 adrenergic receptor agonists. Clenbuterol and isoproterenol have been implicated in accelerating hippocampal and cerebral amyloid production, likely due to the enhanced γ-secretase activity.37,38
The inhibition of β2 adrenergic receptors, for example by its antagonist ICI118551, has also yielded contrasting findings. Studies have reported that ICI118551 can attenuate acute stress-induced Aβ production 37 and even reduce Aβ plaque formation with chronic treatment, 38 suggesting potential neuroprotective effects. In contrast, other investigations have revealed that the antagonist can elevate Aβ levels through increasing amyloidogenic AβPP processing30,39 and induce cognitive deficits in AD model mice by inhibiting dendritic ramification. 39 These discrepancies likely arise from a variety of factors, including differences in AD models used, the dosage and duration of pharmacological treatment, and the stage of disease progression. Addressing these variables through standardized protocols and comprehensive studies will be essential for uncovering the precise role and therapeutic potential of β2 adrenergic receptors in AD.
α2 adrenergic receptors
Research focusing on the interface of Aβ and α2 adrenergic receptor has revealed surprising molecular interactions relevant to AD progression. Specifically, Aβ oligomers can bind to the α2 adrenergic receptor with nanomolar affinity, redirecting NE signaling, triggering the glycogen synthase kinase-3 β (GSK3β) cascade and resulting in tau hyperphosphorylation. 40 Moreover, AβPP also directly interacts with the α2A adrenergic receptor subtype, decreasing receptor internalization and potentially modulating NE signaling. 41 As pharmacological interventions, activation of the α2A adrenergic receptor subtype by clonidine has been shown to exacerbate Aβ production by disrupting the interaction between AβPP and Sorting-related receptor with A repeat (SorLA). 42 Conversely, inhibiting the α2 adrenergic receptor could reduce Aβ generation and rescue Aβ-induced cognition dysfunction.42,43
Beyond β2 and α2 adrenergic receptors, researchers also explored the effect of other subtypes of adrenergic receptor on AD progression related to Aβ. For example, researchers found that treatment of CL316243, a β3 adrenergic receptor agonist, effectively rescued Aβ-induced memory dysfunction and reduced Aβ42/Aβ40 ratio.44,45 Furthermore, by inhibiting α1 adrenergic receptor in AD model mice, BACE1 expression and GSK3β phosphorylation were reduced, which in turn resulted in less Aβ production and better behavior performance. 46 (Table 1).
The effect of adrenergic receptor manipulation in AD.

Interaction between cholinergic receptor activity and Aβ pathology
AChRs are divided into two types: muscarinic (mAChRs), which are G-protein coupled receptors, and nicotinic (nAChRs), which are ionotropic receptors. Muscarinic receptors have five subtypes (M1-M5), 47 among which the M1 subtype is prominently expressed in the central nervous system and is significantly associated with AD. 48 nAChRs are found in various subtypes, each with unique properties and distinct distributions within the brain. 49 Research has demonstrated Aβ effects on both nAChRs and mAChRs.50–52
α7-nACh receptors
The α7 nicotinic acetylcholine receptor is a homomeric receptor composed solely of five identical α7 subunits. 53 This receptor is known for its high calcium permeability, which distinguishes it from many other nAChR subtypes. It is widely distributed in the central nervous system and plays a role in cognitive function, learning, memory, and synaptic plasticity. Research has shown Aβ oligomers could bind to the orthosteric binding site of α7-nAChR with high affinity,54,55 and this binding induces concentration-dependent conformational changes on α7nAChR. 