
Abstract
Phylogenetics uses alignments of molecular sequence data to learn about evolutionary trees relating species. Along branches, sequence evolution is modelled using a continuous-time Markov process characterized by an instantaneous rate matrix. Early models assumed the same rate matrix governed substitutions at all sites of the alignment, ignoring variation in evolutionary pressures. Substantial improvements in phylogenetic inference and model fit were achieved by augmenting these models with multiplicative random effects that describe the result of variation in selective constraints and allow sites to evolve at different rates which linearly scale a baseline rate matrix. Motivated by this pioneering work, we consider an extension using a quadratic, rather than linear, transformation. The resulting models allow for variation in the selective coefficients of different types of point mutation at a site in addition to variation in selective constraints.
We derive properties of the extended models. For certain non-stationary processes, the extension gives a model that allows variation in sequence composition, both across sites and taxa. We adopt a Bayesian approach, describe an MCMC algorithm for posterior inference and provide software. Our quadratic models are applied to alignments spanning the tree of life and compared with site-homogeneous and linear models.
Keywords
Introduction
In statistical phylogenetics, the goal is to learn about the evolutionary relationships amongst a collection of species, generally using DNA or protein sequence data. These relationships are represented through a rooted, bifurcating tree called a phylogeny. Substitutions in the molecular sequence alignment are typically modelled using continuous-time Markov processes, parameterized through an instantaneous rate matrix. Early phylogenetic models were simplistic, generally assuming that the evolutionary process was in its stationary distribution and that substitutions at each site of the alignment could be described by the same underlying rate matrix. Under these models, the probability of change from one character state to another was therefore independent of both organismal lineage and the biochemical function of the nucleotide or amino acid in question. These simplifying assumptions were known to be false, but were made for the sake of mathematical convenience and computational tractability, given the limited computing power for model fitting available at the time. In particular, it was already clear to early molecular evolutionists that rates of evolution vary according to functional or structural pressures acting at a site: Important sites are subject to high selective constraints and evolve slowly because most mutations that arise at those sites are eliminated from the population by negative selection (Fitch and Markowitz, 1970). Uzzell and Zorbin (1971) showed that the numbers of substitutions occurring at different sites could be modelled using a negative binomial distribution. Later, Yang (1993) incorporated the idea into statistical phylogenetics by allowing different sites to evolve at different rates. These rate parameters scaled the underlying Markov process rate matrix and were modelled as multiplicative random effects, with unit mean gamma distribution.
Incorporation of across-site rate variation into standard, stationary substitution models has led to major improvements in model fit and to the accuracy of phylogenetic inference (Yang, 1996). But there are other, pervasive features of molecular sequence data that these models do not accommodate. In particular, nucleotide composition is believed to vary across both sites of the alignment and branches of the phylogenetic tree. For example, the GC-content of ribosomal DNA genes varies from 45% to 74% across the known diversity of cellular life (Cox et al., 2008), implying that the probabilities of each of the four nucleotides can change over time. These compositional shifts might reflect changing biases in DNA repair enzymes (Sueoka, 1988) or, at least for genes encoding structural RNAs, adaptation to different growth temperatures (Galtier and Lobry, 1997). As well as variation in sequence composition across taxa, there is also compositional variation observed among the different sites within an individual protein-coding sequence: Due to functional constraints, most sites can tolerate only a limited, and typically biochemically homogeneous, subset of the 20 amino acids (Fitch and Markowitz, 1970). The result is that, in addition to varying in evolutionary rate, sites can also differ in sequence composition. As with heterogeneity in evolutionary rates, failure to account for variation in composition can lead to model misspecification and, therefore, serious phylogenetic error, as demonstrated by a number of empirical studies Embley et al., 1993; Foster, 2004; Lartillot et al., 2007; Philippe et al., 2011). The phylogenetic literature includes a number of models designed to capture one type of compositional heterogeneity, or the other, that is, either heterogeneity across sites or heterogeneity across branches. In the former case, this is often achieved using mixture models which classify sites into groups, each of which has a different stationary distribution; see, for example, Pagel et al. (2004) or Lartillot and Philippe (2004). To allow heterogeneity across branches, a number of models have been developed which drive the Markov process towards a different stationary distribution at different points on the tree, typically by allowing evolution on different branches to be governed by different instantaneous rate matrices; see, for example, Yang and Roberts (1995), Blanquart and Lartillot (2006), Dutheil and Boussau (2008) or Heaps et al. (2014). Despite the large body of literature focused on modelling compositional heterogeneity of one type or the other, there have been very few attempts to model both jointly. Such efforts are typically based on mechanistic models which allow different rate matrices to govern the evolutionary process on different (site, branch) pairs; see, for example Blanquart and Lartillot (2008) or Jayaswal et al. (2014). Unfortunately, use of these models has been limited due to computational difficulties with model-fitting.
In a simple phylogenetic model, evolution at all sites is controlled by a single instantaneous rate matrix. The across-site rate variation model offers greater flexibility by allowing site-specific linear transformations of the baseline rate matrix, with variation amongst scaling factors dependent on a single-parameter gamma distribution. Owing to the success of this simple modification, the across-site rate variation model has been extended in a number of ways. For example, covarion models (Tuffley and Steel, 1998; Huelsenbeck, 2002; Galtier, 2001) allow the site-specific (linear) scaling factors to vary from branch to branch. This is intended to capture the variation over time in selective constraints that arise as a consequence of earlier substitutions at other sites. In this article, we consider a different generalization of the across-site rate variation model, applying site-specific quadratic, rather than linear, transformations of a baseline matrix. This gives a more flexible model which is dependent on an additional unknown parameter. It has the effect of allowing variation in the selective coefficients—that is, the strength of selection—for different types of point mutation at a site, in addition to heterogeneity in the overall selective constraints across sites. We thereby obtain a more biologically plausible model. Further, we demonstrate that when linear or quadratic across-site transformations are combined with a class of non-stationary Markov processes, we obtain computationally tractable models that allow sequence composition to vary across both branches of the tree and sites of the alignment, addressing the clear need in the literature for models of this type.
The remainder of this article is organized as follows. Section 2 introduces phylogenetic models of sequence evolution and the incorporation of multiplicative random effects to allow rate variation across sites. Section 3 describes our quadratic generalization and its properties. In Section 4, we combine across-site linear and quadratic transformations with a general class of non-stationary substitution models and describe the properties of the resulting Markov processes. Section 5 addresses the issue of inference for models incorporating our quadratic transformation. Specifically, working in a Bayesian framework, we describe the posterior distribution of interest and details of our numerical approach to model-fitting via Markov chain Monte Carlo sampling. In Section 6, we consider analyses of two biological datasets; the first involving a stationary model and the second, a non-stationary model. In each case, we compare the performance of a site-homogeneous model with analogous models incorporating linear and quadratic across-site transformations of the baseline rate matrix. Finally, we summarize our conclusions in Section 7.
Phylogenetic models of sequence evolution
Denote by
Standard phylogenetic models assume that the underlying continuous-time Markov process is time reversible and in its stationary distribution
Classically, the sites of the alignment

