
Abstract
Purpose:
Iscalimab is a fully human, monoclonal anti-CD40 antibody that blocks CD154-induced CD40 signaling.
Method:
In a Phase 2 (2a) study, new-onset stage 3 type 1 diabetes mellitus (T1DM) participants were randomized 2:1 to iscalimab or placebo administered as a single intravenous dose followed by weekly subcutaneous injections for 1 year to evaluate safety and effects on β cell function.
Results:
At 14 centers in 6 countries, 44 participants (29 M/15 F, mean age 16 years [range 12–21 years]) were randomized; 39 completed the study (26 active:13 placebo). Treatment was discontinued prematurely in seven, two of these due to a temporary trial halt during the COVID-19 pandemic. No difference in C-peptide area under the curve (C-peptide(AUC)) during a mixed meal tolerance test was observed after 52 weeks (ratio active:placebo 1.173 [80% confidence interval (CI) 0.94, 1.47], P(one-sided) = 0.18). The yearly rate of change of normalized stimulated C-peptide(AUC) suggests a slower decline of β cell function: iscalimab −0.14 (80% CI −0.23, −0.05) versus placebo −0.33 (−0.42, −0.23) nmol/L per year (P(one-sided) = 0.04). The estimated geometric mean ratio to baseline of hemoglobin A1c at week 52 was lower with iscalimab than placebo (0.95 [80% CI 0.92–0.99] versus 1.05 [80% CI 1.00–1.11], respectively). Leukocytes, neutrophils, and monocytes were lower, whereas T and B lymphocytes were higher in iscalimab-treated participants compared with placebo. Iscalimab was generally safe and well tolerated. Five serious adverse events (AEs) occurred under iscalimab (urinary tract infection, diabetic metabolic decompensation, traumatic fracture, hypoglycemia, and large intestine infection [3.4% each]) and one under placebo (mastoiditis [6.7%]). The most common AEs were hypoglycemia, nasopharyngitis, injection site reaction, COVID-19, and neutropenia. The majority of AEs were mild-to-moderate in intensity and resolved.
Conclusion:
Iscalimab has an acceptable safety and tolerability profile. The sample size limits interpretation of efficacy results. CD40:CD154 inhibition warrants further investigation in T1DM.
Introduction
Over the last 100 years, type 1 diabetes mellitus (T1DM) has changed from an acutely fatal disease to a chronic insulin-dependent disease. However, those living with T1DM continue to experience multiple complications and shortened lifespans, 1 and to date no cure is available. While T1DM is recognized to be an autoimmune disease, immune modulation as a therapeutic approach has been challenging. Cyclosporin was the first immunomodulatory agent shown to reduce or eliminate insulin injection requirements. 2 Although the potential value of immunomodulation was demonstrated, increased infections and kidney damage limited clinical use of this agent. Multiple immunomodulatory approaches have subsequently been investigated targeting differing immune regulatory processes, including T lymphocyte activation inhibitors, regulatory T-lymphocyte inducers, effector T-lymphocyte depletion or exhaustion, and B-lymphocyte antagonists. 3 Teplizumab, a humanized, an Fc receptor–nonbinding anti-CD3 monoclonal antibody is the first disease-modifying therapy to be marketed to delay progression of T1DM from stage 2 to stage 3 T1DM. 4 However, despite this advancement additional investigations are warranted as some patients may not respond, duration of protection may be variable, there is uncertainty on potential emergent resistance to retreatment, and those with more advance disease or progression on teplizumab may require complementary approaches.
While much work on the pathogenesis of T1DM has focused on the T lymphocyte, depletion of B lymphocytes delays disease progression in the nonobese diabetic (NOD) mouse and translates to humans, as seen for example with rituximab. These findings implicate a pathogenic role for B lymphocytes in T1DM pathogenesis and support an intervention that targets T and B lymphocyte interactions. 5
CD154, also known as CD40 ligand, is primarily expressed on activated CD4+ T lymphocytes, but can be found on other immune cells. CD154 protein expression on activated T lymphocytes interacts with CD40 expressed on B lymphocytes and is required for the generation of germinal centers, isotype switching, and sustained Ab production. 6 CD154-CD40 signaling events lead to inflammatory cell proliferation and production and release of pro-inflammatory cytokines and chemokines. Pancreatic β cells also express CD40, 7 which may target activated lymphocytes directly to the islet and promote the insulitis that underlies T1DM.
