
Abstract
Background
Soluble amyloid-β oligomers (Aβo) are considered the most neurotoxic species of Aβ and key drivers of Alzheimer's disease (AD) pathogenesis. Selective targeting of Aβo offers a promising therapeutic strategy. We previously identified an amyloid-binding peptide (ABP) that binds Aβo and engages Aβ deposits in AD mouse and human brain tissue. To facilitate brain penetration, ABP was fused to the blood-brain barrier (BBB) transporter FC5 and an Fc fragment, generating BBB-permeable constructs (mouse FC5-mFc2a-ABP and humanized FC5-hFc-ABP, referred to as BBB-ABP). Previous in vivo studies demonstrated the ability of BBB-enabled ABP to engage and clear Aβ from the central nervous system.
Objective
This study aimed to evaluate the in vitro functionality of BBB-ABP and its ability to prevent Aβo-induced neurotoxicity and synaptic dysfunction.
Methods
Binding specificity was assessed using ELISA and western blot overlay assays. Functional assays were performed in SH-SY5Y cells and primary neurons to evaluate Aβo sequestration, protection against Aβo-induced toxicity, and effects on synaptic activity measured via multi-electrode arrays.
Results
BBB-ABP retained selective binding to Aβo and effectively prevented its interaction with neuronal proteins and its binding to dendritic spines in live primary neurons. BBB-ABP significantly reduced Aβo-induced toxicity in SH-SY5Y cells and primary neurons, including under conditions of NMDA-induced stress. Aβ exposure did not significantly alter spontaneous synaptic activity, precluding assessment of electrophysiological rescue by BBB-ABP.
Conclusions
These findings demonstrate that BBB-ABP maintains Aβo-selective binding and is capable of preventing the interaction of Aβo with neurons, thereby mitigating Aβo-induced toxicity in vitro, supporting its further development as a therapeutic candidate for AD.
Introduction
Soluble amyloid-β oligomers (Aβo) are widely recognized as the primary neurotoxic form of Aβ in Alzheimer's disease (AD), and targeting Aβo has been identified as a promising strategy to prevent both neurodegeneration and cognitive decline in AD.1–3 Selectively targeting Aβo over monomeric and fibrillar Aβ prevents target distraction by minimizing interaction with nontoxic monomers, leaving more free therapeutic to engage Aβo, 4 and may avoid safety liabilities associated with binding to Aβ fibrils. 5 Notably, the ability to selectively target Aβo correlates with the clinical efficacy of anti-amyloid therapeutics.3,4
We have recently found a 40-amino acid peptide (ABP) from the brain pericentriolar material-1 (PCM-1) protein that selectively binds to amyloid-β1−42 (Aβ1−42) oligomers (Aβo) but not to Aβ1–42 monomers or Aβ1–40 in vitro. 6 Moreover, this selective Aβ-binding peptide binds Aβ aggregates in sections of cerebral cortices and hippocampi from APPswe/PS1dE9 AD-transgenic mice and postmortem brains from AD patients ex vivo. Most importantly, ABP bound Aβ deposits when microinjected into the hippocampi of live AD mice, indicating target engagement in vivo. 7 These studies suggested that when delivered to the brain, ABP can target Aβo and potentially facilitate their clearance from the brain. However, ABP has limited ability to cross the blood-brain barrier (BBB) and access brain parenchyma when administered systemically. In order to overcome this limitation, we have recently fused ABP with the BBB carrier FC5, a camelid single-domain antibody that undergoes receptor mediated transcytosis,8,9 along with mouse Fc2a (to enhance serum half-life of the fusion protein) to generate the BBB-permeable fusion protein FC5-mFc-ABP (Figure 1). 10 Using this fusion protein (formerly referred to as KG207 m), we have delivered ABP to the brain, and demonstrated the engagement and clearance of CNS Aβ in a rat model of AD which included translational biomarker analysis. 10 Similar results were obtained with a humanized version of the fusion protein in studies involving aged beagle dogs that develop Aβ pathology 11 and a mouse model of AD (manuscript under preparation).
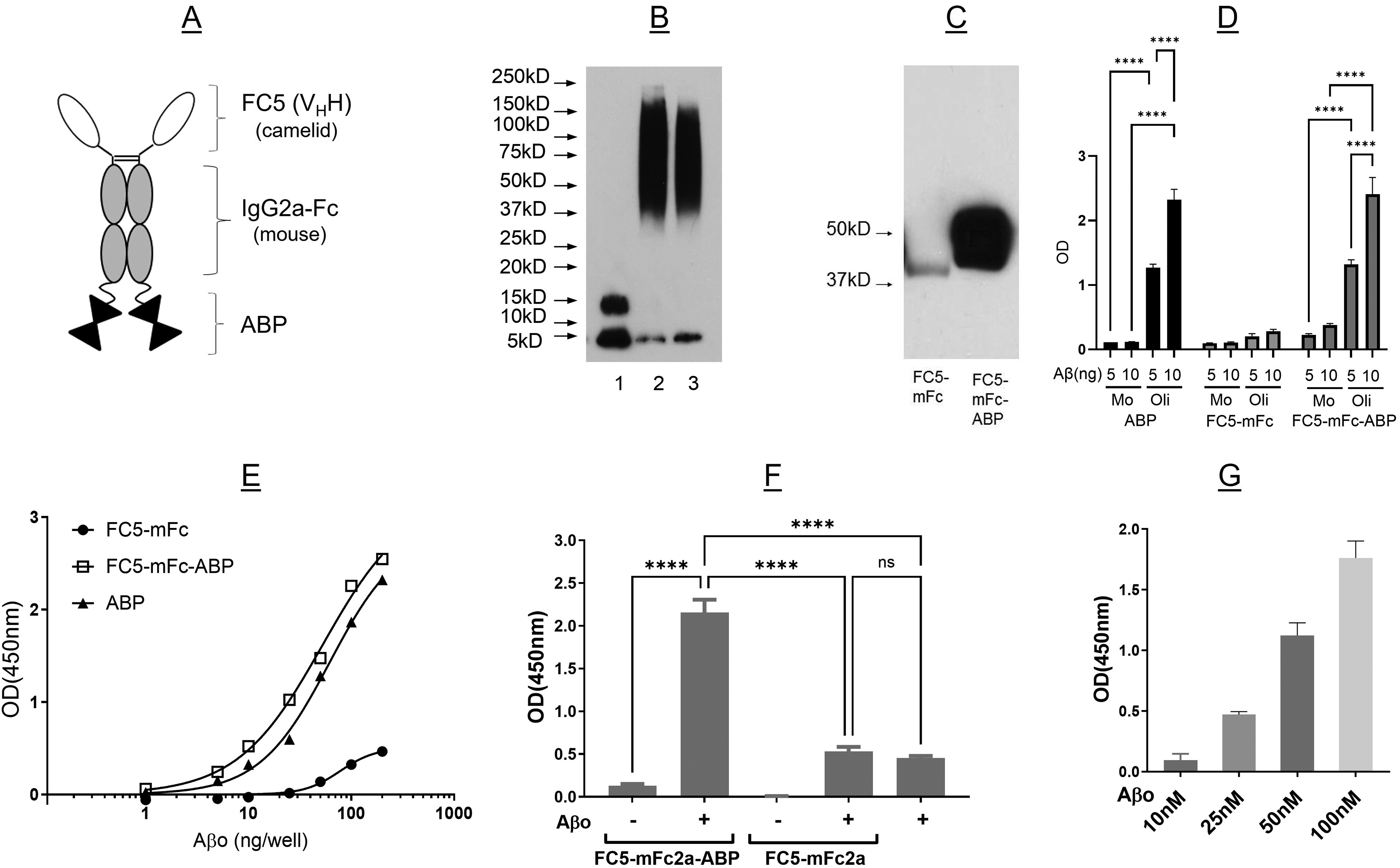
FC5-mFc-ABP fusion protein selectively binds to amyloid-β oligomers (Aβo) as assessed by western blot overlay and ELISA. (A) Schematic representation of fusion protein. FC5: blood-brain barrier crossing single-domain antibody (VHH); mFc: mouse IgG2a Fc fragment; and ABP: amyloid-β binding peptide. (B) Western blot analysis of Aβ monomer (lane 1) and two oligomer preparations made a year apart (lanes 2 and 3) demonstrating consistency. (C) Aβo binding to FC5-mFc2a and FC5-mFc2a-ABP was done by western blot overlay. The blot shows representative results of four independent experiments all demonstrating Aβo binding to FC5-mFc-ABP. (D, E) Binding of fusion proteins to monomeric and oligomeric Aβ was assessed by ELISA. ELISA plates were coated with ABP, FC5-mFc, or FC5-mFc-ABP, incubated with monomeric (Mo, D) or oligomeric (Oli, D,E) Aβ and bound Aβ detected with 4G8-HRP. In (E), curve fit using 4 parameter logistic regression and extra sum of squares test indicates that the curves differ significantly (p < 0.0001). (F) FC5-mFc2a-ABP or FC5-mFc2a (100 nM) were premixed with Aβo then captured on an ELISA plate coated with anti-FC5 antibody. Bound Aβo was detected using 6E10-HRP. One-way ANOVA with post-hoc Tukey's test indicated that FC5-mFc-ABP captured significantly more Aβo than FC5-mFc (p < 0.0001). (G) FC5-mFc2a-ABP was premixed with various concentrations of Aβo then captured on an anti-FC5 coated plate. One-way ANOVA indicated a significant difference between concentrations (overall ANOVA, F3,8-1938; p < 0.0001), with a significant linear trend across concentrations (p < 0.0001). Panel D shows representative results from at least three experiments done in triplicate wells, and Panel F shows representative results from one of two independent experiments done in triplicate wells. Panels E and G each show the results of a single experiment done in triplicate wells.
Notably, in rat studies, treatment with FC5-mFc-ABP was not only able to engage and facilitate the clearance of Aβ as determined by PET imaging, but also impacted downstream events associated with Aβ accumulation, resulting in improved hippocampal volume and neuronal connectivity as assessed by structural and functional MRI. 10 Based on these findings, we hypothesized that BBB-ABP might be capable of preventing the deleterious interactions of Aβo with neurites and dendritic spines, which are believed to impair synaptic and neuronal integrity in AD.3,12 To further explore this potential mechanism of action of BBB-ABP, we initiated a series of in vitro experiments which aimed to determine whether BBB-functionalized ABP could prevent the interaction of Aβo with neurons and dendritic spines, and whether sequestration of Aβo by ABP can offer neuroprotection against amyloid toxicity. As well, based on numerous reports documenting an effect of various forms of Aβ on electrophysiological and other functional properties of neurons, we sought to establish conditions under which Aβ altered spontaneous activity of rodent neurons cultured on Multi-Electrode Arrays (MEAs), to determine whether BBB-ABP can reverse the pathological effect of Aβo.
