
Abstract
Neurovascular dysfunction is increasingly recognized as a central feature of dementia pathogenesis in the early or even preclinical stage, rather than merely a downstream effect of amyloid–tau neurodegeneration. Based on multimodal clinical evidence showing that cerebrovascular dysregulation can precede amyloid-β accumulation, this commentary emphasizes three converging lines: (1) hippocampal blood–brain barrier breakdown as an early, potentially amyloid-/tau-independent event, (2) vascular-first/parallel two-hit frameworks linking vascular risk factors to subsequent proteinopathy, and (3) early neurovascular coupling failure and chronic hypoperfusion as upstream drivers. Integrating blood–brain barrier and hemodynamic biomarkers may facilitate earlier biological subtyping and prevention.
Keywords
Alzheimer's disease (AD) has long been referred to as a prototypical proteinopathy, characterized by amyloid-β (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau. 1 This amyloid–tau neurodegeneration framework has been extraordinarily productive, enabling the development of biomarkers, staging frameworks, and disease-modifying trials.2–5 However, anti-Aβ antibodies, such as lecanemab and donanemab, have demonstrated limited efficacy in improving cognitive function in patients with early AD.4,5 Therefore, pathophysiologies other than amyloid–tau-associated neurodegeneration have been gaining increasing attention. Among these, accumulating clinical and experimental evidence suggests that neurovascular dysfunction is not merely a downstream epiphenomenon but can be an early, potentially initiating component of the disease cascade. 6 A large multimodal human study demonstrated that cerebrovascular dysregulation is an early pathological event, and it presents a temporal ordering in which vascular dysregulation precedes subsequent Aβ accumulation and other pathological changes. 7 Clinically, this framing resonates with the observation that subtle hemodynamic, blood–brain barrier (BBB), and microvascular abnormalities can be detected years before the occurrence of overt dementia and, in some cases, even before the appearance of robust Aβ and tau biomarker positivity.
If cerebrovascular dysfunction is positioned upstream, it provides a mechanistic link between vascular risk factors, lifestyle exposures, and inflammation, and classical AD pathologies. Furthermore, the brain is likely to be more vulnerable in individuals who survive cerebrovascular and cardiovascular disease into old age. Notably, a vascular-first or vascular-parallel framework does not negate amyloid and tau. Rather, it reframes them as partly vascular-conditioned phenomena—for example, via impaired clearance of Aβ along perivascular pathways, endothelial and pericyte injury, neuroinflammation and oxidative stress, altered neurovascular coupling, and chronic cerebral hypoperfusion that promotes oxidative stress and tau phosphorylation. Accordingly, elucidating the pathophysiology of dementia from the perspective of cerebrovascular and neurovascular unit (NVU) dysfunction is clinically strategic. In particular, vascular biology offers modifiable targets and measurable intermediate phenotypes, potentially facilitating earlier identification and prevention.
This commentary focuses on early cerebrovascular and NVU dysfunction as potential drivers of neurodegeneration, although other pathways, including metabolic and mitochondrial dysfunction, may also represent equally promising mechanisms.
1) BBB breakdown, particularly in the hippocampus, as an early event (potentially independent of aβ and tau)
Among neurovascular abnormalities, BBB dysfunction has emerged as a particularly actionable early phenotype. 6 The BBB is not a static structure but a dynamic interface regulated by endothelial tight junctions, pericytes, astrocytic endfeet, basement membrane, and immune–metabolic signaling—for example, the NVU. 6 Disruption of this system can allow the entry of plasma proteins, inflammatory mediators, and immune cells; perturb ionic homeostasis; and promote synaptic dysfunction. 6
The BBB permeability in the hippocampus age-dependently increases in individuals with normal cognitive function, and it is more evident in mild cognitive impairment using an advanced dynamic contrast-enhanced magnetic resonance imaging protocol with high spatial and temporal resolutions. The BBB breakdown in the hippocampus and its CA1 and dentate gyrus subdivisions worsened with mild cognitive impairment that is correlated with injury to BBB-associated pericytes, as shown on the cerebrospinal fluid analysis. Notably, BBB breakdown is an early event in the aging human brain that begins in the hippocampus. Furthermore, it may contribute to cognitive impairment (Figure 1). However, the reasons why BBB breakdown in the hippocampus are the early event remain unknown. 8 Therefore, hippocampal BBB leakage may be observed independent of amyloid and tau biomarker status or arise before the emergence of canonical proteinopathy signatures. Accordingly, BBB disruption may serve as a biomarker with particular relevance to early cognitive trajectories.
