
Abstract
Background
Epigenetic dysregulation is increasingly recognized as a key mechanism in the development and progression of Alzheimer's disease (AD). Herpes simplex virus type 1 (HSV-1) infection has been proposed as a potential biological trigger that may accelerate neurodegeneration through epigenetic modifications. Among HSV-1 structural proteins, glycoprotein B (HSV-gB) may influence host–virus interactions affecting neuronal gene regulation.
Objective
This study aimed to investigate the contribution of HSV-gB to AD-related epigenetic alterations and to determine whether HSV-gB exposure exacerbates epigenetic dysregulation in two in vitro neuronal AD models.
Methods
Human SH-SY5Y neuroblastoma cells were used to establish two AD models: a differentiation-based aging model induced by retinoic acid and brain-derived neurotrophic factor (RA + BDNF), and an amyloid aggregation model induced by amyloid-β 1–42 (Aβ1–42). Cells were treated with HSV-gB (190.5 pg/ml) alone or in combination with each model. Global DNA methylation, histone H3 and H4 acetylation, histone multiplex modifications, and HDAC3 and HDAC8 levels were analyzed using ELISA-based assays.
Results
HSV-gB exposure and RA + BDNF treatment induced global DNA hypomethylation and histone hypoacetylation, accompanied by significant increases in HDAC3 and HDAC8 levels. In contrast, the Aβ1–42 model showed DNA hypermethylation and histone hyperacetylation, indicating distinct epigenetic profiles between differentiation-associated aging and amyloid-driven pathology.
Conclusions
HSV-gB contributes to AD-related epigenetic dysregulation and may amplify neurodegenerative mechanisms through HDAC-mediated chromatin remodeling. The findings highlight divergent epigenetic signatures in different AD models and support a potential role for viral factors in modulating AD-associated epigenetic pathways.
Schematic illustration summarizing the effects of herpes simplex virus type-1 glycoprotein B (HSV-gB) on epigenetic regulation in two in vitro Alzheimer's disease models. HSV-gB induces global DNA hypomethylation, histone H3/H4 hypoacetylation, and increased HDAC3/HDAC8 levels in the RA + BDNF-induced aging-like model, whereas distinct epigenetic patterns are observed in the Aβ₁–₄₂-induced amyloid model. The graphical abstract was created by the authors based on the findings of the present study.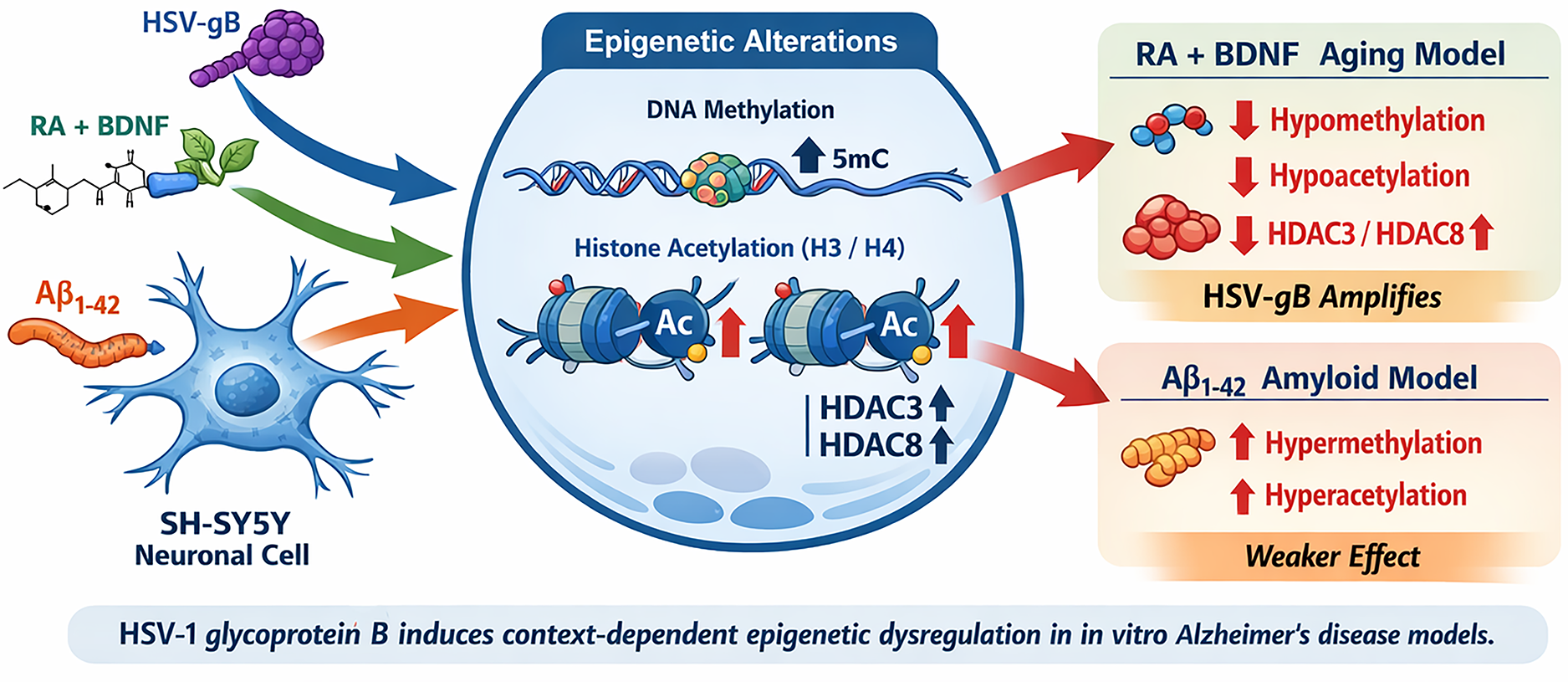
Keywords
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disease characterized by decline in cognitive functions and dementia. 1 Most (>90%) cases of AD are sporadic. However, “familial AD” may also develop because of genetic and epigenetic alterations. The main risk factor for AD is aging. The disease affects 10% of the population over the age of 65 and %50 of the population over the age of 85. Two important pathological hallmarks related to the disease have been identified. These are amyloid-β (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau proteins. Although both play crucial roles in disease initiation and progression, they differ in origin, molecular mechanisms, and spatiotemporal patterns within the brain. 2
AD is a complex disease that involves disruptions in many pathways, and epigenetic changes are among the fundamental mechanisms controlling the regulation of these pathways. Various factors may increase the risk of developing AD. 3 Persistent brain infections, particularly those induced by Herpes simplex virus type-1 (HSV-1), are thought to play a key role in the pathogenesis of the disease. 4 The hypothesis that HSV-1 might play a role in the development of AD was put forward in the early 1980s by Melvyn Ball based on several observations. The sources of this hypothesis are that the similar brain regions are degenerated in AD and HSV-1 induced encephalitis and memory impairments can be observed in some HSV1-induced encephalitis cases. 5 Subsequent molecular studies have determined that HSV-1 infection causes increase of amyloid-β protein precursor (AβPP) transport, hyperphosphorylation of tau protein and intracellular Aβ levels in neuronal and glial cells.6–8 Furthermore, Wozniak et al. (2007) demonstrated that HSV-1 infection directly upregulates β-secretase and γ-secretase activities, promoting amyloidogenesis, while De Chiara et al. (2010) identified that AβPP processing induced by HSV-1 yields neurotoxic AβPP fragments, linking viral replication to AD-related molecular pathology.6,7
Epigenetic regulation plays a critical role in neuronal development, plasticity, and aging. Emerging evidence implicates epigenetic alterations—particularly DNA methylation and histone modification changes—as central drivers of AD-related gene dysregulation. 9 DNA methylation and histone modifications that cause phenotypic changes have been shown to be effective in the aging process of organisms. Similar changes have been observed in aging and AD, suggesting that epigenetic mechanisms play a role in the pathophysiology of the disease. 10 For instance, Chouliaras et al. (2013) reported consistent decreases in global DNA methylation and hydroxymethylation in the hippocampus of AD patients, while Nativio et al. (2018) revealed large-scale disruptions in histone acetylation patterns in AD brains, especially at loci controlling synaptic and immune genes.11,12 These studies collectively suggest that epigenetic instability may bridge environmental exposures (such as HSV infection) with neurodegenerative processes. In this context, in vitro AD models provide a valuable platform to dissect the contribution of viral and epigenetic interactions. Specifically, we aimed to determine how HSV-gB exposure influences global DNA methylation, histone acetylation, and histone deacetylase (HDAC) levels in neuronal cells, and whether these effects amplify AD-related epigenetic alterations.