In order to prevent arbitrary rescaling of the rate matrix
Modelling rate heterogeneity across sites
It has long been recognized that selective pressures vary across sites due to their differing roles in the structure and function of the molecular sequence (Yang, 1996; Simon et al., 1996). This feature is typically captured by a simple modelling device that allows each site j to evolve at its own rate

the likelihood can then be represented as
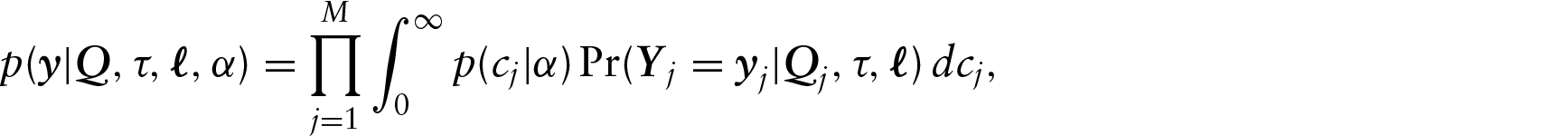
where
In this model, the rate matrix at each site is simply a linearly scaled version of some underlying normalized baseline
Consider a normalized baseline rate matrix

where
Property (ii) is automatically satisfied for any

Therefore, the sum of the elements on row

for any
For property (i) to be satisfied, we need
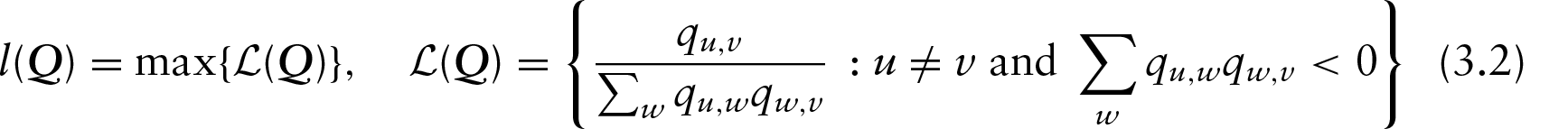
and

By definition,

However,

Because

In contrast, the set
To allow information to be shared between sites, we continue to assume that the coefficients

where

It can easily be shown that the stationary distribution of
If we take the distribution at the root of the tree to be the vector