In preclinical genetic and transgenic models of T1DM, CD154-CD40 signaling participates in the pathophysiology of T1DM. CD40:CD154 is necessary for the initiation of insulitis and diabetes in the NOD mouse. 8 In this model, CD40+CD4+ T lymphocytes are pathogenic and transfer progressive insulitis and diabetes. 9 NOD mice deficient for CD154 (CD154-KO/NOD) do not develop insulitis or diabetes, 10 and therapeutic blockade with the anti-CD154 monoclonal antibody (mAb) MR1 prolongs islet graft survival in NOD mice. 11 Similarly, in NOD mice, CD40 blockade with a targeted small peptide, KGYY15, prevents hyperglycemia and reverses new-onset hyperglycemia. 12 Blocking CD154 restores dendritic cell-mediated induction of tolerance in autoreactive T lymphocytes. 13 In the rip-CD154 transgenic model, pancreatic β cell specific expression of CD154 mediates immune activation, insulitis, and diabetes in a nondiabetes prone background. 14 In the Diabetes-Resistant BioBreeding (DRBB) rat, CD40-CD154 blockade is most effective early in the autoimmune process. 15
Information on the role of CD40 in human T1DM is limited. Both soluble and cellular CD40 are increased in pediatric new-onset T1DM,16,17 and CD40 is expressed in human pancreatic β cells. 7
Together, preclinical data support a role of CD154-CD40 in T1DM, and the presence of CD40 in human pancreas and in new-onset T1DM supports the possibility that findings may translate to people.
Iscalimab is a fully human, IgG1 pathway, blocking anti-CD40 monoclonal antibody that has been modified with a N297A mutation rendering it unable to mediate Fcγ-dependent effector functions. Iscalimab inhibits CD154-induced activation of human leukocytes in vitro, but does not induce human leukocyte activation. 18 In vivo, iscalimab blocks primary and recall T cell-dependent antibody responses (TDARs) in nonhuman primates and abrogates germinal formation without depleting peripheral blood B lymphocytes.
Clinical trials with iscalimab have been conducted in healthy volunteers 19 and patients with kidney transplant, liver transplant, rheumatoid arthritis, 19 Graves’ disease, 20 myasthenia gravis, 21 Sjögren’s syndrome, 22 lupus nephritis, 23 systemic lupus erythematosus, and hidradenitis suppurativa (https://clinicaltrials.gov/study/NCT03663335, NCT03781414, NCT03656562, NCT03827798, and NCT04129528).
We hypothesize that blockade of CD40-CD154 activation with iscalimab in those with new-onset T1DM could halt the autoimmune attack and reduce the insulitis, thus modifying disease progression and improving pancreatic β cell function. This trial was conducted to evaluate the safety, pharmacokinetics, and potential efficacy of iscalimab to preserve pancreatic β cell function in pediatric and young adult people with recent-onset (stage 3) T1DM.
Research Design and Methods
The trial was developed and conducted by Novartis with input from INNODIA, a pan-European infrastructure with investigators focused on novel approaches to treat and prevent T1DM within the Innovative Medicines Iniative - Joint Undertaking (IMI-JU) INNODIA-HARVEST project (https://www.innodia.eu, access June 14, 2025). The protocol generally followed the INNODIA Master Protocol for evaluation of investigational medicinal products in children, adolescents, and adults with newly diagnosed T1DM. 24 Novartis employees designed the trial, analyzed the data, and confirmed accuracy and completeness of data and fidelity of the trial to the study protocol, prepared, reviewed, and approved the article for submission. Trial coordination, laboratory tests, and data management were conducted centrally. The trial was conducted in accordance with the principles of Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council for Harmonization. The protocol was approved by the independent ethics committee or institutional review board at each site. All participants or guardians provided written informed consent, with assent provided by minors.
The Phase 2 (2a) study was conducted from November 2019 to June 2024 at 14 academic clinical research sites in six European countries, including Belgium, Spain, Italy, Slovenia, England, and Germany. The trial was a nonconfirmatory, randomized (2:1), placebo-controlled, participant, and investigator-blinded study in pediatric and young adults with recent-onset stage 3 T1DM evaluating the safety, tolerability, and efficacy for preservation of pancreatic β cell function of iscalimab administered by an initial intravenous dose of 30 mg/kg, followed by weekly, subcutaneously administered weight-based dosing of 300 mg for body weight of 50 kg or more and 195 mg for body weight from 30 kg to less than 50 kg (Clinicaltrials.gov identifier NCT04129528).
Participants were clinically diagnosed with stage 3 T1DM and had evidence of autoimmunity with one or more T1DM autoantibody(ies) against: glutamic acid decarboxylase (anti-GAD), protein tyrosine, phosphatase-like protein (anti-IA-2); zinc transporter 8 (anti-ZnT8); or islet cell (cytoplasmic). In addition, participants had peak stimulated C-peptide levels of 0.2 nmol/L (0.6 ng/mL) or more following standard liquid mixed meal tolerance test (MMTT, Ensure Plus, Abbott, Lake County, IL, USA; 6 mL/kg [maximum 360 mL]); completion of receipt of all locally recommended immunizations; and were able to be administered the first dose of iscalimab or placebo within a 56-day interval from the time of diagnosis. However, the first dose could be extended to 100 days from diagnosis in the event a screening assessment needed to be confirmed or recommended vaccination required before initiation of the study drug (full inclusion and exclusion criteria are provided in the Supplementary Data). After two weekly visits, subsequent visits were scheduled monthly, with MMTT performed every 12 weeks over 52 weeks and at study week 72. Tanner stage and bone age were assessed in participants less than 17 years of age at baseline and week 52, with bone age using the Greulich and Pyle method, and used to evaluate any potential impact on growth in pediatric participants.