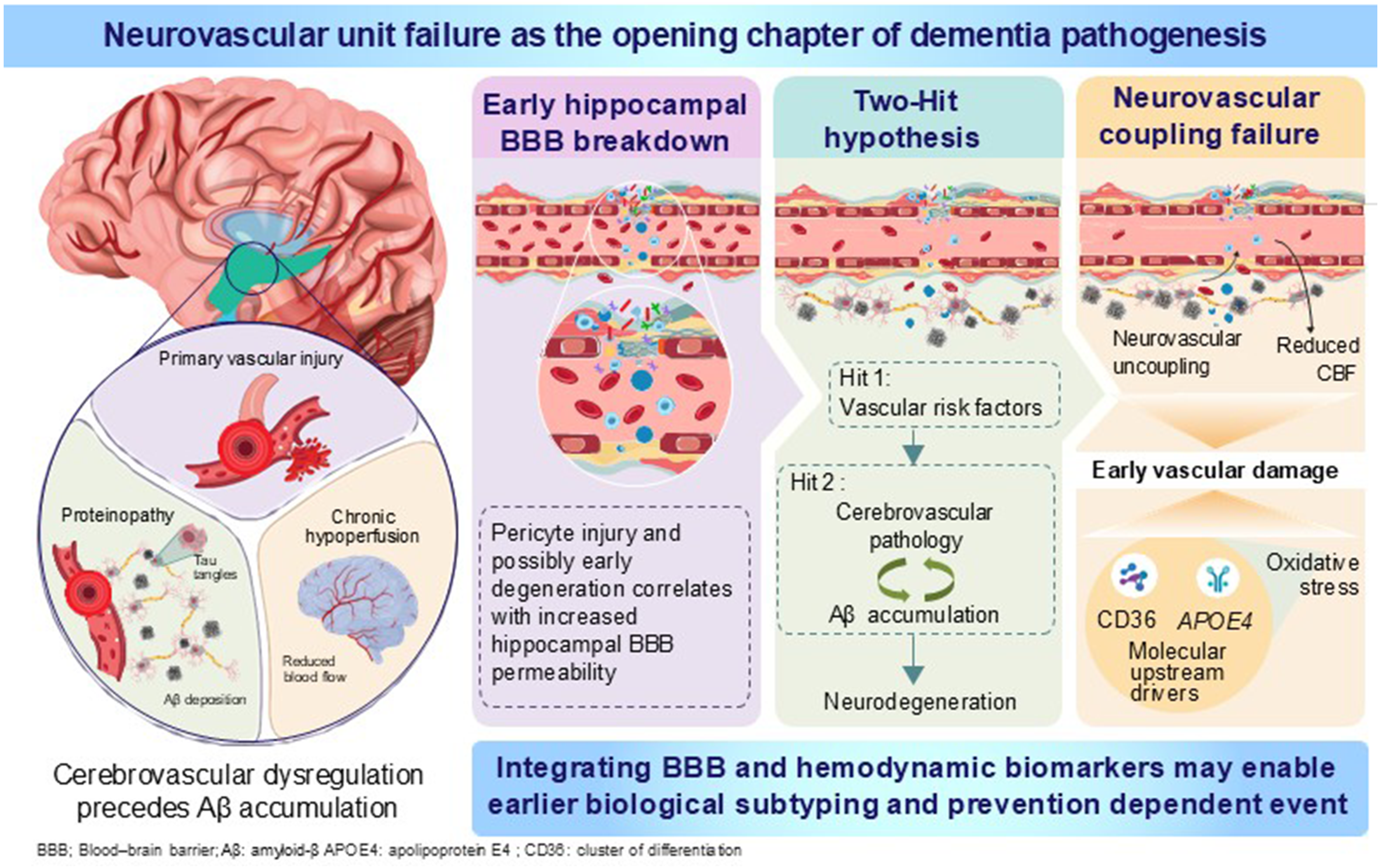
Neurovascular unit failure as the opening chapter of dementia pathogenesis.