Structurally, Herpes simplex virus 1 glycoprotein B (HSV-1 gB) is a class III viral fusion protein that undergoes major conformational transitions to mediate the fusion of the viral envelope with host cell membranes. This fusion step is indispensable for the delivery of viral capsids into host cells, including neurons, which constitute a primary latency reservoir for HSV-1. HSV-1 gB interacts closely with other viral glycoproteins—particularly gD and the gH/gL complex—to coordinate receptor engagement and membrane fusion. Thus, HSV-1 gB is increasingly recognized not merely as a structural protein but as a viral determinant capable of shaping host epigenetic landscapes and promoting neurodegenerative processes. HSV-1 gB can modulate innate immune signaling, influence endosomal trafficking, and interfere with neuronal gene expression. In the context of neurodegeneration, gB has drawn attention due to its ability to induce oxidative stress, mitochondrial dysfunction, and DNA damage responses in neuronal cells. These effects may act as upstream triggers for epigenetic alterations, including aberrant DNA methylation and histone modifications. Importantly, HSV-1 gB has been shown to interact with host chromatin-regulatory enzymes, potentially contributing to the epigenetic dysregulation observed in HSV-1–associated neuropathology and AD. By influencing pathways such as histone deacetylase (HDAC) upregulation, histone acetylation balance, and chromatin accessibility, HSV-1 gB may serve as a mechanistic link connecting viral reactivation cycles with long-term neuronal dysfunction.13–15
By elucidating these molecular patterns, this study seeks to clarify the potential link between HSV-1 infection, epigenetic dysregulation, and neurodegenerative progression by investigating the contribution of HSV-gB to AD-related epigenetic modifications in two in vitro neuronal models and evaluating whether HSV-gB exposure amplifies AD-associated epigenetic instability.
Methods
Chemicals, reagents, and kits
HSV-1 gB (Catalog number: E-PP-1341) was obtained from Elabscience (Houston, TX). Brain derived neurotrophic factor (BDNF) were purchased from Sigma-Aldrich (Mannheim, Germany). Dulbecco's Modified Eagle's Medium (DMEM)-Ham's F12 w/L-Glutamine w/15-mM, hydroxyethyl-piperazineethane-sulfonic acid buffer (HEPES), fetal bovine serum (FBS), and penicillin/streptomycin were obtained from Biowest (Riverside, MO). Retinoic acid (RA), Aβ1–42 peptide and DNA methylation ELISA kit were purchased from Cayman Chemical (Ann Arbor, MI). DNA Extraction kit (Quick-DNA Miniprep) was purchased from Zymo Research (Irvine, CA). Histone H3 and H4 acetylation ELISA kits and The EpiQuik™ Total Histone Extraction Kit (OP-0006) and histone H3 and H4 modification multiplex assay kits were purchased from Epigentek (Farmingdale, NY). Histone deacetylase 3 and 8 (HDAC3 and HDAC8) ELISA kits were purchased from MyBioSource (San Diego, CA). All other chemicals were obtained from Merck (Darmstadt, Germany).
Cell culture and media
Human neuroblastoma SH-SY5Y (ATCC® CRL-2266™, Manassas, VA) cells were grown in DMEM- F12 medium, supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in an incubator with 5% CO2. Cells were split 2–3 times a week after they reached to %80–90 confluency.
Study groups
Two different AD models are used in this study: Retinoic acid (RA) + brain-derived neurotrophic factor (BDNF)–induced differentiation model, which simulates neuronal maturation and age-related epigenetic drift rather than direct neurodegeneration and Aβ1–42 peptide–induced model, which represents amyloid aggregation and toxicity. These models together enable the evaluation of whether HSV-1 glycoprotein B (HSV-gB) modulates epigenetic homeostasis differently in distinct AD-like cellular contexts. While previous research by our group examined HSV-gB's effects on the complement system and cytokine profiles in AD models,
16
the present study extends this line of investigation by focusing on epigenetic regulatory mechanisms.
1. Control 2. Herpes simplex virus-1 glycoprotein B (HSV-gB) group (190.5 pg/ml) 3. RA + BDNF (AD) group (RA: 10 µM; BDNF: 2.5 ng/ml) 4. RA + BDNF + HSV-gB (ADH) group (RA: 10 µM; BDNF: 2.5 ng/ml; HSV-gB: 190.5 pg/ml) 5. Aβ group (10 µM) 6. Aβ + HSV-gB (AβH) group (Aβ1–42: 10 µM; HSV-gB: 190.5 pg/ml)
HSV-gB treatment
Herpes simplex virus causes lifelong infections due to its ability to remain hidden in the infected neurons. 17 In our study, we tried to mimic the chronic effect of HSV-1 infection as this infection flares up from time to time. Recombinant HSV-1 glycoprotein B fragment (residues 497–507; Elabscience) was used to simulate the viral effect. The 497–507 region falls within a well-characterized immunogenic and functionally relevant domain near the C-terminal portion of HSV-1 gB's ectodomain. Peptide fragments are widely used to isolate “signaling functions” from the viral fusion machinery. Full-length HSV-1 gB is large (∼904 aa), membrane-anchored, requires post-translational glycosylation and dependent on viral gD and gH/gL to exert its full fusion activity. Using the whole glycoprotein would require viral expression systems and membrane integration. In contrast, short gB peptides can selectively reproduce the signaling component of gB–host interactions without introducing viral entry or replication. The 497–507 peptide is sufficient to activate stress and immune pathways that can lead to epigenetic alterations and this peptide can have neuronal receptor engagement. Certain segments of gB—including this region—bind host receptors, thereby activating downstream pathways.18–20
Cytotoxicity data from our previous study were used to determine the HSV dose to be used in the study. 16 HSV-1 gB was dissolved in phosphate buffered saline (PBS) at a concentration of 1 mg/ml. This stock solution was aliquoted and stored at −80°C. The inhibitory concentration 20 (IC20) dose of HSV-gB (190.5 pg/ml) was applied to the cells at two different times (days 1 and 4 of the protocol). The applications and the days the chemicals were applied are summarized in Table 1.