Methods
Reagents and consumables
Synthetic Aβ1−42 was purchased from Biopeptide Co. Inc. (San Diego, CA, USA) and Biomatik (Kitchener, ON, Canada). Aβ25−35 was bought from Biomatik. HyLitefluor-488-labeled Aβ1−42 and pyroglutamate (pyr3) Aβ were obtained from Anaspec (Fremont, CA, USA). 6E10 and 4G8 monoclonal anti-Aβ antibodies were from Covance Research Products Inc (Emeryville, CA, USA). HRP conjugated anti-ß-Amyloid 1–16 (clone 6E10) antibody was obtained from BioLegend (San Diego, CA, USA), and HRP conjugated anti-amyloid antibody MOAB-2 was purchased from Novus Biologicals (Bio-Techne Canada, Oakville, ON, Canada). Antibodies for immunocytochemistry were purchased from Abcam (drebrin), Sigma, (MAP2), Novus (AF488-MOAB2), and Invitrogen (Goat anti-Mouse IgG Alexa Fluor-568; Goat anti-Rabbit IgG Alexa Fluor 568). Protein block, antibody diluent and mounting media were purchased from Agilent (Santa Clara, CA, USA).
Eagle's Minimum Essential Medium/F12 (EMEM/F12) was bought from Invitrogen (Burlington, ON, Canada) and from Wisent Bioproducts (St Bruno, QC, Canada). DMEM and DMEM/F12 were from Thermo Fisher Scientific (Whitby, ON, Canada). Bovine serum albumin (BSA), dimethyl sulfoxide (DMSO), 1,1,1,33,3-hexafluoro-2-propanol (HFIP), methylthiazolyldiphenyl-tetrazolium bromide (MTT), RIPA buffer and peroxidase-conjugated anti-mouse and anti-rabbit IgGs were purchased from Sigma-Aldrich (St Louis, MO, USA). SDS-PAGE gels, protein assay reagent, electrophoresis buffers, nitrocellulose (0.2 μm) and polyvinylidene difluoride (PVDF)-membranes were obtained from Bio-Rad Laboratories (Mississauga, ON, Canada). Western Lightning chemiluminescence reagent was bought from Perkin Elmer (Mississauga, ON, Canada). Nunc Maxisorp 96-well plates were from Nunc (Rochester, NY, USA). Phosphate buffered saline (PBS) and Tris-buffered saline-Tween 20 (TBS-T) were purchased from Wisent (St Bruno, QC, Canada). Axygen MAXYMum recovery microcentrifuge tubes and Falcon cell culture dishes were purchased from VWR (Whitby, ON, Canada). Human neuroblastoma cell line SH-SY5Y (CRL-2266) was purchased from ATCC (Manassas, VA, USA). All other chemicals were analytical grade commercial products from Thermo Fisher Scientific (Whitby, ON, Canada).
Fetal bovine serum (FBS) was purchased from Corning (Corning, NY, USA), horse serum (HS), 5-fluoro-2’-deoxyuridine, uridine, polyethyleneimine (PEI) and poly-l-lysine (PLL), were purchased from Millipore Sigma (St Louis, MO, USA). Neurobasal A media, B-27 and GlutaMax were purchased from Gibco (Waltham, Massachusetts, USA). 12 K Eplasia flasks were purchased from Corning (Corning, NY, USA). Cytoview 48 well MEA plates were purchased from Axion Biosystems (Atlanta, GA, USA).
Preparation of Aß1−42 oligomers
Aβo were prepared according to the method of Velasco et al.. 13 A vial containing 1 mg of Aβ1−42 was dissolved in 1 ml of ice-cold hexafluoroisopropanol (HFIP) and incubated at room temperature (RT) for 1 h to allow monomerization and randomization of the peptide structure. The monomerized peptide solution in HFIP was left on ice for 30 min to reduce the evaporation and aliquoted at 100 µl into MAXYMum recovery microtubes. Subsequently, microtubes, with cap open, were centrifuged in a vacuum concentrator (SpeedVacTM or VacufugeTM) for 30 min at RT to ensure complete evaporation of HFIP. The resulting Aβ films (100 µg) were stored at −80°C after capping microtubes. At the time of use, a microtube containing Aβ film (100 µg) was brought to RT, 10 µl DMSO was added to the bottom of the microtube and Aβ was resuspended by vortexing. After bath sonication for 5 min, 434 µl of Dulbecco's Modified Eagle's Medium (DMEM) was added, vortexed and incubated for 24 h at 37°C to form oligomers. After 24 h, the Aβ preparation was centrifuged at 4°C for 10 min at 14,000 x g to remove any insoluble Aβ fibrils. The supernatant containing Aβo was aliquoted into MAXYMum microtubes and stored at −80°C. Aβo concentrations were measured according to the Bradford method using Bio-Rad protein assay reagents with Aβ1−40 as standard. The final concentration of Aβ in the working stock solution ranged between 50 and 100 µM. Fluorophore labelled amyloid-β was prepared as above with a mixture of labeled and unlabeled Aβ at a ratio of 1:4 respectively. 14
Formation of Aβ oligomers was assessed by SDS-PAGE and immunoblotting. Briefly, Aβo was prepared in tricine sample buffer at 5 ng/µl and was separated on 4–20% gradient polyacrylamide precast gels and transferred to nitrocellulose. It should be noted that the Aβo sample was not heat treated prior to SDS-PAGE. Membranes were blocked with 5% nonfat milk in Tris-buffered saline (10 mM Tris pH 8.0 and 150 mM NaCl) containing 0.05% Tween 20 (TBS-T) for 1 h and then incubated with HRP conjugated anti-β-Amyloid 1–16 (6E10) (1:25000) antibody overnight at 4°C. After 3 washes with TBS-T, membranes were incubated with an enhanced chemiluminescence reagent kit. Band profiles were visualized by exposure to X-ray film.
For MEA studies with primary neuronal cultures, stock solutions of Aβ1−42 and Aβ25−35 were prepared at concentrations ∼100 and ∼200 µM, respectively, in DMEM as described above, using as minimal amount of DMSO as possible (2%), in order to minimize any effect of DMSO alone on spontaneous activity (the maximal DMSO concentration in the medium was 0.2% when using the highest 20 µM peptide concentration). Stocks were stored at −80°C and, once opened, were stored at 4°C for a maximum of one month.
Aβ-binding studies
Western blot overlay. For detection of Aβo binding to the fusion protein by western blot overlay, FC5-mFc and FC5-mFc-ABP were prepared in reducing Laemmli buffer and heated at 85°C for 5 min. 50 ng of each were separated by SDS-PAGE on 10% Tris-glycine gel and transferred to a nitrocellulose membrane. The membrane was blocked with 2.5% BSA in TBS-T for 1 h at RT and subsequently incubated in 2.5 µg Aß oligomer in 10 ml of TBS-T containing 0.25% BSA for 1 h at RT. After washing the membrane for 15 min with TBS-T, it was then incubated with anti-Aβ 6E10-HRP (1:10,000) in TBS-T containing 0.5% BSA for 1 h at RT. The excess antibody was washed with TBS-T 3X 5 min. The bound Aβ was detected using a chemiluminescence reagent and exposed to X-ray film.
ELISA. Maxisorp 96-well ELISA plates were coated with ABP, FC5-mFc or FC5-mFc-ABP (500 ng each) overnight at 4°C in PBS. The wells were blocked with 1% BSA in TBS-T for 30 min and then incubated with indicated concentrations of monomeric (Mo) or oligomeric (Oli) amyloid diluted in TBS-T for 45 min with gentle agitation. Following three washes with TBS-T, bound Aβ was detected by incubating wells with 4G8-HRP in TBS-T for 90 min at RT. Bound antibody was detected with SureBlue TMB reagent by colorimetric measurement according to manufacturer's instructions. In other experiments, fusion proteins (100 nM) were premixed with Aβo (10, 25, 50, 100 nM) in TBS-T for 30 min at RT on a rotator, then applied to wells of a Maxisorp ELISA plate pre-coated with anti-FC5 antibody and incubated for 1 h at RT with gentle agitation. After thorough washing, captured Aβ was detected using 6E10-HRP and visualized using SureBlue TMB reagent.
Cell culture
SH-SY5Y human neuroblastoma cells
Stock SH-SY5Y human neuroblastoma cells were grown as adherent cultures in DMEM/Ham's F-12 50/50 mix (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and maintained at 37°C in a humidified 95% air-5% CO2 incubator.
Primary rat mixed cortical/hippocampal or hippocampal neuron preparation
All experiments were approved by the Human Health Therapeutics Animal Care Committee at the National Research Council Canada and carried out in accordance with approved guidelines. Timed pregnant Sprague Dawley rats were purchased from Charles River (St Constant, QC, Canada). Primary neurons were prepared from day 17–18 Sprague Dawley embryonic rats, as outlined elsewhere. 15 In brief, pregnant rats were anesthetized with 4% isoflurane and decapitated. Embryos were promptly transferred to Hank's Balanced Salt Solution and decapitated. Whole cortices with the hippocampus attached (cortical/hippocampal dissociated cultures or 3D neurospheres) or just the hippocampus (hippocampal dissociated cultures) was isolated, mechanically dissociated, and filtered using a 20 µm cell strainer. MEA CytoView 48-well plates were coated with PLL (0.025 mg/ml in 1X PBS) or PEI (0.1% in 25 mM Borate Buffer) for 3 h/2 h respectively at RT. Plates were washed 1X/2X respectively with distilled H2O, allowed to dry, wrapped in parafilm and stored at 4oC until plating of neurons.
Cortical/hippocampal neurons were suspended in plating media (EMEM, 10% FBS, 25 mM D-Glucose) and plated at a concentration of 1.6 × 106 cells/ml (for MEA) or at assay-specific concentrations as indicated below. DNA synthesis inhibitors, 5-fluoro-2’-deoxyuridine (1.5 mg/ml) and uridine (3.5 mg/ml), were added at 4 days in vitro (DIV) and a 50% media replacement was performed at 7 DIV with growth media (EMEM + 5% HS + 25 mM D-Glucose). In order to minimize any sequestration of Aβ1−42 by serum proteins, culture media was replaced with low serum media (EMEM + 1% HS and 25 mM D-Glucose) prior (4–24 h) to exposure to Aβ1−42.
Hippocampal neurons were suspended in Neurobasal A media containing B-27 and GlutaMax, plated at a concentration of 0.4 × 106 cells/ml (for MEA) or at assay-specific concentrations as indicated below, and 33% of the media was replaced 3x per week beginning at 7 days in vitro (DIV).