Here
A biological interpretation for LASH and QuASH models can be obtained by considering how the substitution process in each case might result from the combination of a process of point mutation and a process of selection, where point mutations become fixed in a population.
The fixation rates of point mutations vary across sites according to differences in their structural or functional importance. As a consequence, sites under high selective constraints typically admit fewer substitutions of any type. At a more granular level, different types of mutations at any particular site, that is, mutations between different pairs of nucleotides, may have different selective coefficients. These measure the relative fitness of a particular allele (point mutation), with larger numbers indicating stronger selection for (or against) the allele and hence higher selective pressure. Interpretations of the Markov process arising from the LASH and QuASH transformations are best explained through their representation as jump processes. To this end, consider a baseline, stationary substitution process with rate matrix
Under the QuASH transformation,

for some

for other
Random effect distribution
We model the coefficients
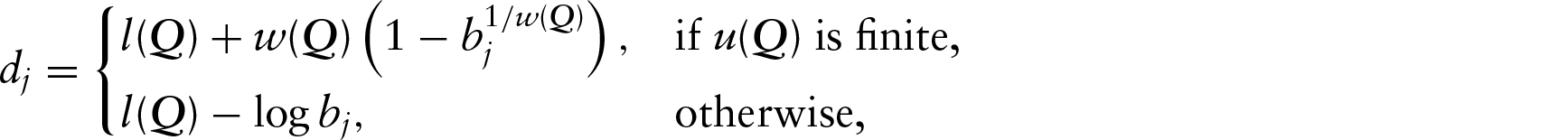
where
The hyperparameter
Conditional mean and standard deviation of
given
, and bounds
and
, plotted with a log-scale on the
-axis

Conditional mean and standard deviation of
given
, and bounds
and
, plotted with a log-scale on the
-axis
The transformations characterizing LASH and QuASH models allow across-site variation in the overall magnitude of the instantaneous rates of change and, for QuASH models, their relative sizes. However, the models discussed so far have been homogeneous across branches, with a single baseline rate matrix
Non-stationary models for sequence evolution can account for differences in composition across taxa by allowing the probability of being in each state (e.g., each nucleotide for DNA data) to change over time. Typically, this is achieved by permitting step changes in the theoretical stationary distribution at different points on the tree. Although these changes do not have to occur at speciation events (e.g., Blanquart and Lartillot, 2006), this assumption is often made (e.g., Yang and Roberts, 1995; Foster, 2004; Heaps et al., 2014; Cherlin, 2016) and we retain it here for simplicity of notation. In general, therefore, consider a rooted topology τ with
Extending the LASH and QuASH transformations to non-stationary models of this form, the rate matrix for site j on branch