Differentiation protocols.

AD model: retinoic acid and brain-derived neurotrophic factor induced Alzheimer's disease model; ADH model: retinoic acid and brain-derived neurotrophic factor induced Alzheimer's disease model; Aβ: amyloid-β 1–42 peptide induced Alzheimer's disease model; AβH: amyloid-β 1–42 peptide induced Alzheimer's disease model + herpes simplex virus type-1 glycoprotein B; BDNF: brain derived neurotrophic factor; DMEM F12: Dulbecco's Modified Eagle's Medium-Ham's F12 w/L-Glutamine w/15-mM hydroxyethyl-piperazineethane-sulfonic acid buffer (HEPES); FBS: fetal bovine serum; HSV: Herpes simplex virus; HSV-gB: Herpes simplex virus type-1 glycoprotein B.
Preparation of AD models
The two different in vitro AD models were created in the same way as in our previous study. 13 The chemicals and the days they were applied are summarized in Table 1.
In the first differentiation protocol, differentiation was made by using RA and BDNF and modified from the method used by Medeiros et al. (2019). 21 Cells were seeded with normal growth medium in 75 cm2 flasks and waited 24 h for complete adhesion. On the first day after culturing, the differentiation protocol was started by reducing the ratio of FBS in the medium from 10% to 1% and adding 10 µM of RA. The cell medium was changed every 3 days and 2.5 ng/ml BDNF was added along with RA from day 4. On the 8th day from the start of differentiation, the protocol was finished and cells and culture supernatants were collected. This RA + BDNF-induced differentiation protocol promotes a neuronal-like, maturation-associated phenotype rather than a degenerative one, providing a model for neuronal aging and vulnerability relevant to AD pathology.
In the second model (Aβ1–42 Peptide Treatment), AD was simulated using synthetic Aβ1–42 peptide. Aβ1–42 was dissolved in dimethyl sulfoxide (DMSO) and then diluted with PBS to make a 350 μM stock solution. The stock solution was incubated at 37°C for 3 days and mixed by pipetting to facilitate the formation Aβ aggregates every day. At the end of the incubation period, the solution containing the aggregates was aliquoted and stored at −80°C. Aβ1–42 aggregates were applied to the cells in a flask at a concentration of 10 μM and incubated for 2 days to establish an amyloid aggregation–based AD model. 22
Cytomorphological evaluation
Phase contrast microscopy. Cells in the flask were photographed by a camera connected to a phase contrast microscope (Leica, Wetzlar, Germany).
DNA methylation
DNA isolation was performed in the study groups using the Quick-DNA Miniprep (Zymo Research) kit according to the instructions. The total amount of DNA was measured using Nanodrop (MaestroGen, Hsinchu City, Taiwan). Global DNA methylation levels in the study groups were measured in isolated DNAs using a commercial competitive ELISA kit. Results are given as pg/µg DNA.
Histone isolation
After the incubation period, cells were washed with pre-cooled PBS and removed by using trypsin. Then cells were suspended in pre-lysis buffer and histone isolation was performed using the The EpiQuik™ Total Histone Extraction Kit kit according to the instructions. At the end of the isolation, protein measurement was performed to determine the total amount of histone protein in the samples. The samples were stored at −80°C until analysis. H3 and H4 acetylation, H3 and H4 multiplex modifications and HDAC 3 and HDAC 8 levels were measured with the isolated histones.
Measurement of histone deacetylase 3 and histone deacetylase 8 levels
Levels of HDAC3 and HDAC8 were measured in the isolated histone extracts by using commercial ELISA kits that use the sandwich-ELISA principle. Results were given as ng/mg protein.
Histone acetylation
Total acetylated histone H3 and H4 were measured in the histone extracts by using commercial ELISA kits that use the sandwich-ELISA principle. Results were given as µg/mg protein.
Multiplex histone modifications
The levels of multiplex modifications in histone H3 and H4 were measured using a commercial colorimetric kits. The kits were designed to measure multiple histone modifications simultaneously. The amount of modified histones in the samples was determined as a percentage of modification by comparing the total amount of histones in the total sample. Modified histone ratios were calculated by measuring the absorbance density at 450 nm with the ELISA reader (SpectraMax M2, Molecular Devices, San Jose, CA).
Statistical analysis
Statistical Package for Social Sciences Program (SPSS) 17.0 (Chicago, IL) was used for statistical analysis. The differences among the groups were evaluated with Kruskal–Wallis one-way analysis of variance, followed by Mann–Whitney U test. Results are expressed as mean ± standard deviation (SD). p values <0.05 were considered as statistically significant.
Results
Cytomorphology of the study groups
SH-SY5Y cells showing a heterogeneous population with epithelial-like cells and neuronal-like cells with short neurites were observed in the control group. In HSV-gB group, a few cells with two nucleus were observed and the inability of cell division supported the senescence while the presence of neurites supported neuronal differentiation in HSV-gB group. The cells were more spindle in shape, possessed longer and slimmer neurites and scattered vacuoles were seen in the cytoplasm of the cells in AD group. Varicosities were prominent along the thin long neurites in this group. In Aβ group, dark accumulations were prominent in the cytoplasm and also around the cells. The neuronal-like cells with irregular contours and numerous round, detached cells forming cell clusters reflected the cell degeneration in this group. Binucleated cells and vacuoles in the soma were observed in ADH group. Binucleated cells with accumulations in the cytoplasm and degenerated cells with prominent multiple vacuoles were examined in AβH group and vacuoles were seen even in the neurites in this group (Figure 1).
The epithelial-like cells and neuronal-like cells with short neurites in the control group. The cell with two nucleus in HSV, AD-HSV, and AB-HSV groups. The pyramidal shaped cells with longer neurites, vacuoles in the cytoplasm of the cells and varicosities along the thin long neurites in AD and AD-HSV groups. Vacoules and small round dark accumulations in the cytoplasm of the cells and also around the cells in AB and AB-HSV group. Many degenerated cells (black arrow) are also seen in these groups. (A) Control; (B) HSV; (C) AD; (D) Aβ; (E) AD-HSV; and (F) Aβ -HSV.