Participants were allocated (ratio 2:1 iscalimab:placebo) in staggered cohorts: Cohort 1: age 15–21 years, inclusive with body weight 40–125 kg, inclusive; then Cohort 2: age12–21 years, with body weight 30–125 kg (Fig. 1). Randomization was managed by Cenduit Interactive Response Technology (Durham, NC, USA). Iscalimab and similar placebo were provided to sites and subsequently to participants in blinded, numbered kits sufficient for 1 month administration. Participants were provided unblinded continuous glucose monitors (Dexcom G6) to use throughout the trial. If they preferred not to use the device, they were requested to perform continuous glucose monitoring at least at baseline, 3, 6, 9, 12, and 18 months. Health care professionals could adjust their insulin according to individual and local guidelines using injection or insulin pump-based approaches.
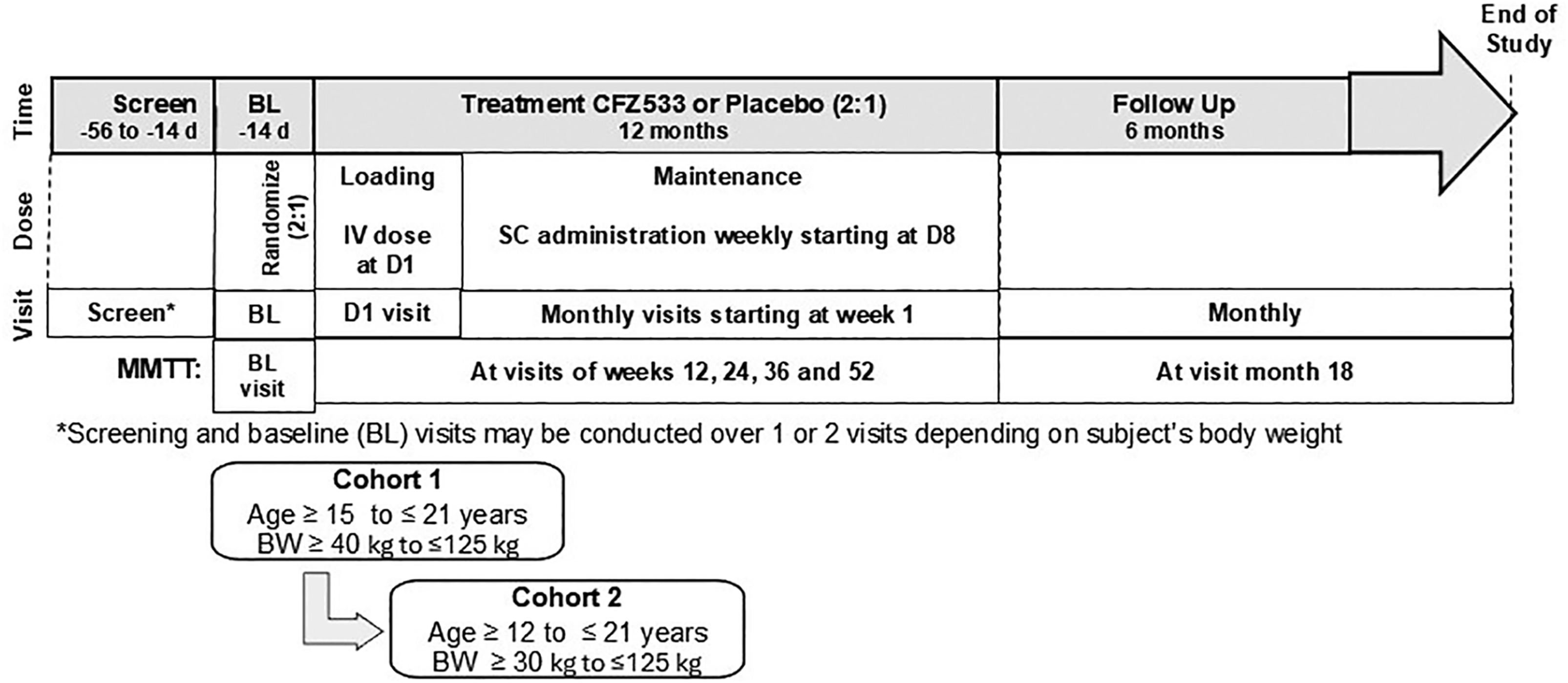
Study schema. Study schema is provided. BL, baseline; BW, body weight; D, day; IV, intravenous; SC, subcutaneous.