where
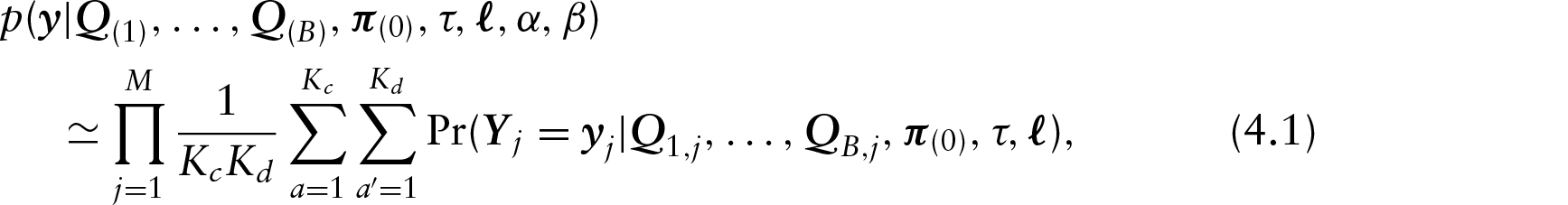
where
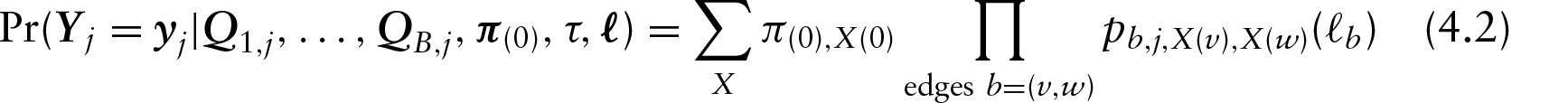
in which
By definition, non-stationary models can allow heterogeneities in sequence composition across taxa. Consider, for example, a simple non-stationary model which allows a single-step change in the stationary distribution at the root of the tree (e.g., Klopfstein et al., 2015; Cherlin, 2016, Chapter 4). In a site-homogeneous version of this model, a single rate matrix
Although more subtle, LASH and QuASH extensions of these non-stationary models additionally allow heterogeneity between sites in the across-taxa variation. For instance, consider the LASH or QuASH extension of the simple non-stationary model described above, regarding
In the application in Section 6.2, we focus on the HB model (Heaps et al., 2014) where each branch of the tree has its own reversible rate matrix
Let
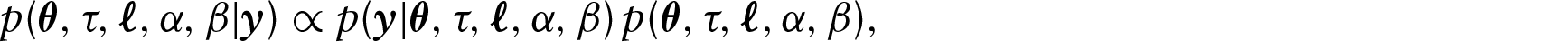
where the likelihood function
Irrespective of the choice of prior distribution
Applications
A controversial issue in evolutionary biology is the deep structure of the tree of life, including the relationships among Bacteria, Archaea and eukaryotes, the three main cellular domains. The balance of evidence favours endosymbiotic hypotheses for the origin of eukaryotes, involving symbiosis between a bacterial endosymbiont (the mitochondrion) and some kind of host cell (Martin