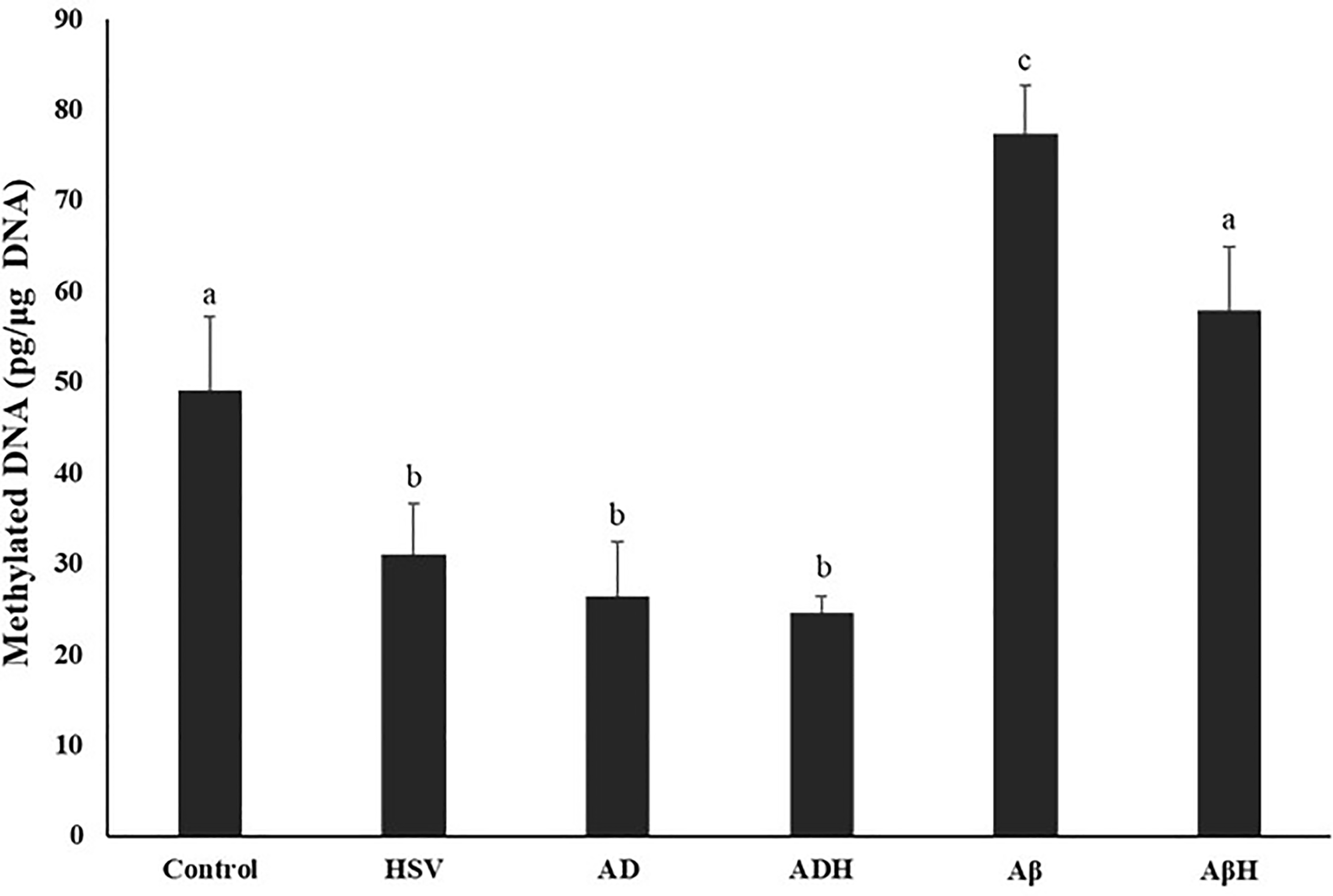
Global DNA methylation
Global DNA methylation levels of study groups are given in Figure 2. When compared with the control group, global methylation was found to be significantly decreased in the HSV-gB, AD and ADH groups (35.8%, 45.9%, 44.8%, respectively; p < 0.05). In the Aβ group, levels increased by 55.5% compared to the control group (p < 0.05). In the AβH group, global methylation level was 16% higher than the control group; however, this difference was not statistically significant (p > 0.05). In addition, the global DNA methylation level in the Aβ group was ∼2.8 fold higher than the AD group (p < 0.05). No difference was observed between the AD and ADH groups, while the global methylation level in the AβH group was 25.3% lower than the Aβ group (p < 0.05).

Methylated DNA levels of study groups. (a,b,c) Bars that do not share same letters (superscripts) are significantly different from each other (p < 0.05).
Histone H3 and H4 total acetylation levels
Histone H3 and H4 acetylation levels of study groups are given in Figure 3. Compared with the control group, histone H3 acetylation was found to be significantly reduced in the HSV-gB, AD and ADH groups (61.5%, 63.9%, 70.44%, respectively; p < 0.05, all). In the Aβ group, histone H3 acetylation level increased by 57.4% compared to the control group (p < 0.05). Moreover, the H3 acetylation levels in the HSV-gB, AD and ADH groups were significantly lower than in the Aβ group (75.5%, 77.1%, 81.2%, respectively; p < 0.05, all). Similar to H3 acetylation levels, H4 acetylation levels were found to be significantly reduced in the HSV-gB, AD and ADH groups compared to the control group (68.9%, 61.5%, 72.5%, respectively; p < 0.05, all). No difference was observed in the Aβ and AβH groups compared to the control group.
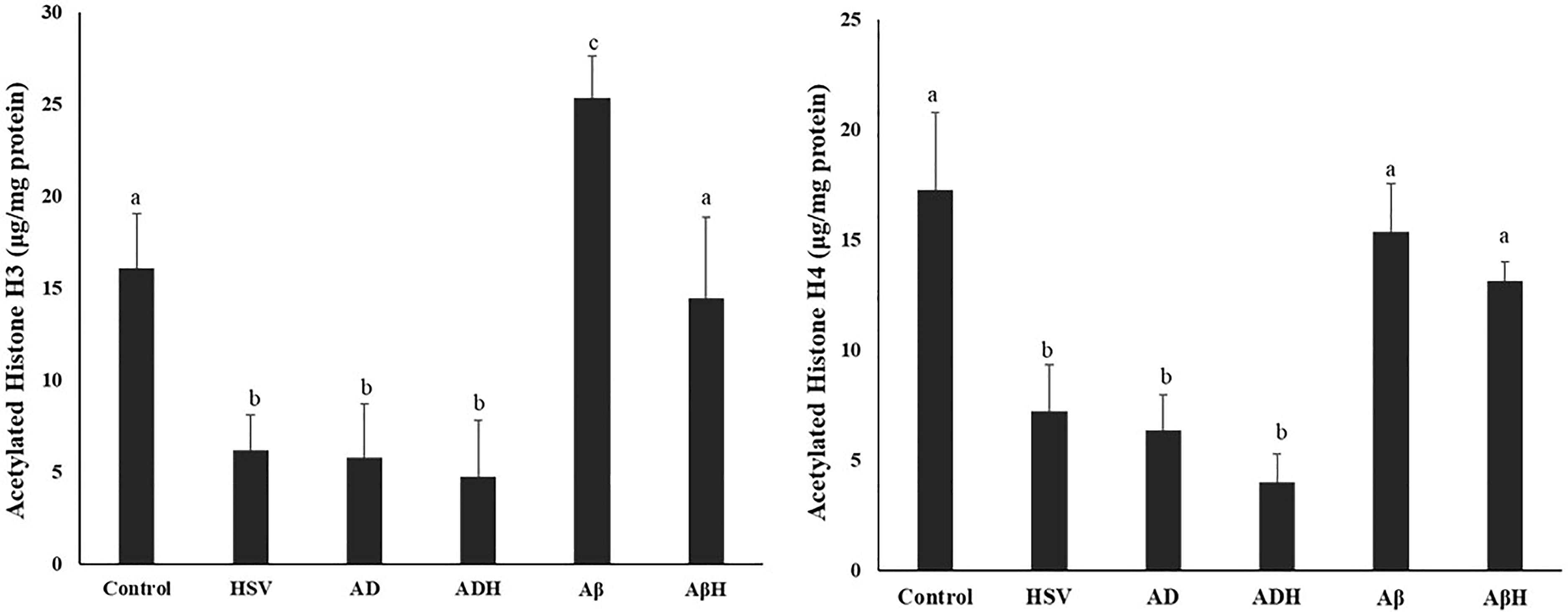
Acetylated histone H3 and H4 levels of study groups. (a,b,c) Bars that do not share same letters (superscripts) are significantly different from each other (p < 0.05).