56 Consistent with results from several studies that have shown the effects of Aβ on neuronal activity and synaptic function, Aβ itself directly influenced α7-nAChR's function by acting as a negative modulator to reduce their activation duration, 56 indicating that Aβ can functionally act as an α7nAChR antagonist.57,58 While Aβ exposure has been shown to lead to unpredictable alterations in membrane potential and decrease in excitatory postsynaptic potentials (EPSPs) through L-type calcium channels, 59 it also resulted in post-translational and functional upregulation of α7-nAChRs60,61 and suppression of nAChR agonist-induced excitation.62,63 Interestingly, early Aβ-induced neuron hyperactivity, mediated by α7-nAChR, usually followed by synaptic inhibition. 64 These results highlighted a complex effect of Aβ on α7-nAChR. Moreover, the interaction of Aβ with α7-nAChRs can alter the dynamic properties of neuronal networks in amyloid overproducing mice. 65 The role of familial AD-associated Arctic Aβ has also been investigated, revealing its ability to bind to the nAChR α7 subunit and inhibit the calcium ion response and ERK1/2 activation. 66
Interestingly, both activation and inhibition of α7-nAChR yielded beneficial outcomes regarding Aβ toxicity. Agonists of α7-nAChR, such as epibatidine, SSR180711, and A-582941, have been shown to protect receptor loss from Aβ42 toxicity, 54 reverse Aβ-induced synaptic transmission deficit, 67 and enhance cognitive functions or induce neuroprotective effects in WT mice or mouse models of AD.68,69 These effects may result from the ability of α7-nAChR agonists to disrupt the interaction between Aβ and nAChR. 70 Conversely, α7-nAChR antagonists, like methyllycaconitine, have also demonstrated inhibitory effects on certain Aβ-induced toxicities, highlighting the complex role of α7-nAChR in AD pathology. For example, methyllycaconitine can prevent and inverse Aβ binding to α7nAChR and prevent Aβ induced neuronal hyperexcitation.55,61 Also, α7-nAChR antagonist cotreated in cell culture with Aβ42 could diminish Aβ42 associated mitochondrial dysfunction. 71 Additionally, genetic approaches, such as the knockout of the α7-nAChR gene, have been shown to trigger age-dependent AD-like pathology. 72 However, some research suggested that this deletion may result in rescuing cognitive deficits and synaptic pathology in certain AD models.61,73
α7β2 nACh receptors
The α7β2 nAChR subtype represents a more recently identified and less understood configuration, comprising both α7 and β2 subunits. 74 This heteromeric assembly diversifies the functional and pharmacological properties of the receptors to other nAChRs. 75 For example, their choline-induced current showed smaller amplitude and a longer duration compared to homomeric α7-nAChR. 75 Researchers also found that α7β2-nAChR exhibits greater sensitivity to Aβ42 oligomers compared to α7-nAChR, as its function can be blocked by lower nanomolar concentrations of Aβ42 oligomers.76,77 This heightened sensitivity may indicate a unique role for α7β2 nAChR in AD pathogenesis. Activation of α7β2-nAChR receptors by Aβ42 oligomers also triggered hyperexcitation and degeneration of basal forebrain cholinergic neurons. 78 At molecular level, Aβ42 oligomers also preferentially extend the open-dwell times of α7β2-nAChR, likely contributing to cognitive decline in AD. 78 In addition, APP/PS1 transgenic mice lacking α7β2-nAChR displayed improved spatial reference memory compared to normal APP/PS1 transgenic mice, further emphasizing the significance of this receptor subtype in AD pathology. 78
α4β2 nACh receptors