For the preparation of 3D neurospheres, cortical/hippocampal neurons were plated onto a 12K-well Eplasia flask at a density of 2.0 × 106 cells/flask, in Neurobasal A media containing B-27 and Glutamax. 3D neurospheres formed within >90% of wells. At 7 DIV, neurospheres were ∼150 µm in diameter, and were extracted by gently shaking the flask, transferring to a 15 ml centrifuge tube and allowing to settle by gravity. The supernatant was removed, and neurospheres were plated on 48-well MEAs in a 3 µl drop over the electrode area, striving for an electrode coverage > 50%. Water was added to the space surrounding the wells to create a humid chamber and prevent evaporative loss. The plate was left undisturbed for 60 min, and 300 µl/well of the conditioned media was added. A 33% media replacement was performed 3x per week.
Aßo binding to SH-SY5Y and primary hippocampal cell proteins and competition with BBB-ABP and FC5(H3)-hFc1×7 (overlay)
Multiple lanes of 1.0 µg of a whole cell lysate from SH-SY5Y cells or primary hippocampal cultures were separated by SDS-PAGE on 4–20% gel and transferred to nitrocellulose membranes. The membranes were cut in lane segments and blocked individually with 2.0% BSA in TBS-T for 1 h at RT. Aβo alone or pre-mixed with BBB-ABP or FC5(H3)-hFc1×7 were prepared to overlay on membranes at 10 nM in 0.25% BSA in TBS-T. The control was 0.25% BSA in TBS-T only. The samples were incubated on the nitrocellulose blots in Ziploc bags for 30 min at RT and then washed for 15 min with TBS-T. The Aβo binding to cellular proteins was detected using 6E10-HRP (1:50,000) or MOAB-HRP (1:10,000) anti-amyloid antibody in 0.5% BSA in TBS-T for 1 h at RT and washed 3X for 5 min with TBS-T. Subsequently the transblots were incubated with ECL and exposed to film.
Aβo binding to live primary hippocampal/cortical cells and competition with BBB-ABP by western blot and immunocytochemistry
For immunocytochemistry, primary rat hippocampal neurons grown in neurobasal media were plated on PLL-coated glass coverslips at a density of 0.035 × 106 cells/ml and treated between 14 and 18 DIV. Conditioned media was used to prepare treatments consisting of vehicle controls, Aβo alone, Aβo + BBB-ABP, and Aβo + FC5-hFc1×7 at 1 µM. Treated media was incubated at RT for 30 min, and then applied to cells for 1 h at 37°C. Following incubation, the media was carefully removed and fresh media added to each well. To fix cells, zinc fixative was diluted to a 2X concentration in Milli-Q water, then added in a 1:1 ratio to the cell culture media in the wells and incubated at RT for 20 min. Cells were washed 3 times with Milli-Q water then stored at 4°C in PBS.
Cells were blocked and permeabilized with Dako serum free protein block containing 0.2% triton X-100 for 1 h at RT, then incubated with Alexa fluor (AF) 488-conjugated MOAB-2 (1:500) and anti-drebrin (1:250) in Dako diluent plus 0.1% triton X overnight at 4°C. The following day, cells were washed with PBS, incubated in anti-rabbit-AF568 (1:500) in Dako diluent plus 0.1% triton X for 1 h at RT. After washing in PBS, coverslips were dipped in distilled water and mounted using Prolong Glass containing DAPI. Images were acquired on a Leica Stellaris confocal platform with DMi8 microscope.
Similar experiments were carried out using HyLite Fluor-488-labeled oligomeric amyloid. In this case, cells were stained with anti-MAP2 (1:500) and anti-mouse-AF568 (1:500).
For western blot, primary mixed cortical/hippocampal cells (1.1 × 106 cells/ml in 24 well plates) were treated as above (except treatment was for 30 min), then washed with PBS and collected in modified RIPA buffer (TBS containing 1% sodium deoxycholate and 1% IGEPAL). Material was pooled across 4 wells, then centrifuged at 10,000 g for 10 min at 4°C. Supernatant was collected and mixed 1:1 with tricine sample buffer. Unheated samples were separated by SDS-PAGE on 4–20% TGX gel and transferred to a nitrocellulose membrane. Aβo was detected using 4G8-HRP anti-amyloid antibody at 1:5000 in 0.5% BSA in TBS-T overnight at 4°C and washed 3X for 5 min with TBS-T. Subsequently the transblots were incubated with ECL and exposed to film.
Aβ toxicity studies
SH-SY5Y neuroblastoma cells
For cell toxicity studies, SH-SY5Y cells were seeded at a density of 1.0 × 104 cells on 96 well culture plates and grown for 3 days. Aβo toxicity and protection with BBB-ABP and FC5(H3)-hFc1×7 in SH-SY5Y cells was evaluated by MTT assay. 16 Twenty-four hours before treatment, the culture medium was changed to DMEM/F12 supplemented with 1% FBS. Conditioned 1% FBS media from spare unused wells was collected and spiked with 1.0 µM Aβo with or without addition of BBB-ABP or FC5(H3)-hFc1×7 at 0.5, 1.0, or 2.0 µM and allowed to incubate for 60 min at RT prior to adding to cells. SH-SY5Y cells in 96 well format were exposed to 100 μl of spiked media for 24 h in triplicate wells. After 24 h, the culture media was removed from the wells and replaced with 100 µl media containing 10 µl MTT reagent, reserving one well without cells for reference. The cells were incubated for 3 h at 37°C/95% air/5% CO2. MTT reagent is taken up by live active healthy cells into mitochondria and reduced by mitochondrial enzymes to form insoluble formazan crystals. The formazan blue crystals formed after 3 h were dissolved by adding 150 µl/well of acidic isopropanol. Each well was mixed well to dissolve all crystals. The absorbance was read at 570 nm with reference at 650 nm. The metabolic activity was averaged across triplicate wells and expressed as % of control within each experiment. The experiment was conducted on a minimum of three different platings, using triplicate wells for each condition.
Primary neurons
Aβo toxicity and protection with BBB-ABP and FC5(H3)-hFc1×7 in primary mixed cortical/hippocampal and pure hippocampal neurons was evaluated by MTT assay as described above for SH-SY5Y cells with minor modifications as described below. Primary mixed cortical/hippocampal neurons were seeded at 100 000 cells/well in 96 well format as described above and used between days 14–18 of culturing. 24 h before treatment, the culture medium was changed to EMEM + 25 mM D-Glucose supplemented with 1% horse serum (HS) for mixed cortical cultures only. Primary hippocampal neurons were seeded at 30 000 cells/well in 96 well format and used between days 14–18 of culturing. No media change was performed 24 h prior to the experiment. Quadruplicate wells were used for hippocampal neurons. In some experiments, hippocampal neurons were pre-treated with 10 or 20 μM NMDA for 30 min in conditioned media, then washed with neurobasal media prior to addition of Aβo with/without BBB-ABP or FC5(H3)-hFc.
MTT assay statistics
OD values were averaged for each treatment condition and normalized to the control condition within each experiment. Normalized data were subjected to one-way ANOVA with post-hoc Dunnett's test comparing Aβo alone to all other treatment conditions. Controls were not included in the ANOVA since variability is artificially 0 in the control group as a result of normalization. For assays which included NMDA, post-hoc testing was done using Tukey's test to compare each treatment condition to all other conditions.
MEA recordings
Spontaneous network activity was measured with an Axion Biosystems Maestro Pro platform (Axion Biosystems, Atlanta, GA, USA). All neuron preparations were plated on 48-well CytoView plates, which contained 16 electrodes per well. The amplifier recorded from all channels simultaneously using a gain of 1200× and a maximal sampling rate of 12.5 kHz/channel. Raw signals were filtered by a bandpass filter (200–33 Hz) and spikes were detected using a threshold of 6 x the S.D. of the noise signal from electrodes.
The effect of long-term exposure to various forms of Aβ on spontaneous activity of neurons grown on MEAs was examined by performing pre- and post-Aβ recordings (5 min duration for all recordings) for 48 h. The recording chamber of the Axion Maestro Pro was maintained at 37oC and under a 5% CO2 atmosphere during measurements. Following transfer of the MEA plate to the Axion Maestro Pro, a 20 min equilibration period and an initial recording was acquired. Media was removed (100 µl/well) and pooled to allow preparation of a 2X final concentration of peptide or vehicle, and then re-distributed (100 µl/well) as required. (Pilot experiments indicated that suctioning of all media from wells caused a substantial spike in spontaneous recordings, precluding evaluation of any early effects of peptides). Recordings commenced immediately, initially more frequently in order not to miss any early changes in activity, and then less frequently, up to 48 h.
The total number of neuron dissections and MEAs employed in this study was 26. Mixed cortical/hippocampal neurons produced a pre-Aβ spiking frequency of 17.0 ± 2.1 Hz, with 35 ± 3 active electrodes, representing an average of 161.6 ± 19.9 spikes/well over the 5 min recording period. Hippocampal neurons produced a pre-Aβ spiking frequency of 39.2 ± 14.1 Hz, with 44 ± 2 active electrodes, representing an average of 320.9 ± 114.9 spikes/electrode over the 5 min recording period. 3D neurospheres neurons produced a pre-Aβ spiking frequency of 28.1 ± 10.7 Hz, representing an average of 675.2 ± 256.7 spikes/electrode over the 5 min recording period.
MEA recording analysis
Raw voltage recordings were analyzed with the Axion AxIS Navigator. Electrodes displaying less than 5 spikes/min and wells with < 25% active electrodes in pre-peptide recordings were excluded from analysis. Due to a wide range of spontaneous activity frequencies and bursting between wells—as well as between electrodes within a well—spontaneous activity frequencies were analyzed on an individual electrode basis. The frequency of spontaneous activity for each electrode from the pre-peptide recording was normalized to 100%, and the effect of a peptide on frequency was normalized relative to this value for each time-point. These ratios of pre-Aβ:post-Aβ frequency computed for each electrode were then averaged for each well. The average frequency of spontaneous activity for each well was then summarized for each condition. Each peptide condition (type and concentration) was examined over a minimum of 2 platings of neurons (N), and each plating contained a minimum of 3 wells (n).
Results
FC5-mFc-ABP fusion protein
The fusion protein consisting of the BBB penetrating single domain antibody FC5, mouse Fc2a fragment (IgG2) and ABP, schematically shown in Figure 1A, was designed and produced in CHOBRI cells, along with FC5-mFc2a lacking ABP. 10
In vitro amyloid binding
We previously reported that ABP bound Aβo in vitro, as well as amyloid deposits in human AD and APPswe/PS1dE9 transgenic mouse brain.6,7 To ensure ABP retained its ability to bind aggregated amyloid after fusion with FC5-mFc2a, the amyloid binding profile of the fusion protein was assessed by western blot overlay and ELISA. FC5-mFc2a and FC5-mFc2a-ABP were run on SDS-PAGE and overlaid with an Aβ preparation consisting predominantly of oligomers (Figure 1B). As shown in Figure 1C, Aβ bound minimally to FC5-mFc2a but showed strong binding to FC5-mFc2a-ABP. Similarly, when immobilized on ELISA plates, both ABP alone and FC5-mFc2a-ABP bound Aβo but showed very little binding to monomeric amyloid, while FC5-mFc2a demonstrated minimal binding to either amyloid preparation (Figure 1D, E). To further demonstrate the interaction of FC5-mFc2a-ABP with Aβo, the full-length fusion protein or FC5-mFc2a (100 nM) were incubated with Aβo (0–100 nM) in solution for 30 min. These were overlaid on ELISA plates pre-coated with anti-FC5 antibody to capture the complex, and bound Aβ was detected using anti-amyloid antibody 6E10. Capture of FC5-mFc2a brought down negligible bound amyloid, while bound Aβo was detected in the samples incubated with FC5-mFc2a-ABP in an Aβ-dose dependent fashion (Figure 1F, G). Together, these data demonstrate that fusion of FC5-mFc2a to the N-terminus of ABP did not alter its amyloid binding activity or its selectivity for oligomeric over monomeric Aβ.