Histone deacetylase 3 and 8 levels
Histone deacetylase levels of study groups are given in Figure 4. Compared to the control group, HDAC3 levels increased >2-fold in the AD and ADH groups (p < 0.05). Although a 31% increase was observed in the HSV-gB group compared to the control group, this increase was not found to be statistically significant (p > 0.05). No significant change was observed in the Aβ and AβH groups compared to control group. Although the HDAC3 level in the ADH group was 28% higher than the AD group, the difference was not statistically significant (p > 0.05). Compared to control group, HDAC8 levels significantly elevated in the HSV-gB, AD and ADH groups (22.5%, 42.6%, 92.1%, respectively; p < 0.05). In the Aβ and AβH groups, HDAC8 levels were found to be 49.7% and 34.3% lower than the control group, respectively (p < 0.05).
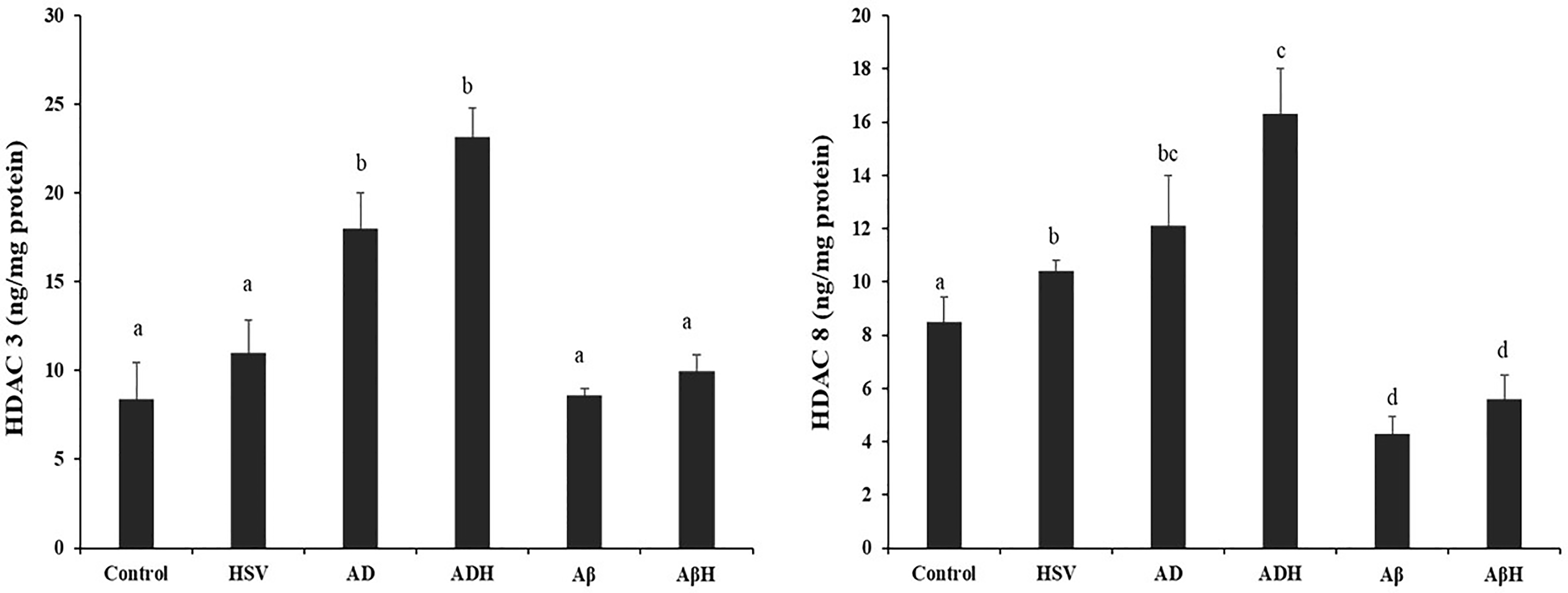
Histone deacetylase (HDAC) 3 and 8 levels of study groups. (a,b,c) Bars that do not share same letters (superscripts) are significantly different from each other (p < 0.05).
Histone H3-H4 multiplex modifications
Histone H3 and H4 modification levels are given in Tables 2 and 3. The results are given as % modification rate according to total H3 and H4.
Histone H3 modifications.