The α4β2 subtype is one of the most abundant nicotinic receptors in the brain. Compared to the α7-nAChR, it has slower activation and desensitization kinetics and lower calcium permeability.79,80 The α4β2-nAChR also plays a critical role in modulating synaptic plasticity and cognitive processes. Essential insights about α4β2-nAChR in AD come from studies that showed Aβ reducing the expression of α4β2-nAChR in cell culture. 81 Epibatidine is a potent agonist of α4β2-nAChR and a less potent agonist of α7-nAChR. 82 Aβ's ability to suppress epibatidine-induced currents in rat hippocampal CA1 pyramidal neurons demonstrated its negative impact on the α4β2- and α7-nAChR subtype. 63 Additional findings revealed that inhibition of Aβ40 on AChR-evoked dopamine release is partly mediated by the extracellular interaction between α4β2-nAChR and Aβ40 molecule. 83 The ability of Aβ on selectively targeting α7- and α4β2-nAChRs attributed to its interaction with arginine 208 and glutamate 211 on the α7 & α4 subunits. 84 Moreover, coactivation of α7- and α4β2-nAChRs reversed AMPAR dysfunction and LTP disruption induced by Aβ. 84
Muscarinic ACh receptors
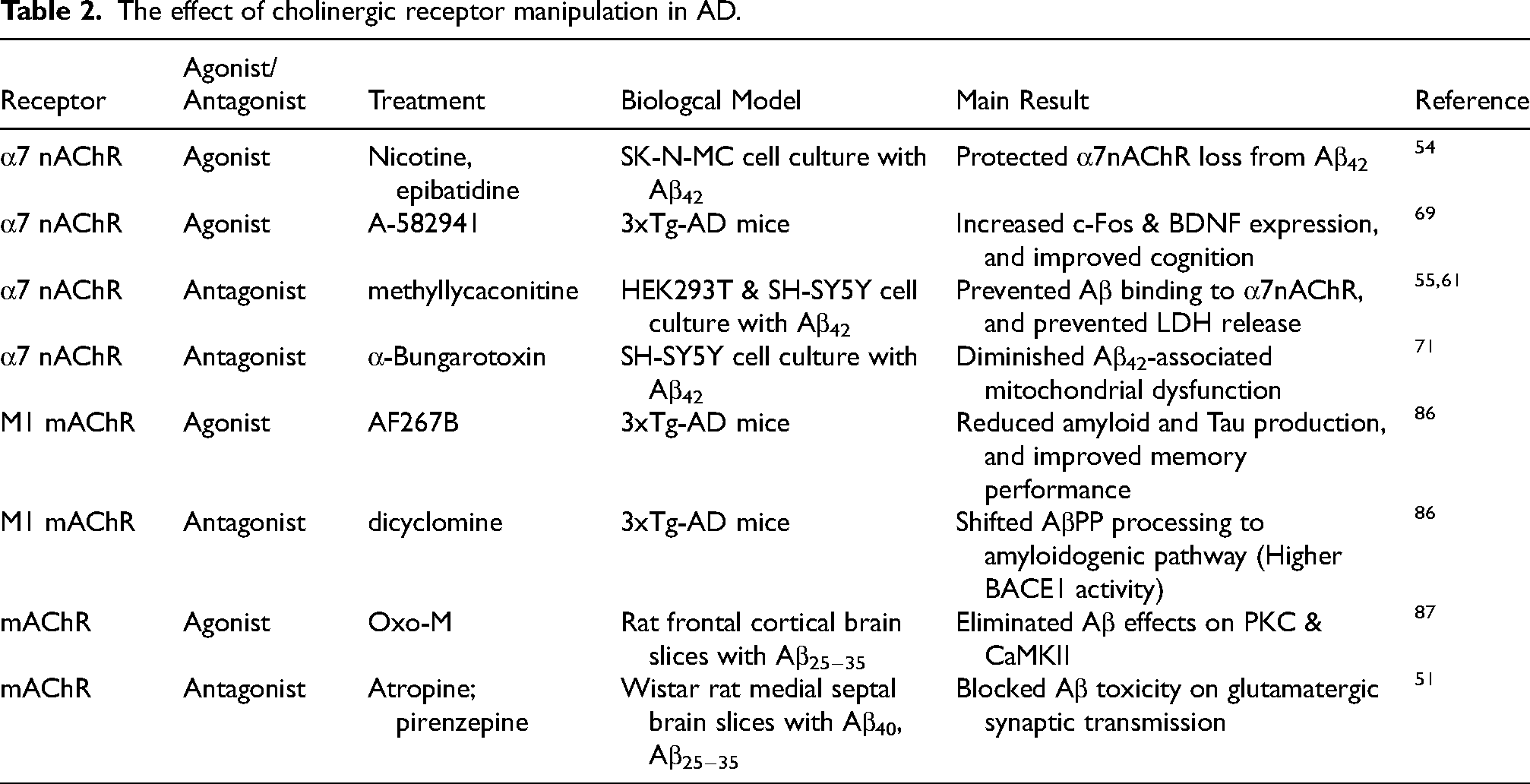
mAChRs are a class of G protein-coupled receptors that respond to the neurotransmitter acetylcholine. Unlike nAChRs, which are ion channels, mAChRs influence cells through a variety of signal transduction pathways. 85 There are five known subtypes of mAChRs, designated M1 through M5, each with distinct functions and pharmacological profiles. A study showed that M1 receptors significantly modulate AD-like pathology in 3xTg-AD transgenic mice, with M1 mAChR agonist AF267B leading to an improvement in memory performance and a reduction in amyloid and tau pathology. 86 This study also found that dicyclomine, an M1 antagonist, yielded opposite results, thereby emphasizing the potential therapeutic importance of M1 mAChR. 86 The interaction of Aβ and mAChR extends to synaptic function as well. It has been demonstrated that Aβ-induced EPSC reduction could be mitigated by atropine, a mAChR antagonist, and calcicludine, a calcium channel antagonist. 51 This finding also indicated that the mAChR plays a more fundamental role than the nAChR in this context since nAChR antagonist has no such effect. Complementing this observation, another study showed that the enhanced activation of intracellular signaling enzymes Protein Kinase C (PKC) and CaMKII by Aβ could be inhibited by oxo-M, an mAChR agonist. 87 However, nAChR agonist had no such effect. 87 Further complicating the Aβ-mAChR interaction, a study suggested that Aβ oligomers interfere with the functionality of the M1 mAChR by altering its interaction with G-proteins 88 or modulate M5 mAChR signal transduction intracellularly. 83 Together, it is clear that the mechanisms through which Aβ and mAChR interact in AD are complicated, and future research is needed to further elucidate these complex interactions. (Table 2).
The effect of cholinergic receptor manipulation in AD.
Interaction between dopaminergic receptor activity and Aβ pathology
In addition to adrenergic receptors, dopaminergic receptors represent another major type of catecholamine receptors. Dopamine receptors are a class of GPCRs and consist of five main subtypes, labeled D1 through D5, which are divided into two main classes based on their pharmacological properties and effects: the D1-like receptors (D1 and D5) and the D2-like receptors (D2, D3, and D4). 89 In the CNS, the most prominent subtypes are D1 and D2 receptors. D1 receptors, primarily excitatory, are coupled to Gs proteins, which increase the cAMP and thus promoting cellular signaling pathways that modulate motor control, cognition, and reward systems. In contrast, D2 receptors are inhibitory and coupled to Gi proteins, which reduce cAMP and modulate neuronal excitability. Because D2 receptors are critically involved in regulating motor functions, mood, and motivation, they serve as essential pharmacological targets in the treatment of disorders like Parkinson's disease and schizophrenia.90,91 In comparison to Parkinson's disease and schizophrenia, research on dopamine receptor involvement in AD is relatively limited. However, altered dopamine receptor levels have been observed in AD patients.92–94 PET imaging studies have demonstrated reduced D2 receptor binding availability in AD brains. 94 Immunohistochemical analyses revealed significantly decreased expression of cortical D1, D3, and D4 receptors, while D5 receptor expression was elevated. 93