The trial was temporarily halted from March to August 2020, as a precautionary measure due to the worldwide COVID-19 pandemic. At that time, two participants had received study drug, had treatment discontinued, and were unblinded as placebo assigned individuals would not require additional monitoring. One was treated with iscalimab who received additional monitoring and one placebo who had a final visit. Countries and sites were reopened based on local readiness. Six amendments to the protocol were implemented, including procedural clarifications, addition of safety measures around viral monitoring, addition of decentralized trial options, and for Novartis strategic reasons, to reduce the number of study participants and duration of follow-up. The Data Monitoring Committee accepted the reduced scope of the trial.
Laboratory
Clinical laboratory assessments, including T and B lymphocytes and natural killer (NK) lymphocytes by flow cytometry, were performed at Q2 Solutions (Edinburgh, Scotland, UK). C-peptide was measured in serum by chemiluminescent immunoassay on Siemens ADVIA Centaur system at Q2 Solutions (Edinburgh, Scotland, UK).
Pharmacokinetics
Plasma samples for the measurement of trough iscalimab concentrations for pharmacokinetic analyses were collected predose baseline and monthly thereafter. Iscalimab concentrations were determined using a validated target-based sandwich ELISA method (BioAgilytix Labs, Hamburg, Germany).
Immunogenicity
Plasma samples were assessed for immunogenicity (antidrug antibodies) using a validated bridging ELISA-based assay.
Sample size
The average MMTT stimulated C-peptide(AUC) in recently diagnosed T1DM is approximately 0.7 pmol·h/mL, with a linear decline resulting in an average C-peptide(AUC) of 0.4 pmol·h/mL at 1 year. 25 Originally a total of 81 participants were planned to be randomized in a 2:1 ratio (stratified by age: 18–21 years, inclusive, or 12 to under 18 years) between iscalimab and placebo to provide 80% power to detect a clinically relevant improvement (i.e., 50% less decrease = 0.55/0.4 = true ratio of 1.375) at 1 year using a one-sided alpha = 0.1. Alpha of 0.1 was chosen as this is an earlier phase trial and to help reduce the type II error rate. After the premature stopping of the trial in the context of company policy decision, retrospective power calculations assuming a larger treatment effect (true ratio of 1.56) indicate 36 completed subjects (of the 44 randomized at the time), which provide 75% power to detect an iscalimab improvement over placebo using a one-sided alpha = 0.1 test. These calculations assume a coefficient of variation (CV) of 70% for the C-peptide(AUC); the standard deviation (SD) of log C-peptide(AUC) was derived from the relationship between the CV and variance parameter for the log-normal distribution. This assumption was the result of the observed CV% of five TrialNet studies in T1DM participants. 26
Statistical analysis
The primary objectives of the study were to evaluate the safety and tolerability of iscalimab in participants with new-onset T1DM and to evaluate the effects of iscalimab on pancreatic β cell function. All participants who received at least one dose of study drug were included in the safety analysis. Stimulated C-peptide(AUC) by the standard MMTT, normalized by the duration of measurements, was analyzed with a mixed model repeated measures analysis using the full analysis set (i.e., participants who study treatment has been assigned by randomization). The natural log of the C-peptide(AUC) was the dependent variable. Independent variables included age group, treatment, visit, and the treatment-by-visit interaction. The natural log of the baseline C-peptide(AUC) was used as a covariate and subject used as a random effect. Least square geometric mean ratio of iscalimab to placebo, the 80% CI of the ratio, and the one-sided P value of treatment benefit are presented for each time point. An 80% CI was chosen to align with the alpha = 0.1 used for the sample size calculations to reduce the type II error rate in this early phase study. The final statistical analysis did not differ from that originally proposed. The statistical analysis was carried out using SAS® procedure PROC MIXED.
Pharmacokinetic (drug concentration) evaluations were based on participants with at least one valid pharmacokinetic concentration measurement (pharmacokinetic analysis set). Arithmetic mean and SD for iscalimab plasma concentrations were provided by weight group and visit/sampling time point. Concentrations below lower limit of quantification were treated as zero.
Human leukocyte antigen risk score
Allele combinations were assigned risk blind to treatment assignment. The human leukocyte antigen (HLA) allele combinations were assigned into haplotypes based on the empirical approach.27–29 Risk was assigned according to odds ratios derived by Sharp et al. 29 with phenotypes ranging from strong susceptibility, moderate susceptibility, moderate protection, and strong protection and summarized for risk estimates with frequency counts and percentages.
All data were analyzed by Novartis.
Results
Participants
Forty-four participants (29 M/15 F, mean 16 [range 12–21] years) were randomized, and all received at least one dose of study medication; 39 completed the study (26 active:13 placebo) (Fig. 2). Participants could remain in study, off treatment. Treatment was discontinued prematurely in seven, two of these (one iscalimab and one placebo) due to a temporary trial halt during the COVID-19 pandemic. These participants were included in analysis. The number of participants who received at least 75% of the planned doses within the 9 months of randomized treatment without any major protocol deviations was 25 (86.2%) for iscalimab and 12 (80.0%) for placebo.