2) Two-hit vascular hypothesis and neurovascular pathways upstream of the neurodegeneration
To integrate disparate findings, the two-hit vascular hypothesis has been proposed: The “first hit” encompasses vascular risk factors such as hypertension, diabetes, aging, and stroke, each of which can impair cerebrovascular integrity and reduce cerebral blood flow. The “second hit” involves two interrelated pathways. Specifically, cerebral hypoperfusion and BBB dysfunction induced by the first hit reinforce one another and promote (1) a cerebrovascular pathway, including microvascular hypoperfusion and neurotoxic injury, and (2) an Aβ pathway, including increased Aβ production and impaired Aβ clearance. Together, these processes are believed to drive neurodegeneration and ultimately lead to dementia. 9 The utility of this model is not merely rhetorical. In particular, it provides a testable ordering and emphasizes feedback loops: vascular injury reduces Aβ clearance and increases production stress, and Aβ itself is vasculotoxic, aggravating endothelial dysfunction and microvascular inflammation (Figure 1).
Comprehensive reviews of vascular contributions to cognitive impairment and dementia indicate that cerebrovascular pathology is a prominent and early feature, followed by neurodegeneration. 10 Moreover, mixed pathologies are the rule rather than the exception in older brains. 11 This is clinically salient: even cases of pure AD often harbor microinfarcts, arteriolosclerosis, cerebral amyloid angiopathy, or capillary rarefaction—lesions that may not cause classic stroke syndromes but can reshape network resilience and cognitive reserve. 11
The two-hit framing is also in accordance with precision medicine: patients differ in terms of the relative weighting of vascular versus Aβ and tau drivers. 12 In some patients, the dominant lever may be vascular health and perfusion. Of the 14 modifiable risk factors for dementia, 7 are vascular: elevated low-density lipoprotein cholesterol, physical inactivity, diabetes, smoking, hypertension, obesity, and excessive alcohol consumption, 13 encouraging regular exercise; reducing smoking through education, price control, smoke-free public policies, and accessible cessation support; preventing and treating hypertension and maintaining systolic blood pressure at 130 mm Hg or below from midlife; detecting and treating elevated low-density lipoprotein cholesterol beginning in midlife; maintaining a healthy weight and addressing obesity; and curbing excessive alcohol intake through pricing policies and improved public awareness of the risks of overconsumption. Collectively, elimination of these seven vascular risk factors could reduce the number of dementia cases by an estimated 17%. 13 For others, proteinopathy predominates, and for many, both processes interact. 12 A neurovascular lens may promote biomarker panels that include BBB permeability, endothelial activation, pericyte injury markers, and hemodynamic measures alongside Aβ and tau, thereby facilitating biological subtyping at the earliest symptomatic stages.
3) Early neurovascular coupling failure and cerebral hypoperfusion as preclinical signals
A third key aspect is dysfunction of neurovascular coupling—the process by which neuronal activity induces a localized increase in blood flow (functional hyperemia) to meet metabolic demand. Iadecola's seminal synthesis positioned the NVU at the center of AD pathophysiology, thereby detailing how oxidative stress, inflammation, and vascular risk factors disrupt coupling and cerebral blood flow regulation, potentially occurring early and contributing to synaptic failure. 14 In 3–4 month-old Tg2576 mice, early neurovascular dysfunction is evident before Aβ accumulation via excess NADPH oxidase-dependent oxidative stress through CD36, which is localized mainly to endothelial cells. 15 Therefore, the identification of CD36 as a key link between Aβ1–40 and vascular oxidative stress offers the prospect of targeted interventions that aim to improve cerebrovascular function by suppressing Aβ-induced CD36 signaling. 15 Furthermore, APOE4 exacerbates neurovascular dysfunction with excess cerebrovascular oxidative stress, which is generated by border-associated macrophages at a younger age.16,17 (Figure 1)
Within this framework, cerebral hypoperfusion is not merely a consequence of atrophy but may act as a central upstream driver of cognitive decline and exacerbation of AD-like pathology. Indeed, chronic cerebral hypoperfusion induced by bilateral common carotid artery stenosis (BCAS) operation 18 in Tg-SwDI mice increases Aβ accumulation. 19
Conclusion: translational strategy
The translational implication is straightforward: if we continue to treat dementia as solely a protein deposition disorder, we risk missing early windows where vascular mechanisms are most modifiable. A neurovascular approach argues for the following: (1) routine incorporation of BBB and hemodynamic biomarkers in prodromal cohorts, (2) mechanistically informed trials targeting endothelial and pericyte health as well as perfusion regulation, and (3) integrated models that consider Aβ and tau not as isolated culprits but as pathologies unfolding within a vascular and metabolic ecosystem. At the earliest stage of dementia, the cerebrovasculature is not merely a bystander—it may represent the opening chapter.
Footnotes
Acknowledgements
The author has no acknowledgments to report.
Author contribution(s)
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Yorito Hattori is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.”