et al., 2015). Woese et al. (1990) proposed that this host cell was part of an independently branching third domain of life, distinct from Archaea and Bacteria. This is often referred to as the three domains hypothesis. On the basis of analyses involving previously unsequenced taxa and more sophisticated evolutionary models (Williams et al., 2013), an alternative view—the eocyte hypothesis—has gained considerable support over recent years. According to this conjecture, the host for the mitochondrial endosymbiont was a fully fledged Archaeon. In addition to uncertainty surrounding the unrooted topology of the tree of life, opinion is also divided on the position of its root. Under the two leading hypotheses, it is either placed on the bacterial branch (Gogarten et al., 1989; Iwabe et al., 1989) or, with fewer proponents, within the Bacteria (Cavalier-Smith, 2006; Lake et al., 2009).
In this section, we consider applications to biological datasets that address these controversial questions. In Section 6.1, we analyse a concatenated alignment of small and large subunit ribosomal RNAs (SSU and LSU rRNAs) sampled from across the tree of life. After alignment using MUSCLE (Edgar, 2004) and editing to remove poorly aligning regions,
In all analyses, mixing and convergence of the MCMC sampler was assessed by comparing the output from multiple chains, initialized at different starting points. In phylogenetics, mixing in tree-space can be problematic due to the low acceptance rates of topological moves. Therefore, in addition to considering the usual numerical and graphical diagnostic checks for continuous parameters, we also examined graphs based on relative cumulative split (Section 6.1) or clade (Section 6.2) frequencies of the chains over the course of the MCMC runs; see (Heaps et al., 2014) for full details of these diagnostics. Here a split refers to a bipartition of the taxa at the leaves of the tree into two disjoint sets, induced by cutting a branch. On a rooted tree, one of the partition subsets of any split is a clade if all the taxa lie on the same side of the root. In biological terms, this corresponds to an ancestor and all its descendants.
Stationary TN93 model
Based on our subjective assessments of the evolutionary process, for the parameters of the S1 model we chose independent gamma
We refer to the output of each complete sweep through the Gibbs steps of our Metropolis within Gibbs samplers as a single draw from the posterior. For each model, the MCMC algorithm outlined in Section 5 was used to generate at least
Top row: Marginal prior and posterior densities for the unknown parameter β in the random effect distribution for the quadratic coefficients dj under the (a) stationary model S3 and (b) non-stationary model NS3. Bottom row: Prior and posterior predictive distributions for dj at an additional site j under the (c) stationary model S3 and (d) non-stationary model NS3