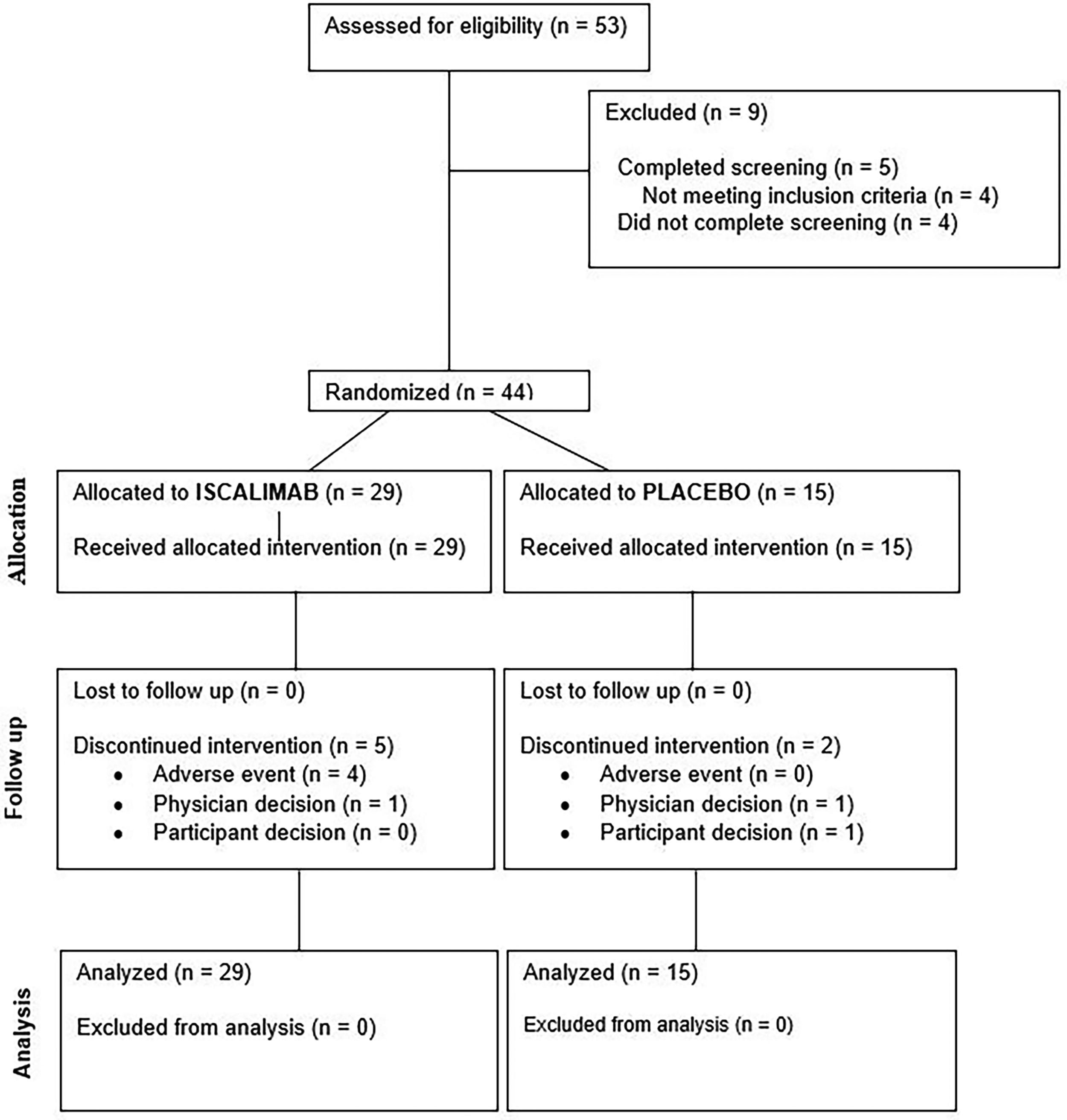
Study consort diagram. Study flow diagram is provided.
Participants had similar baseline characteristics, although stimulated C-peptide(AUC) was lower in the iscalimab compared with the placebo group, and diabetic ketoacidosis at the time of T1DM diagnosis was more common in the placebo group (Table 1). All participants had at least one diabetes autoantibody consistent with the clinical diagnosis and inclusion criteria. HLA risk and diabetes autoimmunity status were balanced between treatment groups (Table 1, Supplementary Data). No participant had a strongly protective HLA genotype, and each of those with indeterminant status had 3 or 4 diabetes autoantibodies.