Based on this in vitro study, we successfully carried out an in vivo efficacy study demonstrating the ability of BBB-enabled ABP to cross the BBB, engage Aβ target in the brain and facilitate its clearance in a rat model of AD (McGill-R-Thy1-APP-tg,. 10 In addition to reduction in brain Aβ load, this study also demonstrated hippocampal volume restoration and improved neuronal connectivity, suggesting therapeutic potential of this fusion protein. Therefore, in anticipation of clinical studies, a humanized version of this fusion protein, consisting of Fc fragment of human IgG1, and modified FC5 and ABP, referred to here as BBB-ABP, was designed (Figure 2A) and produced in CHOBRI cells. 11 Neither BBB permeability (data not shown) nor Aβo binding characteristics were affected by humanization of FC5 and modification of ABP (replacement of 6 arginine and lysine residues with glycine at N- and C-terminus region), respectively, in the humanized fusion protein (Figure 2B, C). Retention of FC5 functionality (BBB-permeability and brain penetration) and ABP activity (brain target engagement) in the humanized version of the fusion protein was further demonstrated in vivo in a mouse model of AD (APPswe/PS1dE9 mice) (data not shown, manuscript in preparation) and aged-dog model of AD. 11
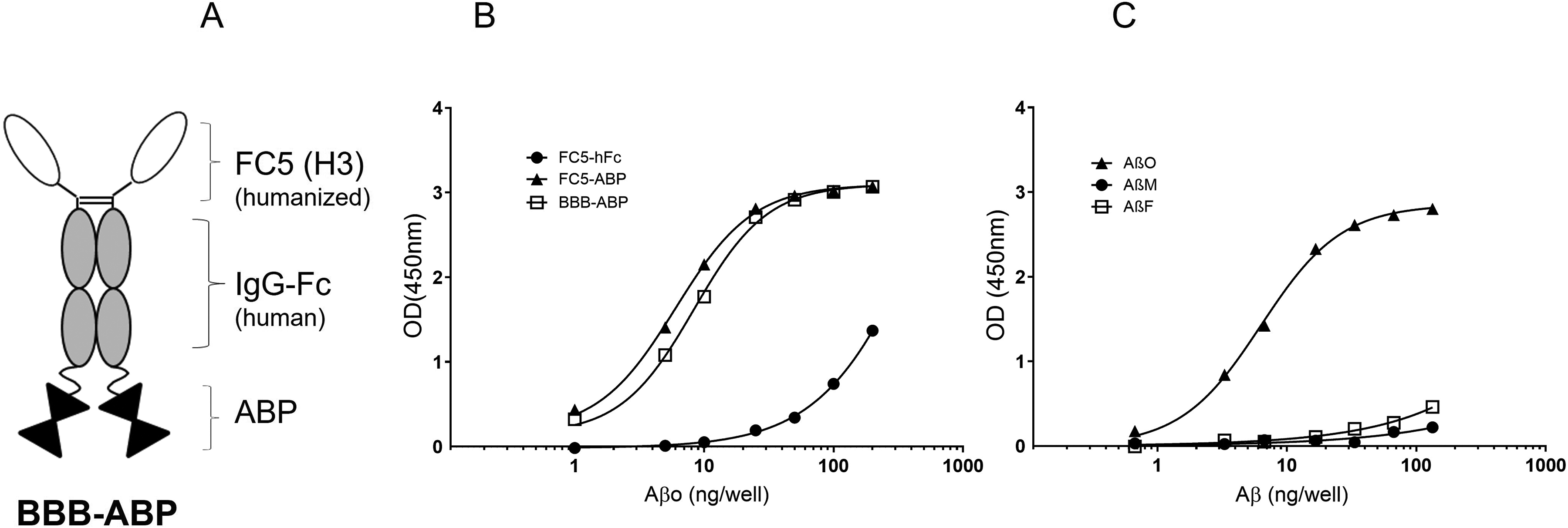
(A) Characterization of humanized BBB-ABP. Schematic representation of BBB-ABP. FC5 (H3): humanized blood-brain barrier crossing single-domain antibody; hFc: human IgG1 Fc fragment; and ABP: amyloid-β binding peptide. (B) Both BBB-ABP and FC5-ABP, but not FC5-hFc bind oligomeric Aβ as determined by ELISA as described in Figure 1. The graph shows representative results of one of three independent experiments, each done in triplicate wells. Curve fit using 4 parameter logistic regression and extra sum of squares test indicate that the curves differ significantly (p < 0.0001). (C) Binding of BBB-ABP to preparations containing predominantly monomeric, oligomeric, and fibrillar Aβ was assessed by ELISA. BBB-ABP retains the ability to bind selectively to oligomeric Aβ. The graph shows representative results of one of two independent experiments, each done in triplicate wells. Curve fit using 4 parameter logistic regression and extra sum of squares test indicate that the curves differ significantly (p < 0.0001).
Aβo binding and toxicity in SH-SY5Y neuroblastoma cells
To further assess other functional characteristics of ABP, besides facilitating Aβ clearance, we focused on whether ABP would affect Aβo's interaction with cellular proteins and its neuro/synaptotoxicity in in vitro cell culture models. To assess Aβo binding to cellular proteins in vitro, proteins in human neuroblastoma cell lysate were separated by SDS-PAGE and transblotted on nitrocellulose membranes. The transblots were exposed to 10 nM monomeric (Aβm) or oligomeric Aβ (Aβo) at 4°C and RT as described in the Methods section. As shown in Figure 3A, there was no observable binding of Aβm to cellular proteins at either temperature whereas there was significant binding of Aβo to cellular proteins, but only at RT. When Aβo was pre-mixed with BBB-ABP before overlay, Aβo binding was significantly reduced, indicating that BBB-ABP interfered with Aβo's interaction with cellular proteins (Figure 3B). Consistent with this, BBB-ABP interference was also seen in Aβo-induced cell toxicity. As shown in Figure 3C, Aβo at 1–10 µM was toxic to undifferentiated human neuroblastoma cultures as measured by MTT assay. This Aβo-induced toxicity was significantly prevented by BBB-ABP when pre-mixed with Aβo before exposing the cultured cells (Figure 3D). This inhibition was apparent at all molar ratios of Aβo:BBB-ABP tested (1:0.5 to 1:2; p < 0.005 compared to Aβo alone). FC5-hFc construct without ABP had very little effect on Aβo-induced toxicity, as expected. BBB-ABP and FC5-hFc did not have any toxic effects by themselves.
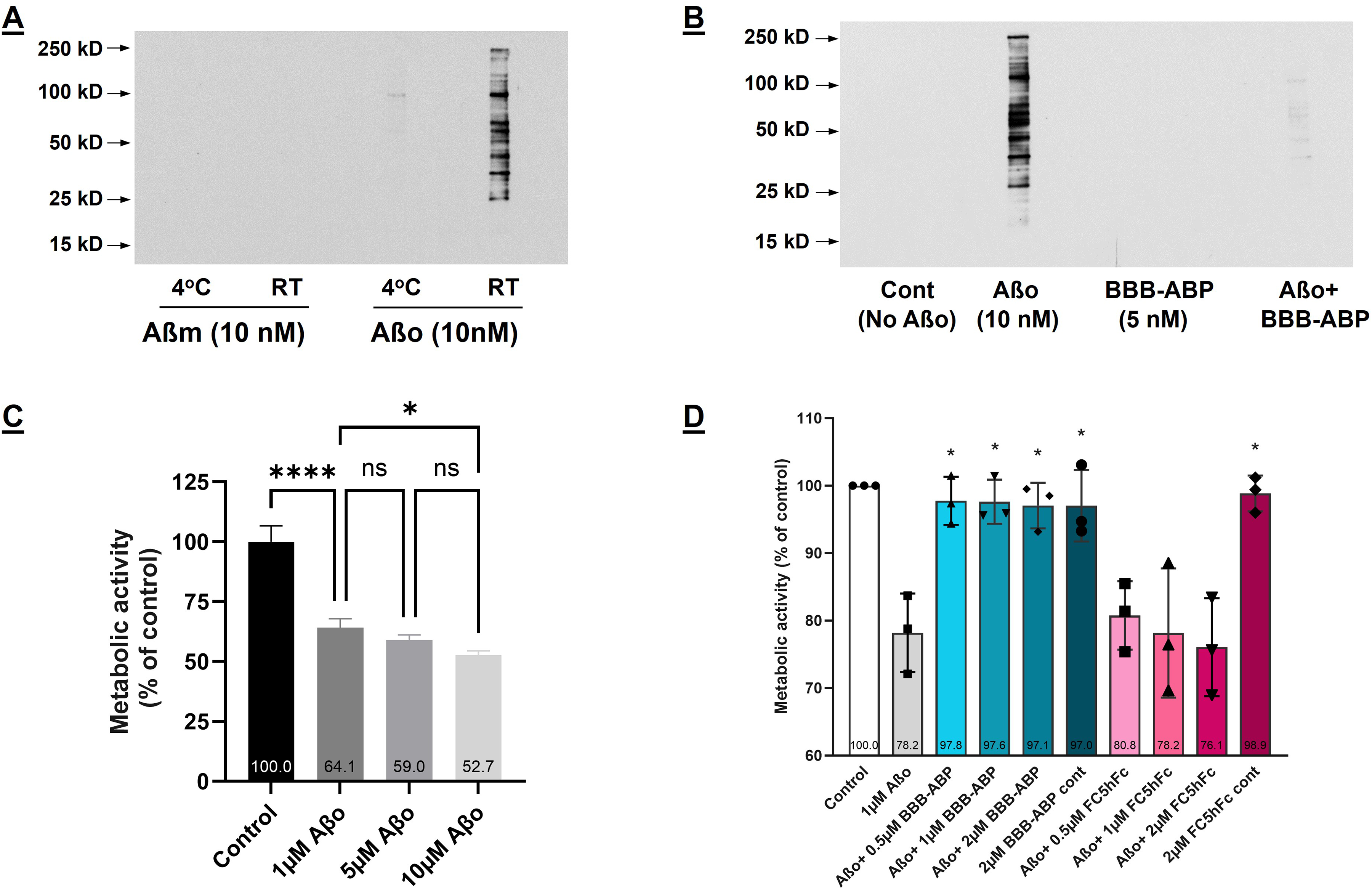
BBB-ABP interferes with AβO-binding to cellular proteins and prevents neurotoxicity in SH-SY5Y cells. (A) Multiple lanes of 1.0 µg of a whole cell lysate from SH-SY5Y cells were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were cut in lane segments which were overlaid with 10 nM Aβm or Aβo at 4°C or room temperature (RT). Bound Aβo was detected using 6E10-HRP. The figure shows the results of a single experiment; however, the main finding of Aβo binding to SH-SY5Y lysates at room temperature is replicated in Panel B. (B) Binding of Aβo to SH-SY5Y cellular proteins (detected with 6E10-HRP) is prevented by pre-incubation with BBB-ABP. Panel B shows representative results from one of three independent experiments. (C) 1 μM Aβo induced substantial toxicity in SH-SY5Y cells (ANOVA with post-hoc Tukey's test; p < 0.0001), while increasing the concentration to 5 μM and 10 μM yielded only slightly greater decrements in metabolic activity; 1 μM was therefore chosen as the dose for further experiments. The graph shows the results of a single experiment done in triplicate wells. (D) Pre-incubation of Aβo with BBB-ABP prevented Aβo-induced toxicity in SH-SY5Y cells at all tested molar ratios of Aβo:BBB-ABP (p < 0.005) which was dependent on ABP since pre-incubation with FC5-Fc failed to prevent toxicity. The graph shows aggregate results of three independent experiments, each done in triplicate wells.