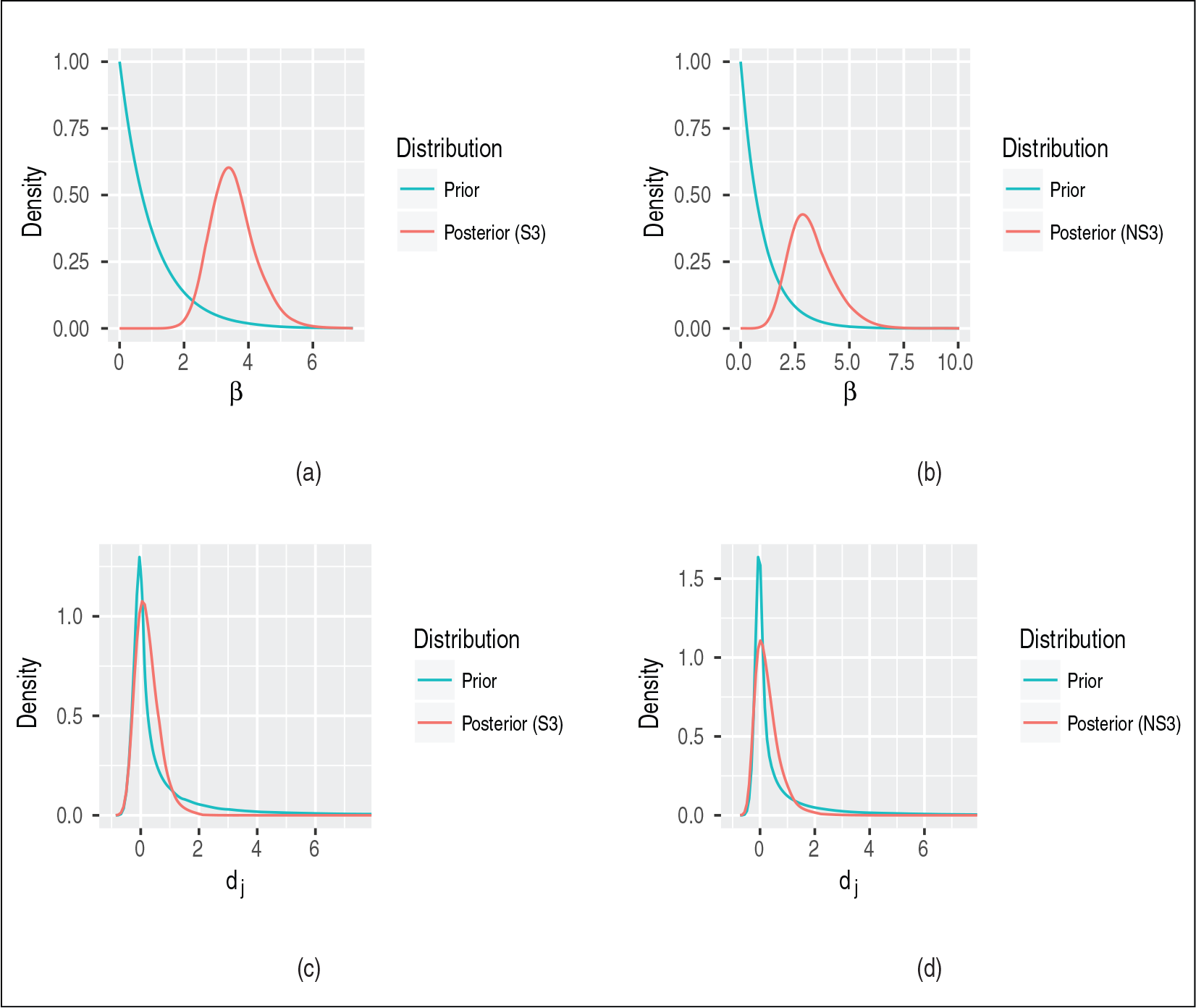
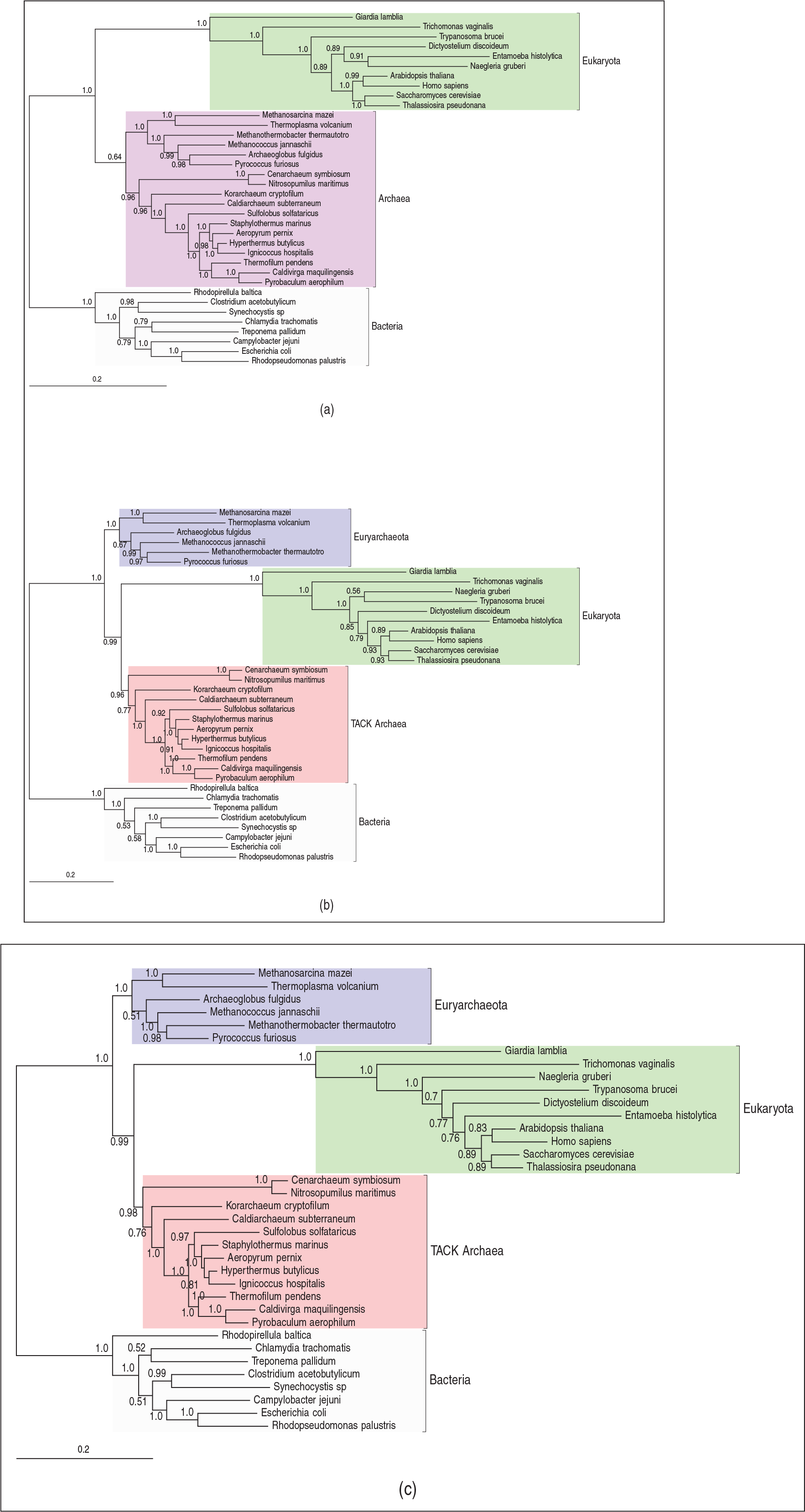
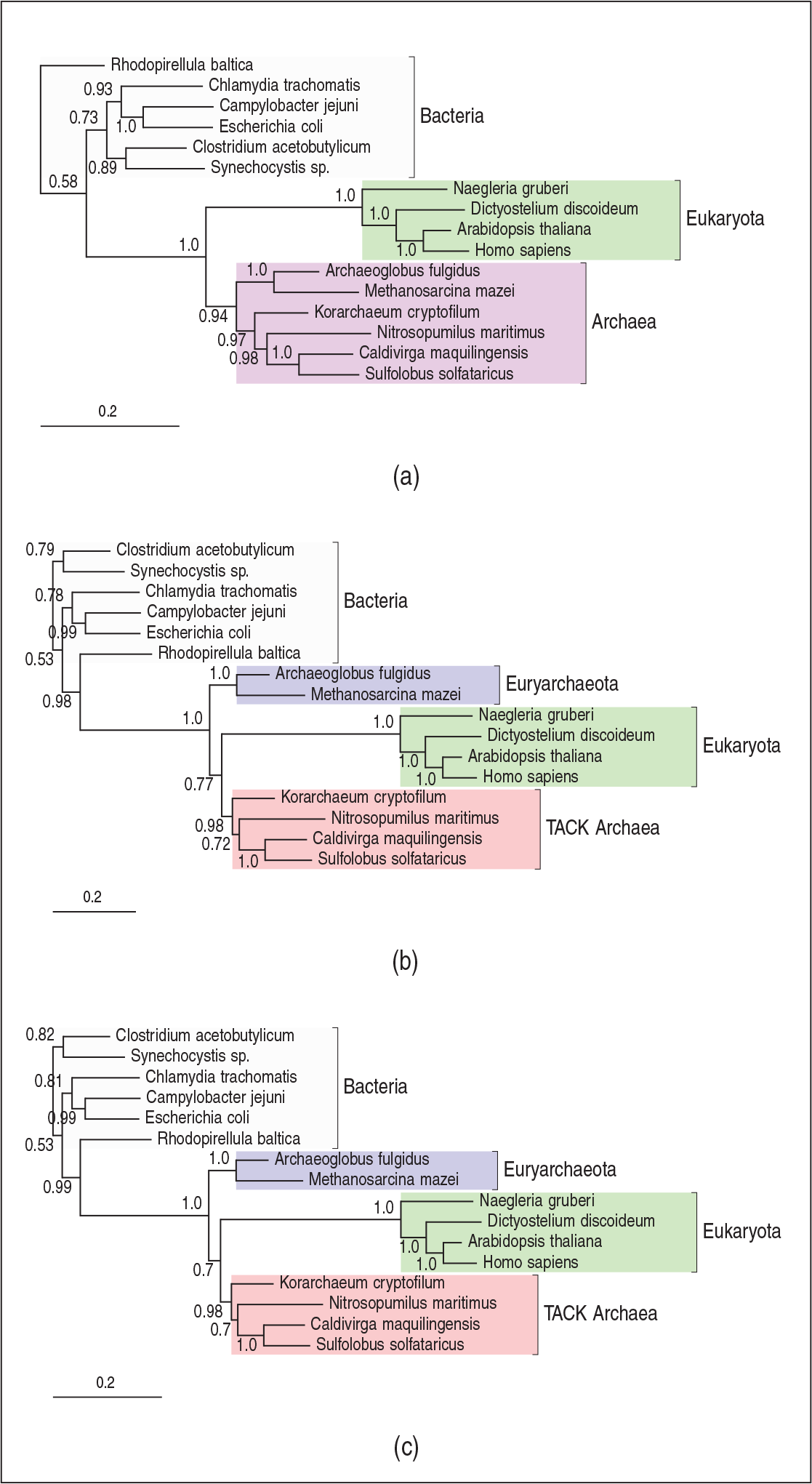
In phylogenetic inference, the majority-rule consensus tree is the most widely used summary of the posterior distribution over tree space. As a summary of a sample of trees, it includes only those splits which appear in over half of the samples (Bryant, 2003), here representing those with posterior probability greater than 0.5. For the analyses under models S1–S3, the consensus trees are shown in Figure 2 in which numerical labels represent the posterior probability of the associated split. To aid comparison, the trees are all visualized with the root on the bacterial branch. The consensus tree under S1 supports the three domains hypothesis, while models S2 and S3 yield eocyte trees, with eukaryotes emerging from within two archaeal clades: The Euryarchaeota and the TACK Archaea. As expected, there is a marked difference in our phylogenetic inferences as we move from the simple TN93 model (S1) to one which incorporates across-site rate heterogeneity. However, there is very little difference in the inferences obtained when extending the LASH model (S2) to the corresponding QuASH model (S3). Comparing the prior and posterior density for