Characteristics of the Participants at Baseline
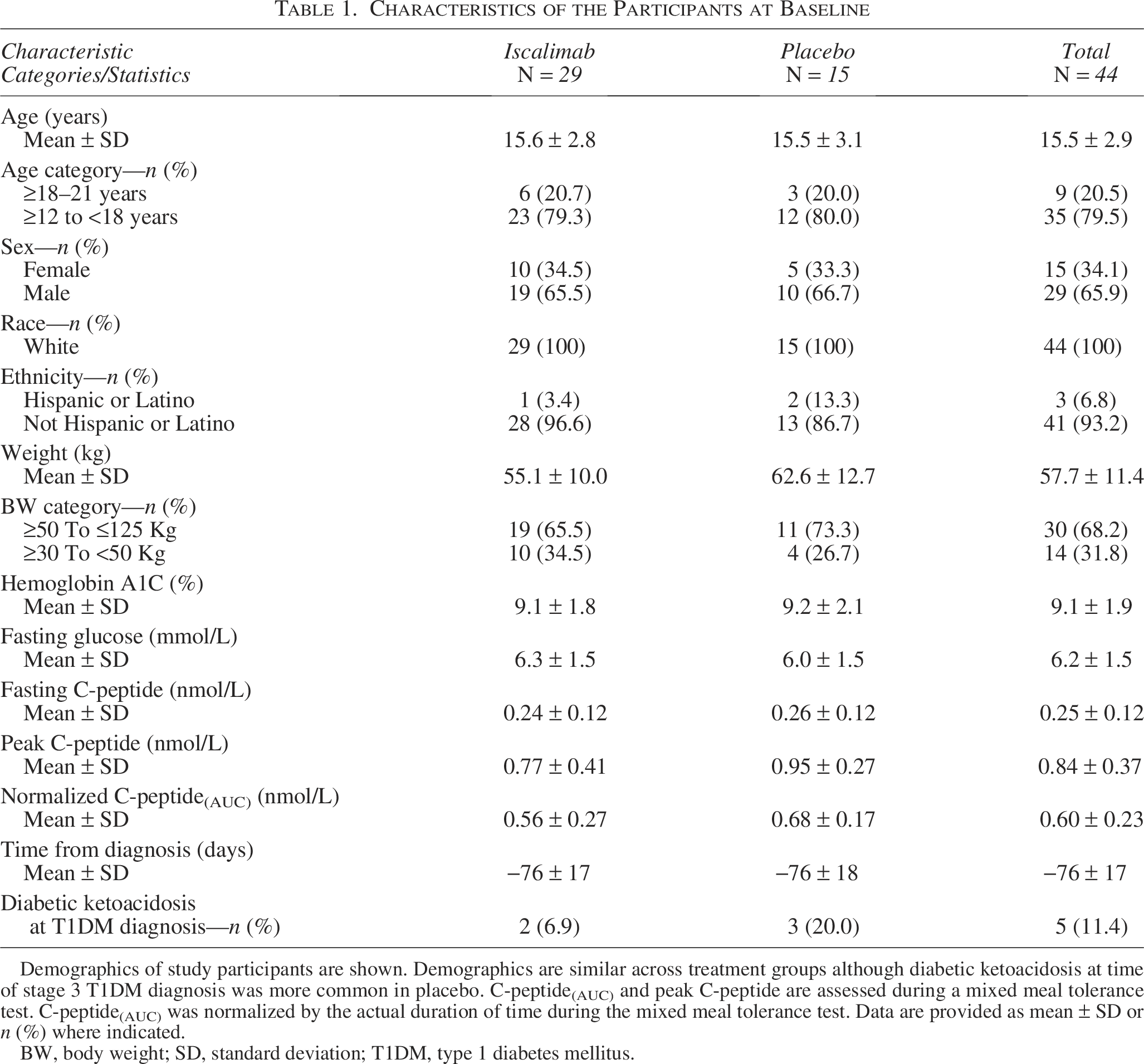
Demographics of study participants are shown. Demographics are similar across treatment groups although diabetic ketoacidosis at time of stage 3 T1DM diagnosis was more common in placebo. C-peptide(AUC) and peak C-peptide are assessed during a mixed meal tolerance test. C-peptide(AUC) was normalized by the actual duration of time during the mixed meal tolerance test. Data are provided as mean ± SD or n (%) where indicated.
BW, body weight; SD, standard deviation; T1DM, type 1 diabetes mellitus.