Aβo binding and toxicity in primary neurons
The effect of BBB-ABP on Aβo binding to primary neuronal cells was tested. As shown in Figure 4A, Aβo bound hippocampal cellular proteins in vitro as assessed by overlay assay. As seen with neuroblastoma cells, BBB-ABP prevented Aβo binding to cellular proteins, whereas FC5-hFc without ABP did not affect this binding (Figure 4A). Similarly, when live primary mixed cortical/hippocampal cell cultures were exposed to Aβo, its binding to cellular proteins was detectable by western blot and this binding was substantially reduced when cells were exposed to Aβo pre-mixed with equimolar BBB-ABP. There was no binding of Aβ monomers. This was supported by immunocytochemical analysis, wherein hippocampal cultures were exposed to Aβo with or without pre-mixing with equimolar BBB-ABP, and bound Aβo was detected with MOAB-2 antibody. As shown in Figure 4C, without BBB-ABP, Aβo bound extensively to cell body and neurites in a punctate manner which was blocked by BBB-ABP when cells were exposed to Aβo pre-mixed with BBB-ABP. FC5-hFc did not prevent Aβo binding to primary neurons. To account for the possibility that BBB-ABP might interfere with Aβo detection by MOAB-2, identical experiments with HyLite Fluor 488-labelled Aβo were conducted. Again, Aβo binding was strongly attenuated by pre-incubation of Aβo with BBB-ABP, but not by FC5-hFc (Figure 4D). Immunostaining for drebrin, an actin-binding protein enriched in dendritic spines, showed extensive binding of Aβo to spines which was substantially reduced by pre-incubation of Aβo with BBB-ABP but not by FC5-Fc (Figure 5).
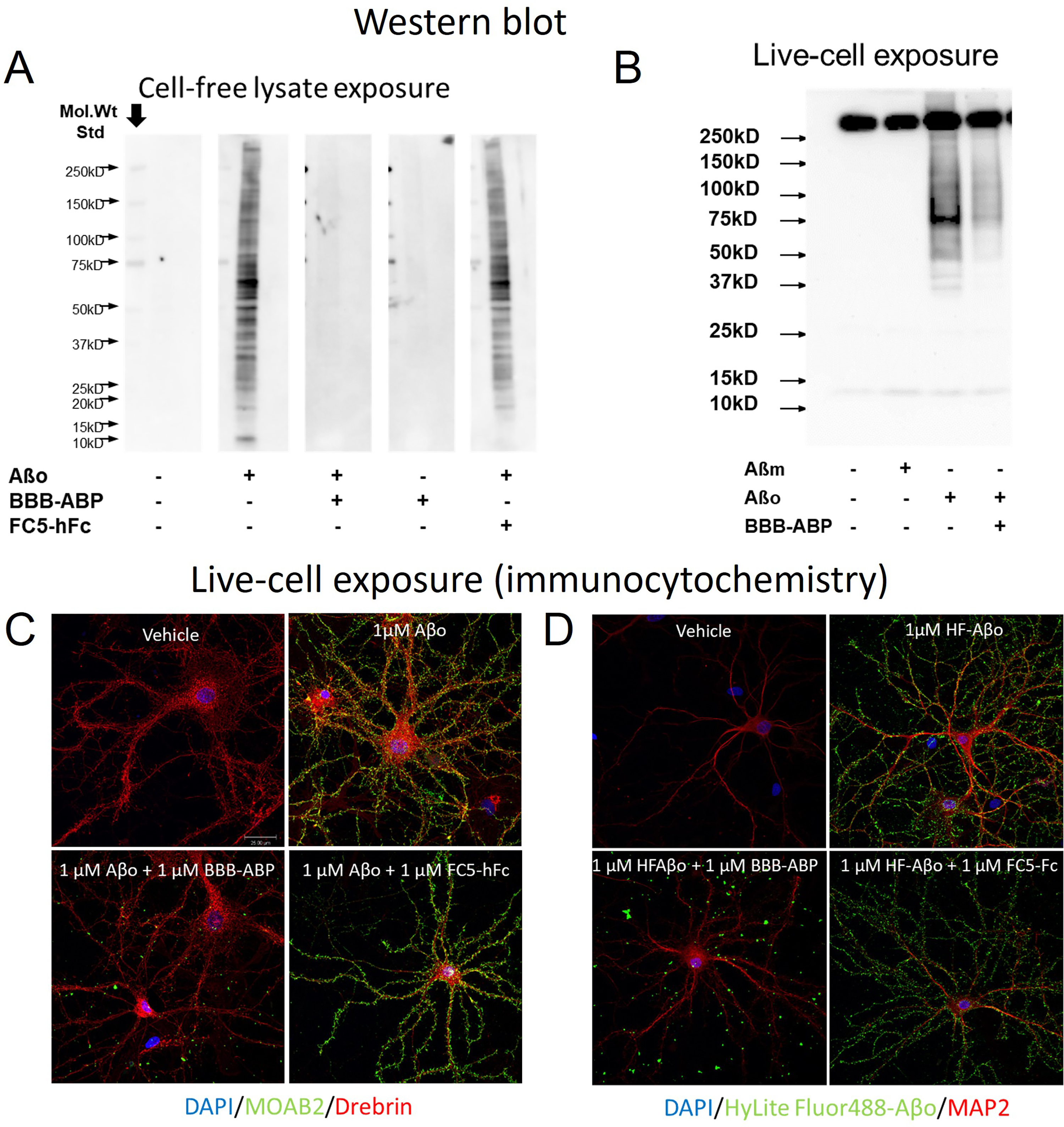
BBB-ABP attenuates Aβo binding to primary neurons. (A) Whole cell lysate from primary hippocampal neurons was separated by SDS-PAGE in multiple lanes and transferred to nitrocellulose membrane. The membranes were cut in lane segments then exposed to Aβo alone or Aβo pre-mixed with BBB-ABP or FC5-hFc. Bound Aβo was detected with MOAB in combination with anti-mouse-HRP. BBB-ABP, but not FC5-hFc prevented Aβo binding to primary neuron lysates. The figure shows representative results from one of two independent experiments. (B) Live primary mixed cortical/hippocampal neurons were treated with Aβo in the presence or absence of BBB-ABP, then lysates were separated by SDS-PAGE, transferred to nitrocellulose membrane and probed with 4G8-HRP. A nonspecific band associated with the detection antibody appears above 250 kD. Similar studies were carried out with hippocampal neurons (data not shown). The figure shows the results of a single experiment, which prompted the corroborating imaging experiments shown in C and D. (C) Live primary hippocampal neurons were treated with Aβo in the presence or absence of BBB-ABP or FC5-Fc, then washed, fixed, and stained with MOAB2 to detect bound Aβo. BBB-ABP but not FC5-Fc strongly attenuated Aβo binding to neurons. (D) Similarly, preincubation of Hylite-fluor-labeled Aβo with BBB-ABP but not FC5-Fc inhibited binding of labeled Aβo to hippocampal neurons. In total, five independent imaging experiments were conducted (using MOAB, 4G8 or Hylitefluor-tagged Aβo), all showing reduced Aβo binding to primary neurons when pre-mixed with BBB-ABP.
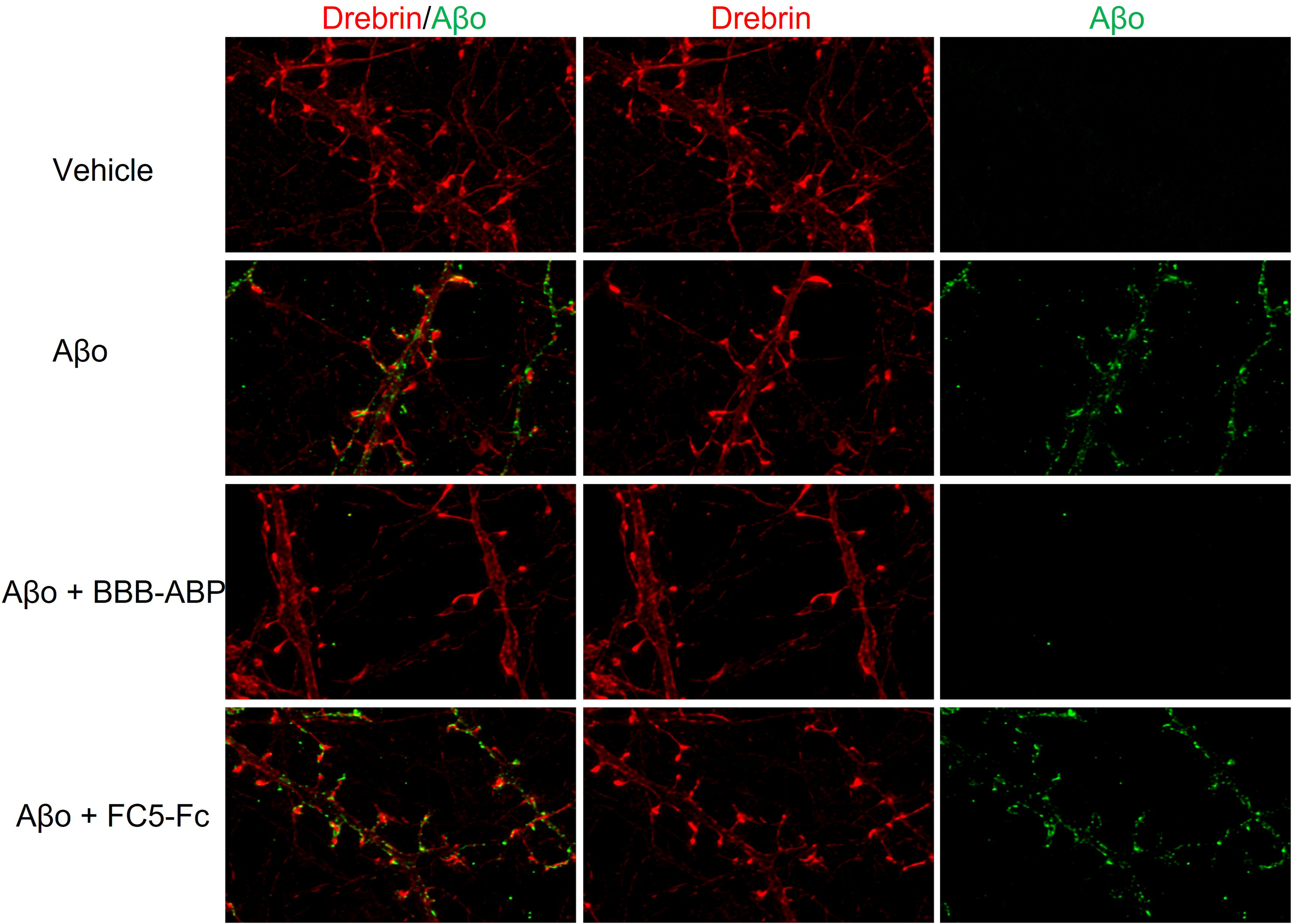
Aβo binding to dendritic spines is prevented by BBB-ABP. Live primary hippocampal neurons were treated with Hylite-fluor-labeled Aβo in the presence or absence of BBB-ABP or FC5-Fc for 1 h then washed, fixed, and stained with drebrin to identify dendritic spines. Aβo binding to dendritic spines was markedly reduced by BBB-ABP but not by FC5-Fc.