Dopaminergic D1 receptors
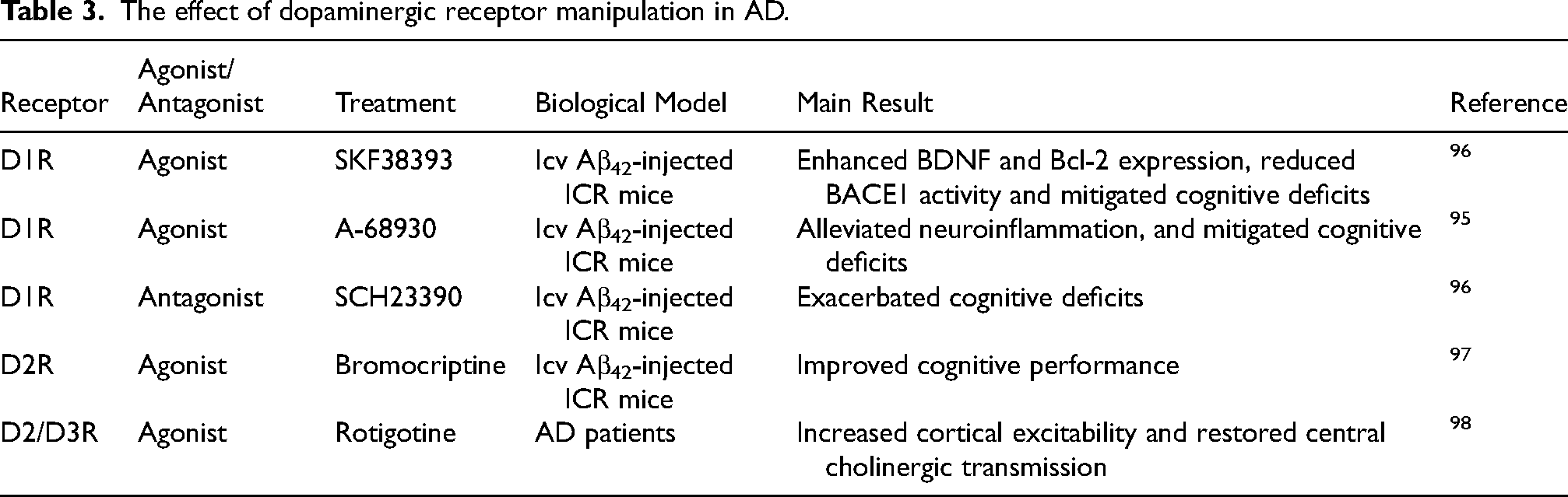
Recent studies have begun to explore the effects of dopamine receptor modulation on amyloid pathology. For instance, D1 receptor agonists such as SKF38393 and A-68930 have shown promise in mitigating cognitive deficits induced by intracerebroventricular (icv)-injected Aβ.95,96 The underlying mechanisms remain under investigation, with researchers identifying different pathways of action. SKF38393 has been found to increase cAMP response element binding protein (CREB) phosphorylation, subsequently enhancing brain-derived neurotrophic factor (BDNF) and B-cell Lymphoma 2 (Bcl-2) expression and reducing BACE1 activity. 96 Meanwhile, A-68930 appeared to alleviate Aβ-induced neuroinflammation via an AMPK/autophagy pathway, promoting NLR family pyrin domain containing 3 (NLRP3) inflammasome degradation and reducing IL-1β and IL-18 levels. 95 Conversely, the D1 receptor antagonist SCH23390 demonstrated opposite effects to SKF38393, further underscoring the therapeutic potential of D1 receptor agonists. 96
Dopaminergic D2 receptors
In studying the effect of dopaminergic D2 receptor activity in AD, bromocriptine, a D2 receptor agonist, demonstrated protective effects against cognitive impairment induced by icv-injected Aβ. 97 Mechanistic studies in both in vivo and in vitro models revealed that bromocriptine, through dopaminergic D2 receptor activation, recruited protein phosphatase 2A (PP2A) and c-Jun N-terminal kinase (JNK) via β-arrestin 2. This action inhibited JNK-mediated transcription of proinflammatory cytokines and prevented NLRP3 inflammasome activation in microglia. Rotigotine, another D2/D3 receptor agonist, has been shown to increase cortical excitability and restore central cholinergic transmission in AD patients. 98 (Table 3).
The effect of dopaminergic receptor manipulation in AD.
Interaction between 5-HT receptor activity and Aβ pathology
The 5-HT (serotonin) receptors are a diverse group of receptors that mediate the effects of serotonin across both the central and peripheral nervous systems. They are classified into seven families, from 5-HT1 to 5-HT7, with most subtypes being GPCRs that modulate intracellular signaling pathways. 99 An exception is the 5-HT3 receptor, which is a ligand-gated ion channel responsible for fast excitatory neurotransmission. The 5-HT1 family, including subtypes like 5-HT1A and 5-HT1B, is primarily inhibitory, reducing cAMP levels and decreasing neuronal excitability. On the other hand, families such as 5-HT2, 5-HT4, and 5-HT6 are excitatory, with 5-HT2 increasing intracellular calcium through Gq signaling and 5-HT4 and 5-HT6 stimulating cAMP production via Gs signaling. 100 Each subtype shows a distinct pattern of expression across brain regions and contributes to a wide range of functions, including mood regulation, cognition, appetite, and circadian rhythms. For instance, 5-HT1AR is highly expressed in the midbrain, limbic system (especially the hippocampus), and cortex, while 5-HT2AR is predominantly located in the cortex, particularly in high-level associative. 101 Research has shown that Aβ affects the serotonergic system, disrupting normal 5-HT receptor signaling. 102 The interaction between Aβ and 5-HT1A, 5-HT2A, 5-HT2B, 5-HT4, and 5-HT6 receptors has been the most extensively studied, revealing important insights into their roles in neurodegenerative processes.