In order to compare the fit of models S1, S2 and S3, we use the framework of posterior predictive checks (Gelman et al., 2013) in which the basic idea is to measure the extent to which a model captures some data summary of interest—a so-called test statistic—by comparing its posterior predictive distribution to the value that was observed. Typically, the posterior predictive distribution is approximated numerically based on an MCMC sample from the posterior of the unknowns in the model by simulating replicated datasets in one-to-one correspondence with the posterior draws. If the model is able to capture adequately the aspect of the data summarized through the test statistic, the observed value should look plausible under its posterior predictive distribution.
As explained in Section 1, functional and structural constraints acting on a particular site can cause it to evolve very slowly. In such cases, we are likely to see little or no variation in the character state at that column of the alignment. Therefore, in fitting to the alignment-wide empirical compositions, models that do not allow variation in, at least, the rate of the evolutionary process across sites tend to overestimate the mean number of distinct nucleotides per column, and underestimate the associated standard deviation. Figure 4a shows the posterior predictive distribution for these test statistics obtained under models S1, S2 and S3, together with the observed values calculated from the alignment. As expected, model S1 markedly overestimates the number of distinct nucleotides per site and underestimates the associated standard deviation. While models S2 and S3 also overestimate the mean, the discrepancies are much less marked, with the QuASH-variant of the TN93 model (S3) being most compatible with the observed data. Interestingly, models S2 and S3 overestimate the standard deviation of the number of distinct nucleotides per site. It is possible that models allowing sequence composition to vary across sites would be required to adequately capture this feature.
For the analyses using the non-stationary models NS1, NS2 and NS3, we adopted the prior distributions outlined in Section 6.1 for the two transition rates
For each model, the MCMC algorithm was used to generate at least
The rooted majority-rule consensus trees for each model are shown in Figure 5. Our conclusions are consistent with those from Section 6.1. Specifically, the model NS1 supports a three-domains tree, while models NS2 and NS3 support very similar eocyte trees with, in this case, the same rooted topology. Although the site-homogeneous HB model (NS1) and the LASH and QuASH variants (NS2 and NS3) support different conclusions about the unrooted topology, they both suggest a root within the Bacteria. The marginal posterior distribution for root splits under the three models is summarized in Table D.3 of our online supplementary materials. Again, the differences between inferences under NS1 and NS2 are much more marked than those between NS2 and NS3. However, in all cases the posterior probability for a root within the Bacteria is 1.0.
The LASH and QuASH variants of the HB model allow sequence composition, as well as the overall rate of evolution, to vary across sites. Therefore, we expect these models to be better equipped to capture the number of distinct nucleotides per site. Posterior predictive densities of the across-site mean and standard deviation are plotted in Figure 4b. For the mean, all three models capture the observed statistic well, with the site-homogeneous model (NS1) offering slightly more support to larger values, as expected. As in the analysis from Section 6.1, the site-homogeneous model very markedly underestimates the standard deviation. The posterior predictive densities under the LASH (NS2) and QuASH (NS3) variants of the HB model are very similar. Although both overestimate the standard deviation, the observed statistic is more plausible than under the NS1 model, and the overestimation seems less marked than the corresponding comparison from Section 6.1. The similarity in both phylogenetic and posterior predictive inferences under the LASH and QuASH models are consistent with the implications of the comparison between the prior and posterior in Figure 4. Figure 3b shows the prior and posterior densities for
Discussion
The introduction of across-site rate heterogeneity into substitution models for sequence evolution led to substantial improvements in model fit and the credibility of phylogenetic inferences. In practice, this feature was incorporated through a set of site-specific rates, modelled as random effects with unit mean gamma distribution, that linearly transformed a baseline rate matrix. Motivated by the advancement gained through this simple innovation, we considered a natural extension of the LASH model based on the incorporation of two sets of random effects, allowing a more flexible site-specific quadratic transformation of the baseline rate matrix. Biologically, this model makes fewer assumptions than the (nested) LASH model and allows for the effects of variation in the selective coefficients of different types of point mutation at a site, in addition to heterogeneity in overall selective constraints across sites. We derived properties of QuASH-transformed rate matrices, showing that they retain the stationary distribution of the underlying baseline matrix, and that the set of reversible rate matrices is closed under our quadratic transformation. In the context of a class of non-stationary models which permit step changes in the theoretical stationary distribution at one or more points on the tree, we demonstrated that both the LASH and QuASH transformations lead to models which allow sequence composition to vary across sites as well as across taxa. This is due to different rates of convergence towards the theoretical stationary distributions at different sites. The QuASH-transformed, non-stationary models therefore provide a parsimonious means of allowing heterogeneity in sequence composition across both alignment dimensions.
We utilized our model and inferential procedures in two biological applications concerning the tree of life. In the first, we compared inferences under a stationary, reversible TN93 model, with those obtained under the LASH and QuASH extensions. In the second, to make computational inference manageable, we considered a smaller dataset and compared inferences under a non-stationary HB model to those obtained under the LASH and QuASH variants. In both applications, we found that the simpler site-homogeneous models supported the three domains hypothesis, with the Archaea, Bacteria and eukaryotes appearing as monophyletic groups. Conversely, the more flexible LASH and QuASH models supported the eocyte hypothesis, with eukaryotes emerging from within a paraphyletic Archaea. The non-stationary models consistently placed the universal root within the Bacteria. The marked differences between inferences obtained under the site-homogeneous and LASH models are similar to other results reported in the literature (Yang, 1996). Both analyses suggested that only a small gain was achieved through the quadratic transformation once a linear mapping was in place. We have drawn similar conclusions from applications to several other datasets not reported here.
Although our analyses have reinforced the importance of allowing heterogeneity in the rate of evolution across sites, it appears that only a modest benefit can be found by using a natural extension which exploits a quadratic transformation of the base rate matrix. This may be because the implications of heterogeneity across sites in the selective coefficients of different types of point mutation are difficult to detect from alignments of sequence data. This might be particularly true of the ribosomal RNA sequences we analysed here, which are under strong selective constraints imposed both by the function of the molecule and by the physical interactions among sites that are separated in the primary sequence. However, in the context of non-stationary models, it is worth emphasizing that even the LASH transformation generates models that allow heterogeneity in sequence composition across sites as well as across taxa. To our knowledge, this is a property that has gone unnoticed in the literature. While a few, more mechanistic models have been proposed to offer this flexibility (e.g., Blanquart and Lartillot, 2008; Jayaswal et al., 2014), their complexity has made model-fitting computationally prohibitive. In contrast, non-stationary LASH and QuASH models provide a more parsimonious, data-driven alternative for which computational inference is substantially more straightforward. Our software implementation, described in the Appendix, provides a tool which allows practitioners to fit these models to their biological datasets.
Supplementary material
Online supplementary material for this article, including a software implementation, is available from http://www.statmod.org/smij/archive.html
Acknowledgements
Suggestions and comments from the Joint Editor, an Associate Editor and three reviewers have improved the article and are gratefully acknowledged.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: SEH, TAW, SC and TME were supported by a European Research Council Advanced Investigator Award (ERC-2010-AdG-268701) and a Programme Grant from the Wellcome Trust (number 045404) to TME.
Appendix
The analyses in this article were performed on a 2.40GHz Dell PowerEdge R410 server with two six-core Intel Xeon E5645 CPUs and 32GB RAM. When fitting the S2 and S3 models to the alignment from Section 6.1, generating 500K MCMC samples took approximately 4 and 16 days, respectively. When fitting the more complex NS2 and NS3 models to the alignment from Section 6.2, it took approximately 2.5 and 10 days, respectively, to generate 500K MCMC samples. In principle, our software could be used to analyse alignments with any number of taxa and any number of sites. However, increasing the number of sites or the number of taxa increases run times. For example, for both sets of analyses detailed earlier in this article, doubling the number of sites or the number of taxa roughly doubled the computational time. Clearly the size of the datasets that we can feasibly analyse is limited due to the computational complexity of the models considered. However, as demonstrated in Section 6 for the tree of life, by fitting more complex, biologically plausible models, even to relatively small datasets, we can challenge biological assumptions that would otherwise remain uncontested.