To determine whether Aβo binding of BBB-ABP reduces Aβo-toxicity as seen in neuroblastoma cells, primary hippocampal cultures were exposed to 5 µM Aβo, which was determined to be the dose required to achieve sufficient toxicity in these cells (Figure 6A), with and without pre-mixing with BBB-ABP. As shown in Figure 6B, Aβo induced significant toxicity as measured by the MTT assay, and this toxicity was significantly reduced when cells were exposed to Aβo premixed with BBB-ABP at 1:1 or 1:2 ratio respectively (one-way ANOVA with post-hoc Dunnett's test, p < .01). This neuroprotection was seen even when neuronal cells were subjected to additional stress by pre-exposure to sublethal levels of NMDA (10 µM) before Aβo (one-way ANOVA with post-hoc Tukey's test, NMDA versus NMDA + Aβo p < 0.005; NMDA + Aβo versus NMDA + Aβo + BBB-ABP p < 0.005). Notably, primary cultures varied in responsiveness to NMDA alone. In cultures that were very sensitive to NMDA, Aβo had little additional effect; in those cultures, the BBB-ABP effect on Aβo-induced toxicity could not be assessed. Therefore, experiments in which NMDA + Aβo resulted in less than 10% greater loss of signal than NMDA alone were excluded from analysis. In cells exposed to a higher concentration of NMDA (20μM) in combination with Aβo, the neuroprotective effect of BBB-ABP was reduced (Figure 6C,D). Another notable observation is that NMDA-treated neuronal cultures were more susceptible to Aβo toxicity, with a significant effect seen at 1 µM Aβo in NMDA-treated cultures compared to the higher dose of Aβo (5 µM) required to observe significant toxicity in non-NMDA treated cultures.
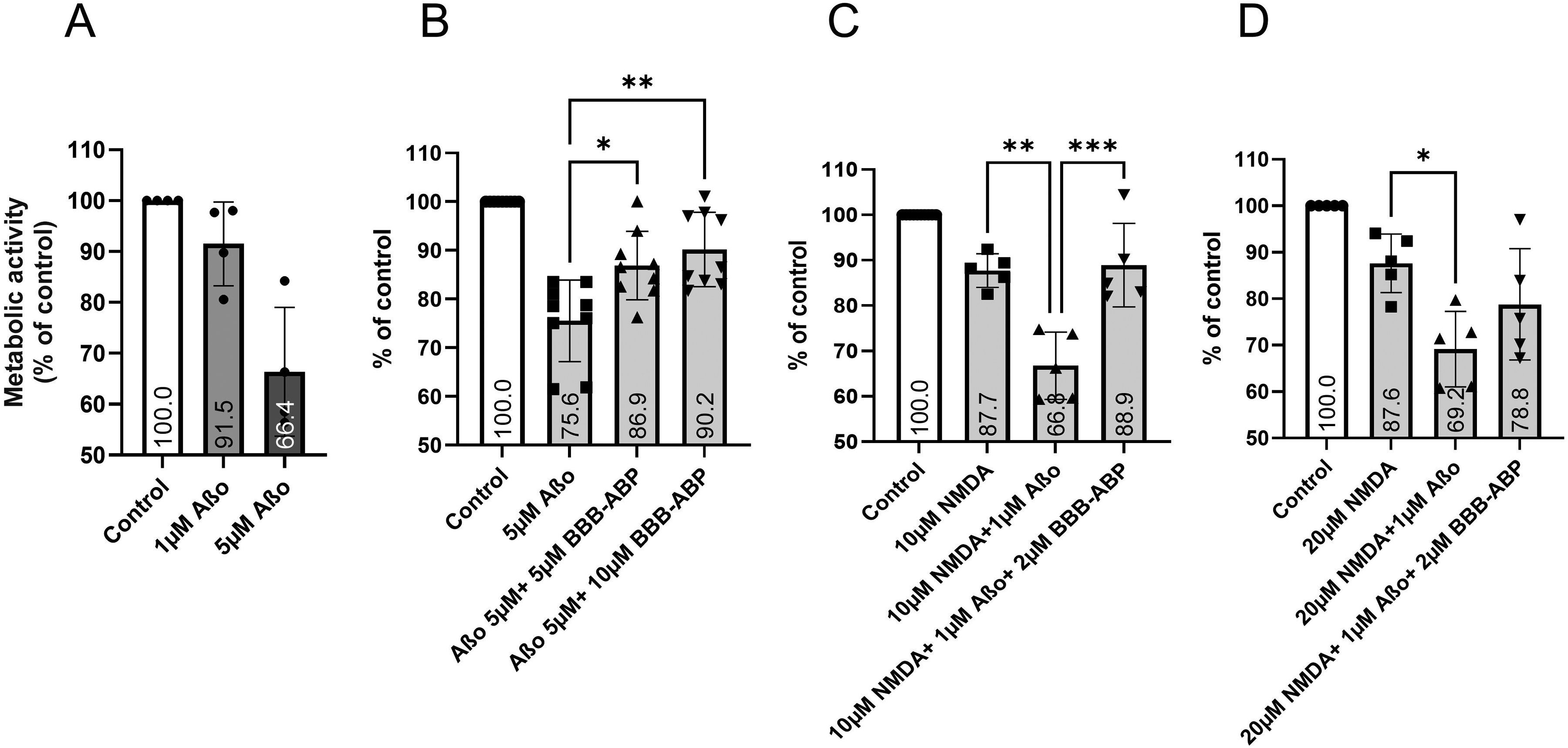
Inhibition of Aβo-induced toxicity by BBB-ABP in rat primary hippocampal neuronal cultures. (A) Primary neurons showed only minimal reduction of metabolic activity in response to 1 μM Aβo, while 5 μM produced robust reduction of metabolic activity. 5 μM Aβo was therefore used to test the ability of BBB-ABP to prevent neurotoxicity. The graph shows aggregate results of four independent experiments done in triplicate wells. (B, C) Pre-incubation of Aβo with BBB-ABP significantly reduced Aβo-induced neurotoxicity in hippocampal cultures (B) even in cells subjected to additional stress by prior exposure to 10 μM NMDA (C). The graph in (B) shows aggregate results of 9 experiments done in quadruplicate. The protective effect of BBB-ABP was less apparent in cells treated with 20μM NMDA prior to Aβo exposure (D). The graphs in C and D show aggregate results from 5 experiments each, done in quadruplicate. *p < 0.05, **p < 0.005, ***p < 0.001.
Neuroprotective effects of BBB-ABP against Aβo toxicity in primary cortical neurons were not as pronounced (Figure 7) as seen with hippocampal neurons.
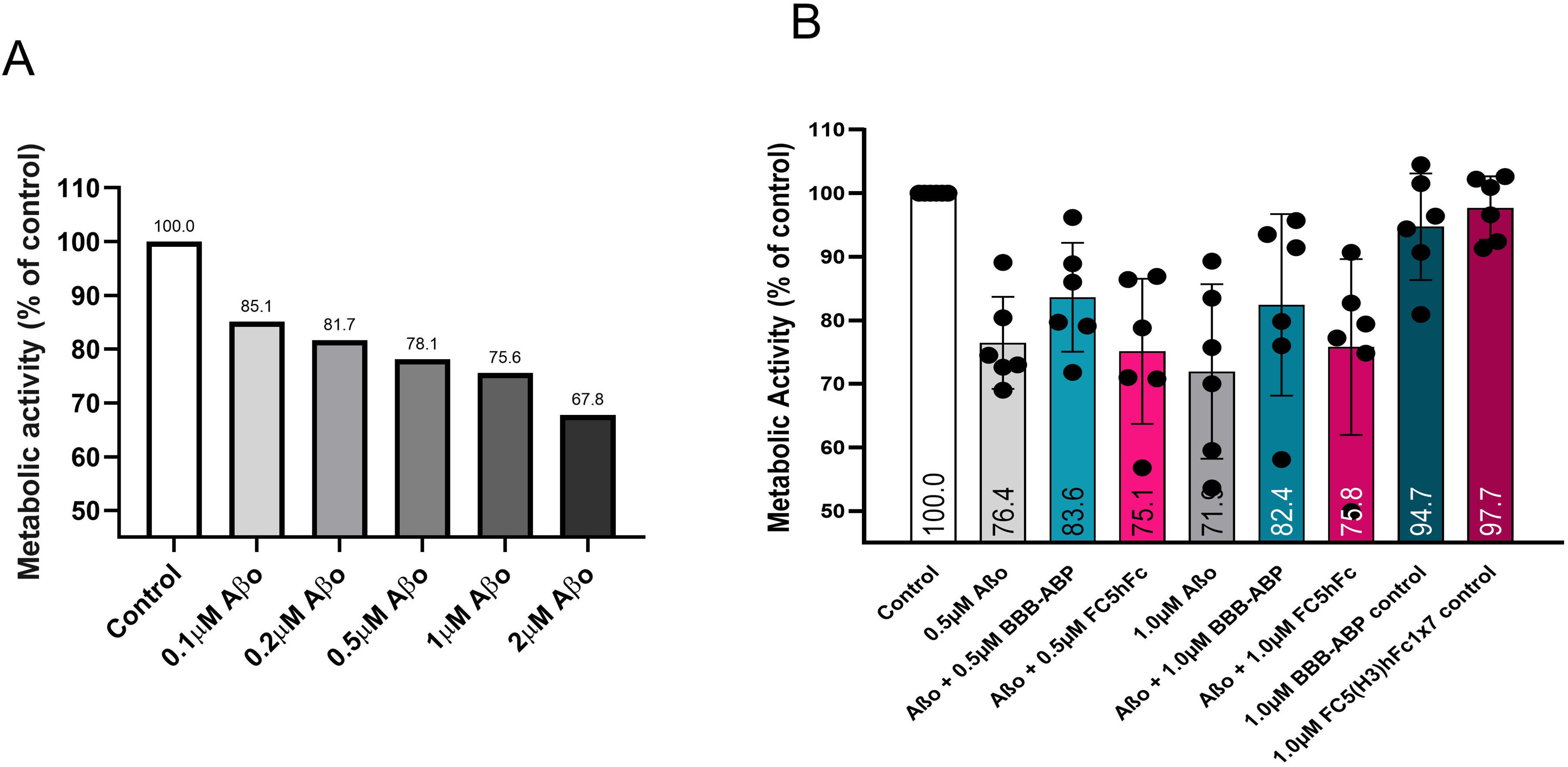
Inhibition of Aβo-induced toxicity by BBB-ABP in rat primary mixed cortical/hippocampal neuronal cultures was not as pronounced as in pure hippocampal cultures. (A) Mixed cortical/hippocampal neurons exhibited increasing reductions of metabolic activity in response to increasing doses of Aβo from 0.1 μM to 2 μM. The figure shows representative results from two independent experiments done in triplicate wells. (B) Reversal of Aβo-induced reductions in metabolic activity by BBB-ABP was not significant. The graph shows aggregate data from 6 independent experiments done in triplicate wells.