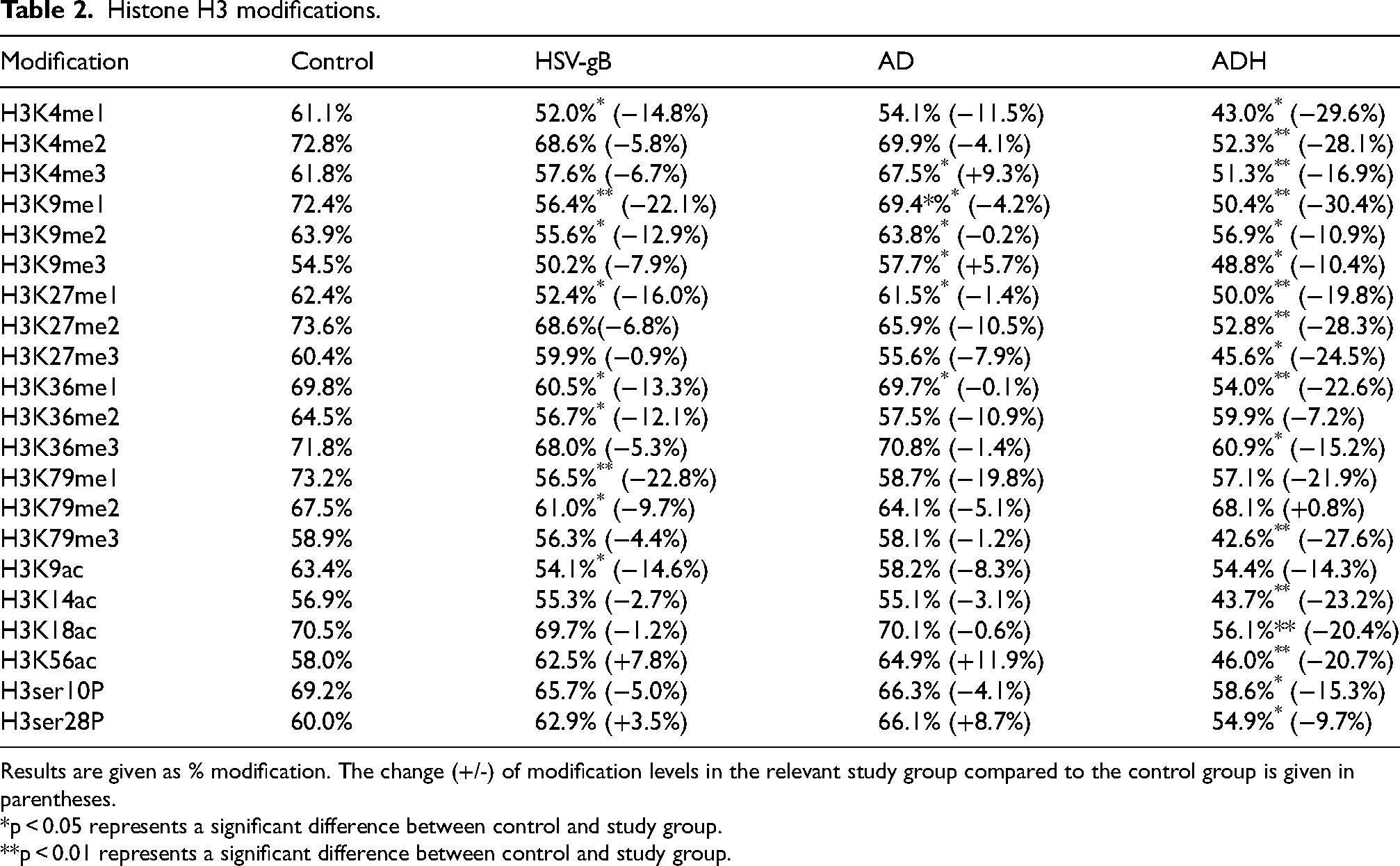
Results are given as % modification. The change (+/-) of modification levels in the relevant study group compared to the control group is given in parentheses.
*p < 0.05 represents a significant difference between control and study group.
**p < 0.01 represents a significant difference between control and study group.
Histone H4 modifications.

Results are given as %modification. The change (+/-) of modification levels in the relevant study group compared to the control group is given in parentheses.
*p < 0.05 represents a significant difference between control and study group.
**p < 0.01 represents a significant difference between control and study group.
HSV: herpes simplex virus type-1 glycoprotein B group; AD: retinoic acid and brain-derived neurotrophic factor induced Alzheimer's disease model; ADH: retinoic acid and brain-derived neurotrophic factor induced Alzheimer's disease model + herpes simplex virus type-1 glycoprotein B; Aβ: amyloid-β 1–42 peptide induced Alzheimer's disease model; AβH, amyloid-β 1–42 peptide induced Alzheimer's disease model + herpes simplex virus type-1 glycoprotein B.
H4K5ac: acetylation at the 5th lysine residue of the histone H4 protein.
H4K8ac: acetylation at the 8th lysine residue of the histone H4 protein.
H4K12ac: acetylation at the 12th lysine residue of the histone H4 protein.
H4K16ac: acetylation at the 16th lysine residue of the histone H4 protein.
H4K20m1: mono-methylation at the 20th lysine residue of the histone H4 protein.
H4K20m2: di-methylation at the 20th lysine residue of the histone H4 protein.
H4K20m3: tri-methylation at the 20th lysine residue of the histone H4 protein.
H4R3m2a: asymmetric di-methylation at the 3rd arginine residue of the histone H4 protein.
H4R3m2s: symmetric di-methylation at the 3rd arginine residue of the histone H4 protein.
H4Ser1P: phosphorylation at the 1st serine residue of the histone H4 protein.
Fifteen different methylation, four different acetylation and teo different phosphorylation modifications on histone H3 were measured in the study groups. Measurements were performed in four study groups: Control, HSV-gB, AD and ADH. All methylation levels were found to be lower in the HSV-gB group than in the control group. Similar to the HSV-gB group, methylation modification levels were observed to be lower in the AD and ADH groups compared to the control group. The decrease in methylation levels, especially the modifications occurring at the 4th lysine residue (H3K4me1, H3K4me2, H3K4me3), were more pronounced in the ADH group compared to the HSV-gB and ADH groups. When HSV-gB, AD and ADH groups were compared with the control group, it was observed that acetylation levels at the histone 3 lysine 9 position (H3K9ac) showed decreases (14.64%, 8.28%, 14.31%, respectively). While no change was observed at the H3 lysine 18th position (H3K18ac) in the HSV-gB and AD groups, acetylation decreased by 20.43% in the ADH group compared to the control group at the H3 lysine 18th position.
Five different methylation, four different acetylation and one different phosphorylation modifications on histone H4 were measured in the study groups. Acetylation modifications were measured as acetylation levels occurring at the 5th, 8th, 12th, and 16th lysine residues of the histone H4 protein. When the acetylation level at the lysine residue in the fifth position was examined, there was a decrease of 14.26% and 8.57% in the HSV-gB and AD groups, respectively, compared to the control group. In the ADH group, which is the combination of the two groups, the acetylation level at this position decreased by 35.95% compared to the control group. It was observed that acetylation levels increased in the Aβ and AβH groups compared to the control group. When the phosphorylation levels at the histone H4 serine position were examined, it was observed that the phosphorylation level in the HSV-gB and Aβ groups decreased compared to the control group (15.68% and 13.76%, respectively). In addition, it was observed that H4K20 m2, H4K20m3, H4R3m2a, and H4R3 m2 s methylation levels decreased by 12.01%, 27.49%, 21.43% and 17.19% in the HSV-gB group compared to the control group, respectively.