5-HT1A receptors
The 5-HT1A receptor, part of the 5-HT1 receptor family, is highly expressed in brain regions such as the hippocampus and amygdala, which are crucial for emotional processing and cognitive functions. It is primarily an inhibitory GPCR that reduces cAMP production by inhibiting adenylate cyclase, therefore decreasing neuronal excitability. PET imaging studies have shown altered 5-HT1A receptor expression during different stages of AD, with some studies reporting a reduction in 5-HT1A density,103,104 while others have observed an upregulation.105,106 The interaction between Aβ and 5-HT1A receptors presented complex effects on neuronal function and cognitive outcomes in AD models. Aβ40 and Aβ42 differentially influenced 5-HT1AR expression: Aβ40 induced receptor overexpression, possibly as a protective mechanism, whereas Aβ42 caused neuronal lesions without affecting receptor levels. 107 In models of memory loss induced by streptozotocin (STZ), which mimic memory impairments in AD, the 5-HT1AR antagonist NAD-299 mitigated memory deficits, reduced oxidative stress, and decreased neuronal loss.108,109 Additionally, the 5-HT1AR antagonist WAY100635 reduced neuroinflammation and improved cognitive performance in Aβ42-injected mice, possibly through the NF-κB pathway. 110
5-HT2 receptors
Research has shown both AD mouse model and human patients exhibit a loss of 5-HT2A receptors in various brain regions, including the hippocampus, medial prefrontal cortex (mPFC) and cerebral cortex.111–115 Studies using PET imaging in AD patients have revealed a reduction in cortical 5-HT2AR binding, independent of serotonergic neuron loss. This finding suggested that 5-HT2AR loss may be an early feature of AD. 111 Injecting Aβ42 into the hippocampus led to reductions in BDNF, memory deficits, and a loss of hippocampal 5-HT2AR.114,115 Both 5-HT2AR agonists and antagonists showed therapeutic potential in AD treatments. In a STZ-induced rat model of memory loss, the 5-HT2AR agonist TCB-2 has been shown to alleviate memory deficits, reduce oxidative stress, and mitigate neuronal loss, suggesting a neuroprotective effect.108,109 Moreover, Desloratadine (DLT), a selective 5-HT2AR antagonist, reduced Aβ plaque deposition in AD model mice by facilitating microglial phagocytosis of Aβ. 116 Unlike the 5-HT2AR, pioneering research indicated that patients with sporadic AD exhibit elevated expression of the 5-HT2BR in the cortex. 117 Additionally, there was an increase in 5-HT2BR mRNA expression associated with Aβ accumulation. 118 The selective 5-HT2BR antagonist, MW701, has demonstrated promising results in mitigating Aβ42-induced impairments in LTP and memory deficits. 117
5-HT4 receptors
The 5-HT4 receptor is another excitatory serotonin receptor. It activates the cAMP-PKA pathway, increasing neuronal excitability. Predominantly expressed in the hippocampus, 5-HT4 receptors are widely recognized for their involvement in learning and memory. Treatment with the 5-HT4R agonist RS-67333, both short-term (two weeks) and long-term (four months), improved memory in AD model mice while reducing Aβ load and neuroinflammation.119–121 Moreover, treatment with both RS-67333 and Usmarapride (a 5-HT4R partial agonist) increased soluble AβPPα, indicating the possible involvement of α-secretase activation.121,122 Research has further suggested that 5-HT4R agonists modulate AβPP processing to favor the non-amyloidogenic pathway.123–125 Activation of α-secretase may occur through the ERK signaling pathway via cAMP and PKA signaling, which activates MEK and ERK, enhancing α-secretase ADAM10 activity and reducing Aβ levels.123,125 Evidence also suggested that 5-HT4R may activate α-secretase through a G-protein and Src-dependent activation of PLC, bypassing cAMP and PKA signaling. 124 These findings highlighted the complexity of GPCR signaling in AβPP processing and Aβ metabolism.
5-HT6 receptors
The 5-HT6 receptor, primarily expressed in the hippocampus, is important in mediating learning and memory. Treatment with 5-HT6R antagonists, such as AVN-211 and SB-25858, has shown positive effects in attenuating Aβ-induced memory loss,120,126–128 likely by regulating the morphology and function of neuronal primary cilia. 129 There is also evidence that 5-HT6R agonists can decrease amyloid pathology and prevent memory loss.123,130 One potential mechanism is that 5-HT6R activation would increase α-secretase activation through the PKA-ERK pathway. However, further research is needed to further clarify the mechanisms by which 5-HT6R modulates amyloid pathology.
Research on the interaction between Aβ and other types of 5-HT receptors is limited, but there is potential for discovering novel therapeutic targets for AD. For instance, the 5-HT7R agonist AS19 has shown promising effects in reducing Aβ plaque deposition, preventing neuronal apoptosis, and improving memory performance in rat AD models.131,132 (Table 4).
The effect of 5-HT receptor manipulation in AD.

Interaction between glutamate receptors and Aβ pathology
Glutamate is the primary excitatory neurotransmitter in the central nervous system. Given the importance of the glutamate system in memory formation, the interaction between Aβ and glutamate receptors has been heavily studied. Glutamate receptors are broadly categorized into ionotropic and metabotropic types. The ionotropic receptors include NMDA (N-methyl-D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors, which are directly involved in fast synaptic transmission. Metabotropic glutamate receptors (mGluRs) are G protein-coupled receptors that modulate neuronal and synaptic function through secondary messenger systems. It can be further divided into 3 groups. Group I mGluRs, including mGluR1 and mGluR5, activate phosphoinositide hydrolysis, leading to increased intracellular calcium levels and activation of PKC. Group II mGluRs, such as mGluR2 and mGluR3, and Group III mGluRs, which include mGluR4, mGluR6, and mGluR7, inhibit adenylate cyclase activity, resulting in a reduction of cAMP levels. 133 These distinct signaling mechanisms allow mGluRs to play diverse roles in modulating neuronal excitability and synaptic plasticity.