MEA experiments
The goal of this series of experiments was to first establish conditions under which Aβ altered spontaneous activity of rodent neuron cultures on MEAs and, secondly, to determine whether BBB-ABP reversed the effect of Aβ. To do this, the effect of long-term exposure to various forms of Aβ on spontaneous activity of neurons grown on MEAs was examined.
Mixed cortical/hippocampal neurons cultured on 48-well Cytoview MEA plates (Figure 8A shows a representative image) were subjected to a dose-response of oligomeric Aβ1−42 (1, 2.5, 5, 10, and 20 µM) over 48 h. No significant differences in frequency were detected in response to exposure to any concentration of Aβ1−42, using a mixed-effects analysis (Figure 9A).

Representative images of (A) mixed cortical/hippocampal; (B) hippocampal and; (C) mixed cortical/hippocampal 3D neurospheres. Shown are neuronal dispersions plated on 48-well Axion CytoView MEA plates (not all of the 16-electrode field is shown in each image). Images were acquired at 14, 11, and 14 DIV, for cortical/hippocampal, hippocampal neurons and 3D neurospheres respectively. At these stages of development, the 2D neuronal cultures were sub-confluent, and show numerous projections. The 3D neurospheres show some single cell off-shooting, as well as some aggregation of neurospheres themselves.
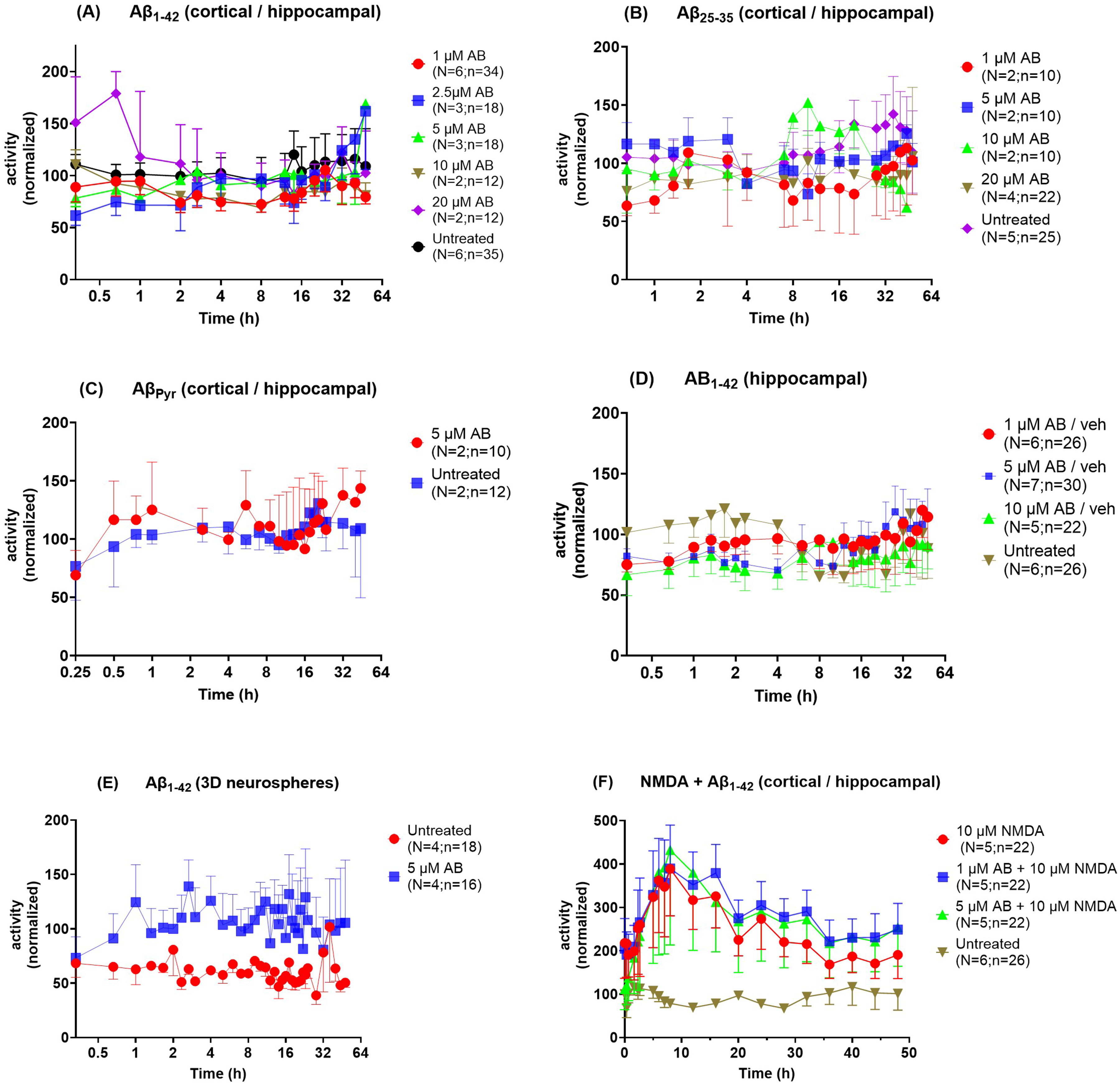
Lack of effect of Aβ on spontaneous activity in neurons cultured on MEAs. Spontaneous activity was monitored with 5-min recording sessions before and following exposure of various neuronal dispersions to different preparations of Aβ for 44–48 h. Data were processed as described in the methods section and analyzed using mixed-effects analysis. No significant differences in frequency were detected in response to exposure to any concentration of Aβo (A), Aβ25−35 (B), or Aβpyr (C) in mixed cortical/hippocampal neurons. In A, N/n were 6/34, 3/18, 3/18, 2/12 and 2/12 for 1, 2.5, 5, 10, and 20 µM Aβo, respectively, and 6/35 for the untreated condition. In B, N/n were 2/10, 2/10, 2/10, 2/12 and 4/22 for 1 5, 10 and 20 µM, respectively, and 5/25 for the untreated condition. In C, N/n were 2/10 and 2/12 for 5 µM Aβpyr and untreated wells, respectively. No effect of Aβo on spontaneous activity was detected in hippocampal neurons (D) or in 3D cortical/hippocampal neurospheres (E). In D, N/n were 6/26, 7/30 and 5/22 for 1, 5 and 10 µM for Aβo, respectively, and 6/26 for untreated wells. In E, N/n were 4/16 for 5 µM Aβo and 4/18 for untreated wells. (F) Spontaneous activity was monitored in cortical/hippocampal neuron cultures following exposure to NMDA ± Aβo for 48 h. Significant differences in frequency were detected between vehicle and the NMDA ± Aβo conditions, however, no significant difference was found between 10 µM NMDA, 1 µM Aβo + 10 µM NMDA and 5 µM Aβo + 10 µM NMDA. N/n were 5/22, 5/22, 5/22 for 10 µM NMDA, 1 µM Aβo + 10 µM NMDA and 5 µM Aβo + 10 µM NMDA, respectively, 6/26 for untreated wells.
Given the lack of effect of Aβ1−42 on spontaneous activity, we expanded this study to evaluate different forms of Aβ in different neuronal dispersions. Spontaneous activity was monitored before and following exposure to Aβ25−35 for 48 h, using the same procedures as followed for evaluation of Aβ1−42, and analyzed using mixed-effects analysis. No significant differences in frequency were detected in response to exposure to any concentration (1, 2.5, 5, 10 and 20 µM) of Aβ25−35 in cortical/hippocampal neuron cultures (Figure 9B). No significant differences in frequency were detected in cortical/hippocampal neurons in response to exposure to AβPyr (5 µM) over 48 h (Figure 9C). An early target of Aβ is believed to be hippocampal neurons, however, no significant differences in frequency were detected in hippocampal neurons alone in response to exposure to any concentration of oligomeric Aβ1−42 (1, 5, and 10 µM) (Figure 9D; Figure 8B shows a representative image of hippocampal neurons). We also considered the possibility that Aβ1−42 might have an effect on 3-dimensional cortical/hippocampal neuron cultures (Figure 8C shows a representative image of 3D neurospheres); however, no significant differences in frequency were detected in neurospheres in response to exposure to oligomeric Aβ1−42 (5 µM) (Figure 9E).