Efficacy end points
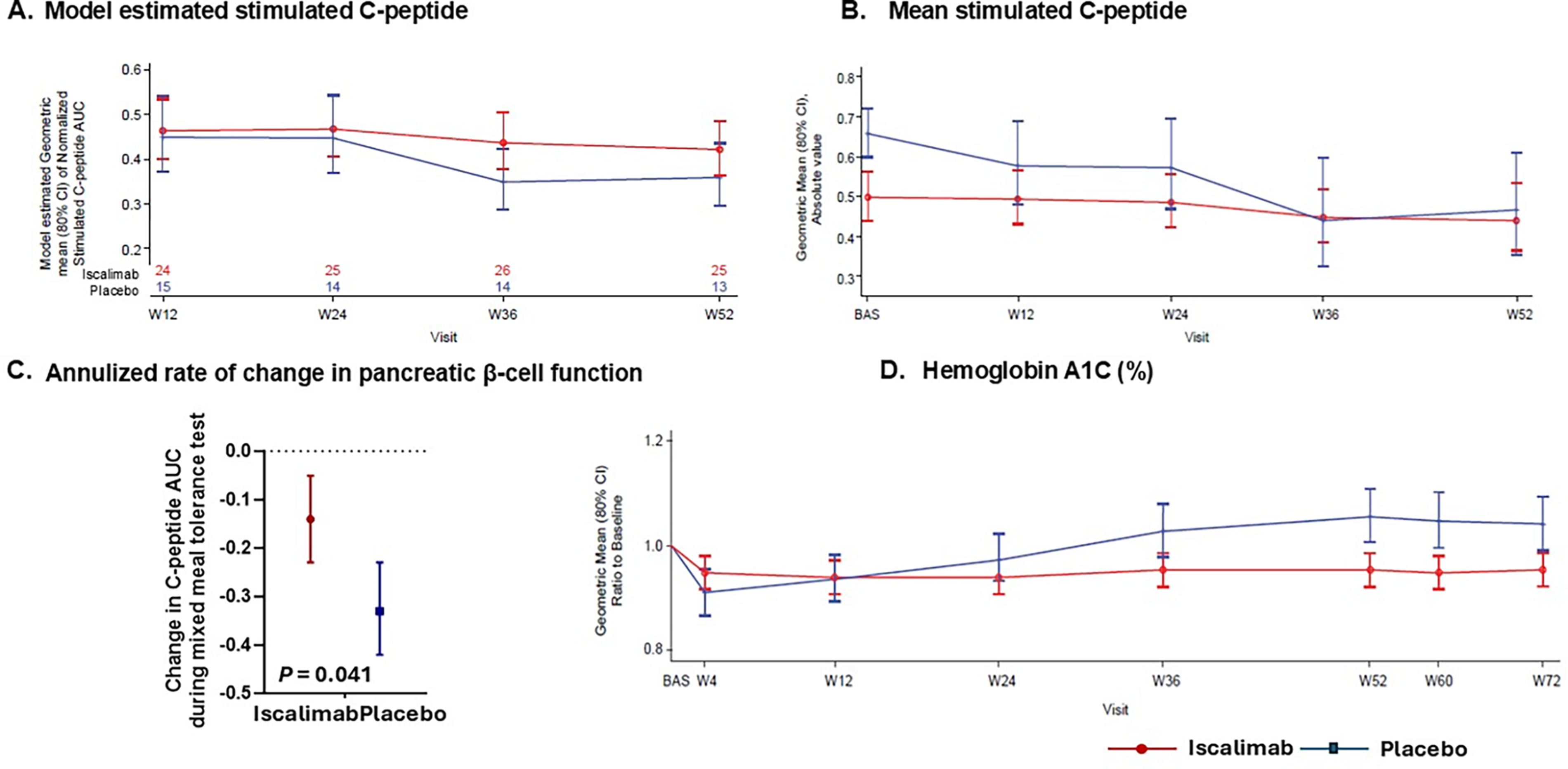
C-peptide(AUC) during MMTT at 52 weeks (primary efficacy end point) was similar between iscalimab versus placebo (mixed model repeated measure [MMRM] analysis of geometric mean of meal stimulated C-peptide [geometric mean ratio of iscalimab versus placebo was 1.173, 80% CI 0.94–1.47, P(one-sided) = 0.182]) (Fig. 3A). The MMRM least square geometric mean C-peptide(AUC) (80% CI) decreased to 0.42 (0.36, 0.49) at week 52 with iscalimab and to 0.36 (0.30, 0.44)% with placebo. Baseline mean stimulated C-peptide(AUC) was lower in iscalimab than placebo, with less decline from baseline over the year (Fig. 3B). The annualized rate of decline in β cell function by stimulated C-peptide(AUC) was slower with iscalimab −0.14 nmol/L per year (80% CI −0.23 to −0.05) than placebo −0.33 (80% CI −0.42 to −0.23) (P(one-sided) = 0.041) (Fig. 3C). Change in fasting C-peptide was similar for both groups. Hemoglobin A1c (HbA1c) was lower with iscalimab than placebo at week 52, which generally persisted post-treatment to week 72. The estimated geometric mean ratio to baseline of HbA1c at week 52 was 0.95 (80% CI 0.92–0.99) for iscalimab versus 1.05 (80% CI 1.00–1.11) for placebo by MMRM (Fig. 3D). No participants achieved remission, defined as requiring no exogenous insulin use.