NMDA receptors
NMDARs are distinguished by their voltage-dependent activation, requiring both glutamate binding and membrane depolarization to relieve Mg2+ block of the ion channel. This makes them highly sensitive to the synaptic activity and capable of detecting coincident pre- and postsynaptic activity, fundamental for the synaptic plasticity processes like LTP. The relationship between Aβ and NMDAR is most studied among all glutamate receptors. Aβ was shown to decrease the surface expression of NMDAR.134,135 More specifically, Aβ oligomers colocalized with GluN1 and GluN2B, further leading to the loss of GluN2B subunit.136,137 and a shift in NMDAR composition from GluN2B to GluN2A. 138 NMDAR played an important role in Aβ-induced spine loss and LTP impairment.139–142 One possible reason for Aβ oligomers-induced spine loss could be their role in reducing calcium influx into active spine through NMDAR, 143 or it could be the hyperactivation of caspase-3. 144 All the subunits, GluN1, GluN2A and GluN2B, were required for Aβ-induced spine loss. 135 As for LTP impairment, Aβ has been shown to inhibit LTP by enhancing extrasynaptic NMDAR-mediated function, but not via synaptic NMDAR.140,141 Among all NMDAR subunits, GluN2B is particularly significant in Aβ-related pathology. Aβ oligomers mainly targeted hippocampal extrasynaptic NMDAR containing GluN2B 141 and resulted in loss of GluN2B response. 138 In addition, the dephosphorylation of GluN2B at Tyr1472 was found to correlate with Aβ-mediated NMDAR endocytosis. 145
Some research indicated NMDAR activation can increase the activity of α-secretase and thus inhibit amyloid pathogenesis. 146 However, a different viewpoint claims that after separating the functions of extrasynaptic and synaptic NMDAR, extrasynaptic NMDAR activation can shift AβPP isoform from AβPP695 to Kunitz protease inhibitory domain (KPI) containing AβPP (KPI-AβPP). 147 KPI-AβPP has a higher amyloidogenic potential, meaning KPI-AβPP could inhibit α-secretase pathway and enhance β-secretase processing. 148 In parallel, the calcium pathway played an important role in Aβ-NMDAR interaction. Studies suggested that Aβ oligomers directly reduce NMDAR-mediated calcium influx, particularly in highly active synapses.134,143,149 Furthermore, the dose of Aβ needed to block calcium entry for NMDAR is much lower compared to the dose needed to induce NMDAR endocytosis. 149 This result indicated a new early-stage impairment of Aβ on NMDAR metabolism. However, other evidence suggested that instead of inhibiting calcium entry, Aβ oligomers may enhance calcium influx through the activation of NMDAR, ultimately leading to mitochondrial dysfunction and neuronal loss. 150
Inhibition of NMDAR for AD treatment has already been successful on the bedside. Memantine, a non-competitive NMDAR antagonist, has been approved by FDA for AD treatment.151,152 One potential mechanism of action of memantine therapy might be its ability to block extrasynaptic NMDAR-mediated currents without affecting the normal synaptic NMDAR-mediated currents. 153 Studies also found memantine inhibited NMDAR-mediated KPI-AβPP production, hence inhibiting Aβ generation, in a dose-dependent manner. 147 Chronic treatment of memantine not only reduced Aβ oligomers and prevented Aβ-induced LTP impairment, 154 but also reduced neuronal loss and prevented memory dysfunction.155,156 Beside memantine, AP5, another broad NMDAR antagonist, also showed similar effects on preventing Aβ-induced LTP impairment 157 and calcium level imbalance. 158 On the contrary, 3-(2-carboxypiperazin-4yl) propyl-1-phosphonic acid (CPP), also a NMDAR antagonist, increased Aβ load in the brain and triggered spine loss.143,159 People has now been focusing on the manipulation of GluN2B subunit. Ifenprodil and Ro 25-6981, both GluN2B antagonist, showed to prevent Aβ oligomer-induced deficit in LTP and NMDAR impairment.149,160,161 These findings indicated the significance of GluN2B in Aβ pathological impact.
AMPA receptors
AMPA receptors are the main mediators of fast excitatory synaptic transmission inside the brain. Their rapid activation by glutamate allows for quick changes in membrane potential via the influx of sodium. Research has shown Aβ decrease the density of AMPAR in the cortex. 162 One possibility is that Aβ induced AMPAR ubiquitination via increasing AMPAR E3 ligase Nedd4 and decreasing AMPAR deubiquitinase USP46 expression. 163 This process depended on the presence of GluR1 subunit, whose synaptic expression was decreased with the presence of early Aβ pathology,164,165 but the detail mechanism has been controversial. Some argued that it is due to GluR1 phosphorylation, since GluR1 ubiquitination-deficient could increase GluR1 phosphorylation, and further prevent Aβ-induced AMPAR endocytosis. 166 However, some believed Aβ-induced caspase-3 activity enhancement would cause dephosphorylation of GluR1 via calcineurin and thus resulted in AMPAR degradation. 167 The Aβ effect on GluR1 subunit may be an explanation of why GluR1 knockdown, but not GluR2 knockdown, could prevent Aβ toxicity on AMPAR-mediated EPSC enhancement. 168 Beyond GluR1 subunit, Aβ has also shown effect on GluR2 and GluR3 subunits. Human AD samples had fewer GluR2/3 subunits on neuronal membrane. 169 What's more, the phosphorylation of GluR2 170 and GluR3 by Aβ is essential for its role in causing synaptic impairment and memory dysfunction. 171 CaMKII may also play an important role in Aβ-AMPAR interaction, since CaMKII expression enhancement could rescue Aβ-induced AMPAR deficit on its ionic current and response. 165 Similar to NMDAR, research has shown Aβ oligomer could induce overactivation of AMPAR, leading to a substantial calcium influx and subsequent neuronal oxidative stress. 150 Furthermore, intracellular Aβ oligomers have been shown to induce neuronal hyperexcitability through AMPAR-mediated current. 172 In addition to Aβ changing AMPAR downstream pathway, AMPAR also mediates Aβ metabolism. Research has shown steady-state AMPAR activity could increase ISF Aβ level. 173 However, evoked AMPAR activity could reduce Aβ level in a dose-dependent manner via a pathway including NMDAR and IL-6. 173 Although treatments based on the manipulation of AMPAR are relatively limited, previous work has shown that hippocampal neurons lacking the GluR3 subunit were protected from Aβ-induced synaptic depression, spine loss, and impairment of LTP. 171 The behavioral outcome matched this finding, as GluR3-deficient APP mice maintained normal memory despite Aβ overproduction. 171 The inhibition of AMPAR desensitization, which is the ability of AMPARs change their conformation under prolonged exposure to glutamate to prevent neuronal overexcitation, by cyclothiazide rescued synaptic plasticity in AD model mice. 162 The research on behavior level is lacking regarding AMPAR regulation.