Discussion
DNA methylation
DNA methylation occurring in the promoter regions of genes directly leads to the termination of gene expression by inhibiting the binding of transcription factors to DNA. 23 In AD, decreases in DNA methylation have been observed, particularly in hippocampal and cerebral cortex cells. For example, 5-methyl cytosine (5-mc) levels were found to be lower in cortical neurons from postmortem AD patient brains compared to healthy controls. Similarly, low 5-mc levels have been reported in the hippocampus, cerebral cortex, and cerebellum of AD patients. Additionally, some methylation maintenance factors, such as DNA methyltransferase 1 (DNMT-1) and methylated DNA-binding domain protein 2, were found to be reduced in the hippocampus of AD patient brains.11,24 In one study, increasing the expression of DNMT3a2 in the hippocampus of old mice increased the general methylation level and improved memory. 25 Hypomethylation of promoter regions of genes such as Presenilin-1 (PSEN-1) results in increased production of products such as Aβ. 26 In a study comparing postmortem normal brain samples with AD patient brain samples, the promoter region of the breast cancer type 1 susceptibility protein (BRCA1) gene, which encodes the DNA repair protein BRCA1, was found to be hypomethylated in AD patient brains. 27
In our study, significantly decreased DNA methylation levels were observed in the HSV-gB, AD and ADH groups compared to the control. The global hypomethylation seen in the first AD model is consistent with data from previous studies.25,27–29 The observation of a similar methylation pattern in the HSV-gB group as in the AD group indicated that HSV-1 infection may contribute to cell aging and AD pathogenesis through the hypomethylation mechanism. On the contrary, it was found that the level of DNA methylation in the Aβ group, the second AD model, was significantly increased compared to the control group and other study groups. Thus, it was observed that two different AD models showed different DNA methylation profiles. In an in vitro study, Taher et al. (2014) showed that global DNA methylation did not change as a result of exposure of cholinergic IMR-32 human neuroblastoma cells to Aβ1–40 peptide. However, both hypermethylation and hypomethylation were observed in gene regions associated with neural differentiation and apoptosis. 30
The difference in DNA methylation profiles between the two AD models can be interpreted as the fact that the first model (AD group) is based on cell aging and similar to hypomethylation studies that are observed with aging31,32; and in the Aβ group, which was developed by applying Aβ1–42 peptide to the cells, hypermethylation that may occur in the promoter regions of genes that encode enzymes that are necessary for normal physiological conditions such as α-secretase and play a role in neurogenesis may increase the total DNA methylation rate. The in vivo data of Bie et al. (2014) support this hypothesis as hypermethylation was observed in the promoter regions of the neuroligin-1 (NLGN1) gene in the brains of rats injected with Aβ1–42. This caused a decrease in NLGN1 expression and decreased synaptic plasticity, resulting in impaired memory and learning. 33
Histone modifications
Histone modifications are involved in broad neurobiological processes such as central nervous system (CNS) development, post-traumatic stress disorders, memory and addiction. In addition, histone modifications have important roles in many specific physiological events such as neuronal differentiation, regulation of choline acetyltransferase activity, BDNF transcription, microglial apoptosis, axon formation and synaptic pruning.34,35 It has been observed that histone modifications may change at different levels and in different directions with aging. In animal model studies, a decrease in H3K9me2, H3K9me3, and H3K27me3 levels and an increase in H3K36me3 and H3K27ac levels were observed in the cerebral cortex and hippocampus with aging. 36 It was found that H3K27ac levels decreased and H3K27me3 levels increased in the BDNF gene promoter regions in old mice. 37
Post-translational modifications such as histone acetylation are highly recognized for their roles in gene activation and repression in the brain. 38 In mouse models of AD, hypoacetylation of H3 and H4 is often observed. A recent study has shown that hypoacetylation of histones H3 and H4 causes a decrease in NLGN1 expression. 39 Healthy aging is associated with increases in H4K16ac levels in aged cortex, whereas H4K16ac is dramatically lost in the cortex of individuals with AD. 12 Marzi et al. (2018) examined the acetylation of H3K27 (H3K27ac), a critical marker of gene expression in the entorhinal cortex of individuals with AD. In this study, it was observed that acetylation levels changed in gene regions associated with AD pathology (APP, PSEN1, PSEN2, etc.). 40 Similarly, Klein et al. (2019) investigated the acetylation of H3K9 (H3K9ac) in gene regions important for Aβ or Tau protein expression in a large cohort of 669 individuals. In particular, they observed large genomic changes in H3K9ac levels in gene regions related to tau pathology. 41
In our study, we found that global acetylation levels in histone H3 and H4 proteins in HSV-gB, AD and ADH groups decreased >2-fold compared to the control group. The observation of histone H3 and H4 hypoacetylation is consistent with the data obtained in some other AD studies. The fact that the hypoacetylation level observed in the HSV-gB group was similar to that of the AD group suggests that, as with other parameters, HSV-gB may affect the histone acetylation level, leading to cell aging and contributing to AD pathology. In addition, acetylation levels decreased more in the ADH group than in both the HSV-gB and AD groups, indicating that HSV infection may also worsen disease progression. In the Aβ group, the second AD model used in our study, it was observed that the acetylation levels in histone H3 and H4 increased, although not at a significant level. In the AβH group, the acetylation level was determined to be similar to the control group. Therefore, the difference in acetylation patterns between the first model, which was induced by RA and BDNF and operates through the cell senescence mechanism, and the second model, which is directly induced by Aβ1–42 peptide, indicates that AD may occur with different pathologies in the two models. In the AβH group, the observation of a similar total acetylation level to the control group can be explained by the fact that HSV-gB causes hypoacetylation at some positions of the histone 4 (especially 5th and 16th lysine residue of the histone H4 protein), while the Aβ1–42 peptide causes hyperacetylation at some positions of the histone 4 (especially 8th lysine residue of the histone H4 protein). Important findings were also obtained from the data obtained by measuring multiple modifications of histone H3 and H4. Acetylation (H3K9ac) levels at the H3 lys-9 position were found to be reduced in all measured study groups compared to the control group. When H3K14ac and H3K18ac levels were measured, it was observed that acetylation did not change in the AD and HSV-gB groups alone, but acetylation decreased significantly in the ADH group. The differences observed in these modifications may be considered as the reasons for the lower acetylation level observed in the ADH group than the total H3 acetylation level measured in the HSV-gB and AD groups.
When the levels of acetylation (H4K5ac) at the histone H4 5th lysine position were examined, it was observed that the acetylation level decreased in the HSV-gB and AD groups compared to the control group. Moreover, in the ADH group, acetylation levels decreased, and this decrease was more than twice the decrease observed in the HSV-gB and AD groups. This suggests that the mechanism by which HSV-gB infection worsens the course of AD may be through H4K5ac modification. In a study it was found that H4K5 hypoacetylation was associated with decreased memory function in mice. 42
The most studied histone methylation types in AD studies are tri-methylation of lysine at positions 4 and 27 of H3 (H3K4me3 and H3K27me3), which are markers of gene expression and repression, respectively. 43 In our study, both H3K4me3 and H3K27me3 levels were found to decrease in all measured study groups compared to the control group. When both modifications were examined, it was seen that the decrease observed in the ADH group was more pronounced than the decrease observed in the HSV and AD groups alone. Based on this information, it can be thought that HSV-gB application may induce hypomethylation at these positions and affect the course of AD. In the Aβ group, H4K20m1 and H4K20 m2 levels were observed to increase compared to the control group. These data indicate that the two different AD models used in our study affect histone methylation levels through different mechanisms, leading to different AD pathologies.