Finally, given the lack of effects of various forms of Aβ on various neuronal dispersions, we determined whether stressing neurons produced a discernable effect by Aβo. The rationale for this approach is that several groups have shown no17–19 or minor 20 neurotoxicity of exposure to Aβ25−35 or Aβ1−42 alone, but exacerbation of neurotoxicity induced by glutamate. Yanker et al. 20 reported that Aβ25−35 exacerbated neurotoxicity caused by exposure to glutamate, and similarly, Aβ exacerbated glucose-free toxicity in cortical neuron cultures. 21 Aβ1−40 (1 µM) daily addition to cortical neuron cultures for one week exacerbated a 10 µM glutamate-induced efflux of K+ and Ca2+ influx (although the implications of these findings are uncertain, since neurotoxicity induced by glutamate was not exacerbated by this Aβ1−40 pre-treatment regime). 22
Spontaneous activity was monitored in cortical/hippocampal neuron cultures following exposure to 10 µM NMDA ± 1 or 5 µM Aβo for 48 h. This concentration of NMDA was chosen to intensify neuronal activity, which was not neurotoxic as judged by a live:dead assay (data not shown). NMDA alone induced a ∼4-fold significant increase in activity by 8 h, followed by a slow decline that was nonetheless still supra-physiologically elevated by 48 h. Inclusion of 1 or 5 µM oligomeric Aβ1−42 with NMDA did not significantly alter the profile (Figure 9F). This indicates that Aβo does not increase neuronal activity beyond that induced by NMDA alone.
The lack of any effect of Aβo on spontaneous activity prevented an assessment of the effect of BBB-ABP on Aβo-induced changes in activity.
Discussion
The goal of this study was to assess the functionality of BBB-functionalized ABP (BBB-ABP) and evaluate its ability to reduce Aβo-induced toxicity and changes in spontaneous synaptic activity in cultured neurons. Both ELISA and western blot overlay assays demonstrated that fusion proteins containing ABP, Fc, and the BBB transporter FC5 retained preferential in vitro binding to oligomeric amyloid relative to preparations of amyloid containing predominantly monomeric or fibrillar species. This was true for both the mouse Fc-containing construct (FC5-mFc2a-ABP) and the humanized construct with modified ABP [FC5(H3)-hFc-ABP].
Incubation of BBB-ABP with Aβo sequestered Aβo and prevented its interaction with proteins in whole cell lysates, and when applied to live neurons. While we did not determine the identity of cellular proteins bound by Aβo, many potential neuronal surface receptors for Aβo have been identified, including Prpc, α7 nicotinic acetylcholine receptor, p75 neurotrophin receptor, ephrin receptors, Nogo receptor 1 and others. 23 Indeed, multiple bands were apparent in the Aβo-overlay western blot, suggesting multiple binding partners for Aβo under these conditions. BBB-ABP reduced Aβo binding across the entire blot, suggesting BBB-ABP was capable of preventing interaction of Aβo with many partners.
Prevention of Aβo binding to live neurons was also observed using immunofluorescence (immunocytochemistry). As expected,24,25 Aβo bound extensively to the neurites of primary hippocampal neurons, and binding was nearly completely prevented by preincubation of Aβo with BBB-ABP. FC5-hFc did not prevent Aβo binding, confirming the need for the ABP portion of the molecule. We considered the possibility that BBB-ABP might simply interfere with detection of bound Aβo by anti-amyloid antibodies, thus resulting in apparent blocking of Aβ binding to neurons. We therefore repeated the live cell binding experiment using fluorophore-tagged Aβo and observed the same results, corroborating BBB-ABP's ability to prevent Aβo binding to neuronal cells. Staining for the actin-binding protein drebrin, which is enriched in dendritic spines, 26 confirmed that spines were targeted by Aβo. Intriguingly, binding of Aβo to individual spines/synapses has been shown to act locally to induce downstream effects on spine structure and plasticity. For example, in primary hippocampal neurons exposed to Aβo, changes in F-actin content were observed specifically in dendritic spines that stained positive for Aβo, while Aβo-negative spines on the same neuron did not show changes in F-actin content. 27 Similarly, Aβo exposure has been shown to act at the level of individual spines to impair NMDA function 28 and to induce synaptic plasticity impairment specifically at Aβo-bound synapses, but not at nearby Aβo-free synapses. 29 The ability of BBB-ABP to inhibit Aβo binding to dendritic spines suggests that once in the brain, BBB-ABP should be able to sequester Aβo, preventing the initiation of deleterious downstream effects induced by the interaction of Aβo with spines. This may explain the preservation of neuronal connectivity previously observed in rats treated with FC5-mFc2a-ABP. 10
Consistent with the ability of BBB-ABP to prevent Aβo from binding to neural cells (SH-SY5Y) and primary neurons, BBB-ABP reduced the toxicity of Aβo in these cells as assessed by the MTT assay. While SH-SY5Y cells were more sensitive to Aβo, exhibiting a 35% reduction of metabolic activity in response to 1 μM Aβo, primary neurons were more resistant, requiring a higher dose of Aβo (5 μM) to achieve significant reduction of metabolic activity (25% reduction). To render cells more vulnerable to Aβo, primary neurons were pre-exposed to a low dose of NMDA for 30 min prior to Aβo exposure. 10 μM NMDA exposure induced some toxicity on its own (<15%) but adding Aβo induced significantly more stress to NMDA-treated cells than NMDA alone. BBB-ABP prevented the exacerbation of NMDA-induced toxicity by Aβo. As expected, while BBB-ABP prevented the response to Aβo in NMDA-treated neurons, it did not fully restore metabolic activity to control levels; that is, it did not prevent the part of the toxicity that was induced by NMDA alone. Although a similar trend was seen for the 20μM dose of NMDA, BBB-ABP was less effective in preventing the Aβo-induced exacerbation of NMDA effect at this dose, even though the effects of NMDA and NMDA + Aβo were similar at both concentrations of NMDA. Although the reasons are not clear, it is possible that the cells are more vulnerable to any Aβo not sequestered by BBB-ABP under these conditions. Also, it is not clear why the neuroprotective effect of BBB-ABP against Aβo toxicity was not as robust in mixed cortical neuronal cultures.
The ability of BBB-ABP to prevent Aβo-induced reduction of metabolic activity is similar to the effects of anti-amyloid antibodies crenezumab, mAb158 (murine lecanemab) and PMN310 in reducing amyloid-induced toxicity in PC12 cells, 30 neuro2A cells 31 and primary neurons.32,33 We had previously shown that ABP (free from FC5-Fc) prevents insulin resistance induced by endogenous Aβ produced by murine neuro2a cells overexpressing human AβPP gene, most likely by sequestering Aβ secreted into the medium. 34 These results collectively suggest that, when delivered to the brain BBB-ABP can not only engage the toxic Aβ oligomers and potentially facilitate their clearance, but also can prevent their binding to cellular proteins by sequestration and thereby reduce down-stream toxic effects.
Our BBB-enabled peptide approach has a number of potential advantages over monoclonal antibody-based therapeutics. The small size of the peptide, even after incorporating the BBB carrier and Fc (total <90 kDa compared to ∼150 kDa for monoclonal antibodies), may further facilitate its delivery across the BBB, and its movement within the interstitial spaces of the brain parenchyma. Furthermore, ABP is derived from a naturally occurring protein, and is therefore expected to have very low immunogenicity. Finally, selectivity of BBB-ABP for oligomeric Aβ and the very low affinity of BBB-ABP for fibrillar forms of Aβ may reduce the likelihood of adverse effects associated with binding to fibrillar Aβ (as observed with monoclonal antibodies aducanumab, 35 lecanemab, 36 gantenerumab,37,38 and donanemab39,40). BBB-ABP is also differentiated from the above monoclonal antibodies because it takes advantage of a well-defined receptor mediated transcytosis pathway for brain delivery; while in contrast the mechanism by which monoclonal antibodies penetrate the BBB and enter the brain is not clearly understood.
Lack of effect of Aβ on spontaneous activity
Based on Aβo binding to dendritic spines, we anticipated some effect of Aβo on synaptic function assessed using MEA. However, despite using various Aβ preparations at a range of concentrations in different types of neuronal cultures, we did not detect Aβo-induced changes in spontaneous synaptic activity. Since no effect of Aβ was observed, we could not determine whether BBB-ABP would reverse whatever effect Aβ had on synaptic activity. Electrophysiological responses that are quite divergent in nature and scope have been elicited by Aβ in neurons (reviewed in 41 ). A very recent review by O’Connell 42 reinforces the notion that considerable variability exists between studies on the effect of Aβ preparations on electrophysiological properties of neurons grown on MEAs. 42 For example, for neurons grown on MEAs, Aβ oligomers can cause an initial increase in spontaneous activity (although studies diverge regarding activity levels after this surge)43–47; or a decline in activity.48–55
Our results correlate most closely with one study reporting that chronic treatment of cortical neuron cultures with Aβ42 oligomers (10 µM) caused a slight increase in spiking but not bursting frequency. 56 Thus, our finding of no effect of various forms of Aβ is generally at odds with most studies. We extended our testing to higher concentrations and different forms of Aβ in different types of neuronal dispersions, just to ensure that any effect was not missed. MEAs represent a very sensitive non-invasive indicator of synaptic activity. Nonetheless, despite this comprehensive analysis, we are left to conclude that Aβ alone does not alter synaptic activity over a 48 h time-frame in neuronal cultures.
Our results are generally consistent with numerous studies reporting Aβ-induced neurotoxicity, assessed using the MTT assay and other methods (Supplemental Table 1). Moreover, given our unique approach of employing MTT and MEA-based assays, our results suggest that MTT is a more sensitive indicator of the effect of Aβ than synaptic activity. Iversen et al. have reported that MTT can detect an effect of Aβ at concentrations that are 3–4 orders of magnitude lower than those required to kill cells. 57 They suggest that this exceptional sensitivity may be attributed to MTT reduction being a very specific indicator of the mechanism of Aβ interference in neurons. In any event, our results suggest that Aβ-induced MTT reductions can be withstood without any effect on spontaneous synaptic activity, even over a prolonged period of time.
In summary, this study demonstrates that BBB-functionalized ABP effectively binds and sequesters Aβo, thereby preventing its interaction with neuronal proteins and reducing its associated toxicity in cultured neurons. These findings collectively highlight the potential of BBB-ABP to mitigate the early, synapse-targeting mechanisms of Aβo toxicity and support the continued development of BBB-ABP as a therapeutic candidate capable of engaging toxic Aβ species and preserving neuronal integrity in the context of AD pathology.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261453068 - Supplemental material for The blood-brain barrier-penetrating fusion protein BBB-ABP selectively binds amyloid-β oligomers and prevents in vitro neurotoxicity
Supplemental material, sj-docx-1-alz-10.1177_13872877261453068 for The blood-brain barrier-penetrating fusion protein BBB-ABP selectively binds amyloid-β oligomers and prevents in vitro neurotoxicity by Kerry Rennie, Joseph S. Tauskela, Michel Ménard, Amy Aylsworth, Rosa Comas, James Whitfield and Balu Chakravarthy in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
The authors would like to thank the members of the protein production and purification teams and quality attributes team at the NRC as well as members of the Animal Resources group.
Ethical considerations
All procedures were approved by NRC's Animal Care Committee in accordance with guidelines set out by the Canadian Council on Animal Care.
Consent to participate
Not applicable.
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data used in this study is available upon request from the authors.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.