mGluR5 receptors
mGluR5 is a subtype of metabotropic glutamate receptors that plays a crucial role in modulating neuronal excitability and synaptic plasticity across various brain regions. 174 Cellular prion protein (PrPC) plays a center role in the interaction between Aβ and mGluR5. Experiments have shown Aβ can interact with mGluR5 through a connection mediated by the PrPC.175,176 The Aβ-PrPC-mGluR5 complex not only activated Fyn kinase which led to dendritic spine loss, 175 but also mediated synaptic plasticity alterations. Its effect on synaptic plasticity particularly suppressed LTP and facilitated LTD.139,176 By preventing Aβ binding to PrPC, the Aβ-induced LTD facilitation could be blocked. 176 On the other hand, research has shown mGluR5 binds to PrPC via intracellular protein mediators, including Homer1b/c, CaMPKII and tyrosine kinase 2β. Aβ oligomers have been reported to increase the phosphorylation level of these intracellular protein mediators and thus induce deficit of synaptic plasticity. 177 Furthermore, Aβ showed high binding affinity to mGluR5 via PrPC only in male mice and human brain samples, but not in female samples. 178 This sex-specific characteristics of Aβ-PrPC-mGluR5 complex may be an explanation of sex difference in AD. In addition to its interaction with PrPC, Aβ oligomer itself can also change the configuration of mGluR5 by promoting their clustering, which will lead to higher intracellular calcium concentration and further synaptic impairments. 179
The manipulation of mGluR5 has shown promising results in AD treatment. 2-Methyl-6-(phenylethynyl)pyridine (MPEP), a commonly used mGLuR5 antagonist, has shown to prevent Aβ oligomer-induced impairment in LTP induction161,180; while MTEP, another mGluR5 antagonist, rescued spine loss and memory dysfunction in AD model mice. 175 In addition, chronic treatment of 2-Chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP), a long-lasting metabotropic mGluR5 inhibitor, reduced Aβ level and rescued cognition dysfunction of AD model mice. 181 However, this effect may be sex-specific, as a recent study suggested that the effect of CTEP only works on male mice. 178 Enhancing mGluR5 activity by CDPPB, a mGluR5 positive allosteric modulator, can prevent Aβ-induced neuronal loss, but unfortunately had little effect on rescuing memory deficit on 14-month-old AD model mice. 182 (Table 5).
The effect of glutamatergic receptor manipulation in AD.

Interaction between GABA receptors and Aβ pathology
Gamma-aminobutyric acid (GABA) receptors are the primary inhibitory neurotransmitter receptors in the central nervous system. There are two main types of GABA receptors: GABA-A receptors and GABA-B receptors. Each type plays a critical role in neural inhibition but operates through different mechanisms. GABA-A receptors are ionotropic receptors and ligand-gated ion channels. Their response is fast, allowing chlorine ions flowing into their central pore composed of 5 subunits from seven subunit subfamilies (i.e., α, β, …). 183 This fast inhibitory neurotransmission makes them crucial for maintaining the balance between neuronal excitation and inhibition, influencing everything from encoding sensory signals to cognitive processing. GABA-B receptors, on the other hand, are G-protein coupled receptors consisting of B1 and B2 two subunits. Their response is slower, but more prolonged compared to GABA-A receptors. 184
GABA-A receptors
In the context of AD, Aβ is found to modify the subunit composition of GABA-A receptors. This modification included down-regulation of the α1 and γ2 subunits, along with an upregulation of α2, β1, and γ1 subunits in the AD brain of mouse model and human patients.185,186 This change in composition correlated with an increased EC50 of the GABA-A receptor for GABA in the AD brain, 185 implicating a reduced receptor sensitivity. APP-PSEN1 mice exhibited neuronal hyperexcitability in the locus coeruleus, which may result from impaired function and reduced expression of the GABA-A receptor α3 subunit. These changes may be attributed to Aβ toxicity, as the GABA-A receptor α3 subunit has been shown to overlap with Aβ oligomer expression in both APP-PSEN1 mice and AD patients. 187 Aβ enhanced the inhibitory GABAergic tonic conductance and decreased the inhibitory postsynaptic current mediated by GABA-A receptors, leading to hippocampal dysfunction.186,188 Further, Aβ oligomer disrupted the balance between glutamatergic and GABAergic systems, inducing neuronal hyperexcitation by increasing extracellular glutamate levels, likely through impaired uptake, which heightened glutamatergic activity and spontaneous EPSC frequency. Simultaneously, Aβ reduced the effectiveness of GABAergic inhibition, as shown by its effects being reversed by the GABA-A receptor antagonist picrotoxin. 157
The interaction between Aβ and GABAergic system has also been studied with GABA receptor modulators. 189 As for the agonist, chronic stimulation of GABA-A receptors with muscimol showed neuroprotective effects against Aβ-induced neurotoxicity, but this protection was lost when co-treated with the GABA-A receptor antagonist bicuculline.190,191 Similarly, α5IA, an agonist for GABA-A receptors containing the α5 subunit, decreased Aβ-induced cell loss and restored the expression change of different subunits in GABA-A receptor. 192 In AD model mice having GABAergic system deficit, early treatment with gaboxadol, another GABA-A receptor agonist, could reverse hippocampal neuronal hyperexcitation and mildly restore cognitive function. 186 In addition, Aβ was shown to inhibit GABA-induced Cl− current, suggesting another mechanism for Aβ-induced neuronal hyperexcitation. 193 These investigations clearly highlighted the therapeutic potential of GABA-A receptor agonists against Aβ-induced impairment.