In the study conducted by Ogawa et al. (2003), it was observed that H3 phosphorylation increased in neurons in hippocampus tissues obtained from individuals with AD compared to the control group. Particularly, noteworthy is that the increases in phosphorylation at the H3 ser-10 position (H3Ser10P) are almost identical to the distribution of phosphorylated tau. 44 In our study, when the H3Ser10P levels were examined, no change was observed in the HSV and AD groups compared to the control group, while a decrease in the phosphorylation level was observed in the ADH group. Our findings are different from the findings of the previous study we mentioned. 44 One reason for this may be that we focused on serines 10 and 28 while looking at the total H3 phosphorylation level in that study. Moreover, one study found that H3Ser10P levels increased during mitosis and that folic acid administration to SH-SY5Y cells stimulated cell division by increasing H3Ser10P phosphorylation. 45 In our study, it can be thought that the decreasing H3Ser10P levels may indicate a decrease in mitosis due to cell aging.
Histone deacetylase levels
Histone deacetylase expression and activity can be used as a direct biomarker related to histone acetylation. Increased HDAC2 levels have been associated with cognitive impairment in CK-p35 mice models. Post-mortem studies in mice brains have shown that HDAC2, but not HDAC1 or HDAC3, is increased in the hippocampus. This suggests that different histone deacetylase may have different effects. 46 Guan et al. (2009) revealed that overexpression of HDAC2 reduced dendritic spines, synaptic density, synaptic plasticity and memory function. 47 In addition, in a study with monozygotic twins, HDAC2 and HDAC9 expression levels in peripheral blood cells were found to be higher in twins with AD compared to healthy ones. 48 Ding et al. (2008) showed that an increase in HDAC6 in human embryonic kidney cells caused tau hyperphosphorylation and suppression of HDAC6 expression in the same cells reduced tau phosphorylation. This information indicates that increases in HDAC enzyme expression and activity may underlie histone hypoacetylation associated with AD pathogenesis. 49
In our study, it was observed that HDAC3 levels increased significantly in the AD and ADH groups compared to control group (p < 0.05); there was a less significant increase in the HSV group (p > 0.05) while there was no change in the Aβ and AβH groups. When HDAC8 levels were examined, we determined significant increases in the HSV, AD and ADH groups and marked decreases in the Aβ and AβH groups compared to the control group. When HDAC levels and histone H3-H4 acetylation levels were examined together, these results supported each other. The AD and ADH groups with the highest increase in HDAC levels were also the groups with the highest decrease in histone acetylation. This correlation suggests that the mechanism of observed hypoacetylation may be due to increased HDAC levels. In addition, the increase observed in both the HSV group and the increase in HDAC levels in the groups where AD models were combined with HSV-gB (ADH and AβH) application were greater than the AD and AβH groups separately, indicating that HSV-gB application may cause epigenetic irregularities in cells by independently increasing HDAC levels.
In a multifactorial disease such as AD, determining the contribution of each factor and the underlying mechanisms is critical to developing new therapeutic strategies to prevent the disease and/or slow its progression. Considering the findings of our study, it can be said that epigenetic irregularities may be effective in the development of AD. Identification of epigenetic marks associated with AD development will provide excellent diagnostic markers for AD progression and suggest a new epigenetic model of how AD initially initiates. In addition, the presentation of data on a reversible process such as DNA methylation in AD may contribute to the development of epigenetic therapeutics aimed at preventing or even reversing age-related changes in the epigenome. It seems that more studies on targeting different epigenetic markers are needed. The evaluation of the data obtained from this study may enable the development of new therapeutic approaches aimed at regulating epigenetic modifications in the future. In addition, the data obtained may pave the way for new studies to slow down and improve neurodegeneration in AD.
The present study suggests that HSV-gB induces significant alterations in epigenetic regulatory markers in neuronal cells. Specifically, HSV-gB triggered global DNA hypomethylation, histone hypoacetylation, and a concomitant increase in HDAC3 and HDAC8 levels, suggesting that viral structural components are capable of directly modulating host chromatin-regulatory machinery even in the absence of whole-virus infection. In the RA + BDNF differentiation-based AD model, representing neuronal maturation and aging, HSV-gB further enhanced the existing hypomethylated and deacetylated epigenetic signature. In contrast, in the Aβ model, which mimics Aβ aggregation–induced stress, HSV-gB exposure resulted in global hypermethylation and hyperacetylation. This divergence indicates that the epigenetic consequences of HSV-gB are highly dependent on the underlying cellular context, particularly whether neurons are in a differentiation-associated “aging-like” state or in a stress-driven degenerative environment. The differential effects observed between non-differentiated and RA + BDNF-differentiated SH-SY5Y cells further support this context dependency. Differentiated neurons possess distinct basal epigenetic landscapes, including reduced global DNA methylation, altered chromatin openness, and lower proliferative capacity compared to undifferentiated cells. Consequently, HSV-gB–mediated engagement of cell-surface receptors such as toll-like receptor 2 (TLR2), Nectin-1, or Herpes virus entry mediator (HVEM) likely activates different intracellular pathways depending on the differentiation state. For example, undifferentiated cells may favor mitogen-activated protein kinase/extracellular-signal-regulated kinase (MAPK/ERK)-driven responses, whereas differentiated cells—characterized by heightened inflammatory sensitivity—may shift toward nuclear factor kappa B (NF-κB)–dominant signaling. Such divergence could account for the more pronounced deacetylation and hypomethylation induced by HSV-gB in RA + BDNF-treated cells.
Although direct mRNA or downstream protein expression profiles were not assessed in this study, the observed epigenetic changes point to several plausible molecular mechanisms. HSV-gB–activated signaling pathways [NF-κB, MAPK/ERK, cyclic AMP-response element binding protein (CREB)] are known regulators of epigenetic enzymes: NF-κB can recruit HDAC3 to inflammation-responsive promoters, while MAPK signaling modulates DNMT3A nuclear localization and chromatin association. Thus, HSV-gB may initiate a feed-forward cascade in which receptor-mediated signaling alters DNMT and HDAC/histone acetyltransferase (HAT) activity, leading to sustained epigenetic repression of neuronal genes and amplification of neuroinflammatory pathways. Such mechanisms provide a compelling link between viral structural proteins and AD-relevant epigenetic remodeling. 50
In conclusion, this study demonstrates that HSV-1 gB is capable of inducing substantial epigenetic alterations in neuronal cells, including global DNA methylation, histone oacetylation, and alterations in HDAC activity. These effects differ markedly between differentiation-based (RA + BDNF) and Aβ-induced AD models, indicating that HSV-gB modulates epigenetic pathways in a context-dependent manner. We can state that mechanistic studies are still needed although our findings support a growing body of evidence suggesting that viral structural components may contribute to AD-related epigenetic instability and highlight the importance of further exploring host–virus interactions in neurodegeneration.
Footnotes
Acknowledgements
This study was supported by Hacettepe University Scientific Projects Coordination Unit [Project no: THD-2021-19660].
Ethical considerations
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Hacettepe University Scientific Projects Coordination Unit [Project no: THD-2021-19660].
Hacettepe University Scientific Projects Coordination Unit, (grant number THD-2021-19660).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are available within the article.