GABA-A receptor antagonists, such as pentylenetetrazole (PTZ), picrotoxin (PTX), and bicuculline, have been implicated in modulating Aβ-induced neuronal and cognitive deficits. Research showed that these antagonists can rescue Aβ-induced LTP deficits,157,194 and restore the morphology and functionality impairment of adult-born granule cells by hAPP. 195 In particular, prolonged PTX administration prevented memory deficits and mitigated the Aβ-induced upregulation of postsynaptic density protein 95 (PSD95), GluN2B and GABA-A α1 subunit in AD mouse models. 194 However, these therapeutic implications should be interpreted cautiously, given the antagonists can also exacerbate Aβ-induced seizures 196 and neutralize the neuroprotective effects of melatonin. 197
GABA-B receptors
Unlike the GABA-A receptors, GABA-B receptors do not form ion channels but instead influence cells through secondary messengers. These receptors typically function as heterodimers, consisting of GABA-B1 and GABA-B2 subunits, where GABA-B1 binds with GABA and GABA-B2 couples the receptor to G proteins. 198 Activation of GABA-B receptors leads to the inhibition of adenylate cyclase, decreased cAMP levels, and opening of potassium channels, which further contributes to neuronal hyperpolarization. Their modulation of synaptic transmission is vital for controlling neuronal excitability over longer periods compared to the fast-acting GABA-A receptors.
The density and composition of GABA-B receptor were found to be altered in AD mice and human samples. Both postsynaptic and presynaptic densities of the GABA-B receptor were reduced in the hippocampus of AD model mice, 199 possibly contributing to the cognitive dysfunction associated with AD. On molecular level, a novel non-coding RNA termed 17A, upregulated in AD patients, increased Aβ synthesis and Aβ42/Aβ40 ratio. 200 17A also promoted the expression of an alternative GABA-B receptor isoform by altering RNA polymerase splicing. Research showed that human AβPP can interact with presynaptic GABA-B receptor and lead to GABA release inhibition. 201 Research showed that inhibiting GABA-B receptors could alleviate Aβ-induced neuronal toxicity. For instance, the use of GABA-B receptor antagonists, such as CGP35348, has shown promising results in AD models. In a rat model, CGP35348 treatment alleviated memory impairment induced by acute Aβ toxicity. 202 Similarly, CGP36216, another antagonist acting on presynaptic GABA-B receptor, was able to mitigate the neuronal hyperexcitation induced by hAPP overexpression. 201 However, researchers found no neuronal protective effect on Aβ25−35-induced toxicity with the treatment of baclofen, a GABA-B receptor agonist. 190 (Table 6).
The effect of GABAergic receptor manipulation in AD.

Conclusion
AD, the leading cause of dementia, is a significant global health challenge with no definitive cure. It is characterized by the pathological accumulation of Aβ and tau, which lead to neural network imbalance, synaptic dysfunction and cognitive decline. Emerging research underscored the critical role of Aβ interactions with neural receptors in exacerbating these pathological processes. While some neuronal receptor-targeted treatments showed promise, the complexity of AD pathology demanded a comprehensive understanding of how these interactions influence disease progression.
This review aimed to highlight existing knowledge on the interactions between Aβ and neuronal receptors, focusing on the physiological, cognitive, and clinical consequences of receptor-targeted pharmacological interventions. By examining receptor-specific mechanisms—including adrenergic, acetylcholine, dopamine, 5-HT, and GABA receptors—this paper highlighted the potential of targeting receptor pathways to mitigate Aβ-induced pathology. Moreover, the review introduced various receptor manipulations with therapeutic potential, underscoring the necessity for continued research to optimize these strategies for clinical application. Future efforts could focus on understanding the complex dynamics of neurotransmitter receptor-Aβ interactions and developing neural device-based therapies for AD that modulate receptor activity without disrupting overall neural information processing. 203
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Ethical considerations
This study was based on previously published data and did not involve any new experiments requiring ethical approval.
Author contributions
Funding
This work was supported by NIH R01AG075114, R01NS119813, and U01AG066722.
Declaration of conflicting interests
Q.W. is the co-founder of Sharper Sense, a company developing methods of enhancing sensory processing with neural interfaces.
Data availability statement
This study did not involve the collection or generation of new data.