Glycemic end points:
Continuous glucose monitoring
The duration of continuous glucose monitoring (CGM) use, mean glucose, variation, time in range, above, and below range were comparable between treatment groups (Table 2 and Fig. 1, Supplementary Data). The frequency of CGM-measured hypoglycemia was low in both groups.
Continuous Glucose Monitoring End points
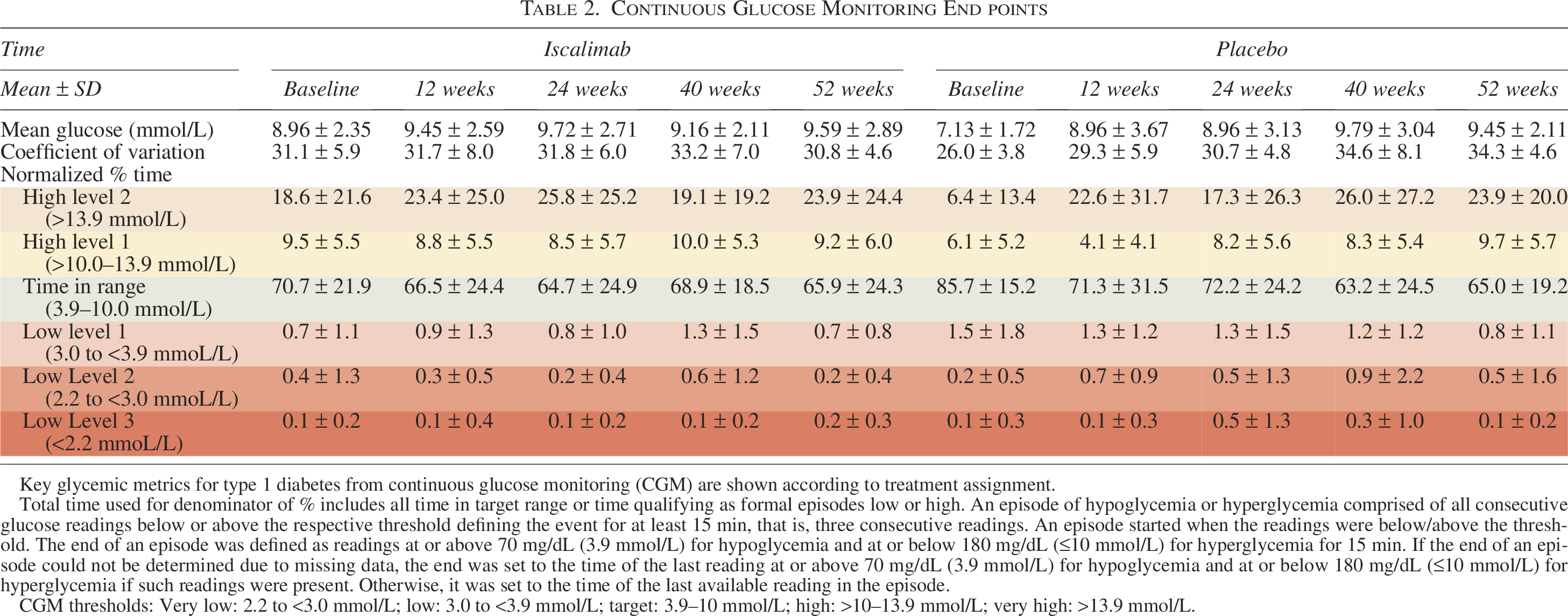
Key glycemic metrics for type 1 diabetes from continuous glucose monitoring (CGM) are shown according to treatment assignment.
Total time used for denominator of % includes all time in target range or time qualifying as formal episodes low or high. An episode of hypoglycemia or hyperglycemia comprised of all consecutive glucose readings below or above the respective threshold defining the event for at least 15 min, that is, three consecutive readings. An episode started when the readings were below/above the threshold. The end of an episode was defined as readings at or above 70 mg/dL (3.9 mmol/L) for hypoglycemia and at or below 180 mg/dL (≤10 mmol/L) for hyperglycemia for 15 min. If the end of an episode could not be determined due to missing data, the end was set to the time of the last reading at or above 70 mg/dL (3.9 mmol/L) for hypoglycemia and at or below 180 mg/dL (≤10 mmol/L) for hyperglycemia if such readings were present. Otherwise, it was set to the time of the last available reading in the episode.
CGM thresholds: Very low: 2.2 to <3.0 mmol/L; low: 3.0 to <3.9 mmol/L; target: 3.9–10 mmol/L; high: >10–13.9 mmol/L; very high: >13.9 mmol/L.
Leukocytes
Emergent differences observed in leukocytes, neutrophils, and T lymphocytes (CD3+CD4+) were sustained, whereas early differences in lymphocytes, B lymphocytes, and monocytes were not sustained (with no difference in NK cells) (Fig. 4).
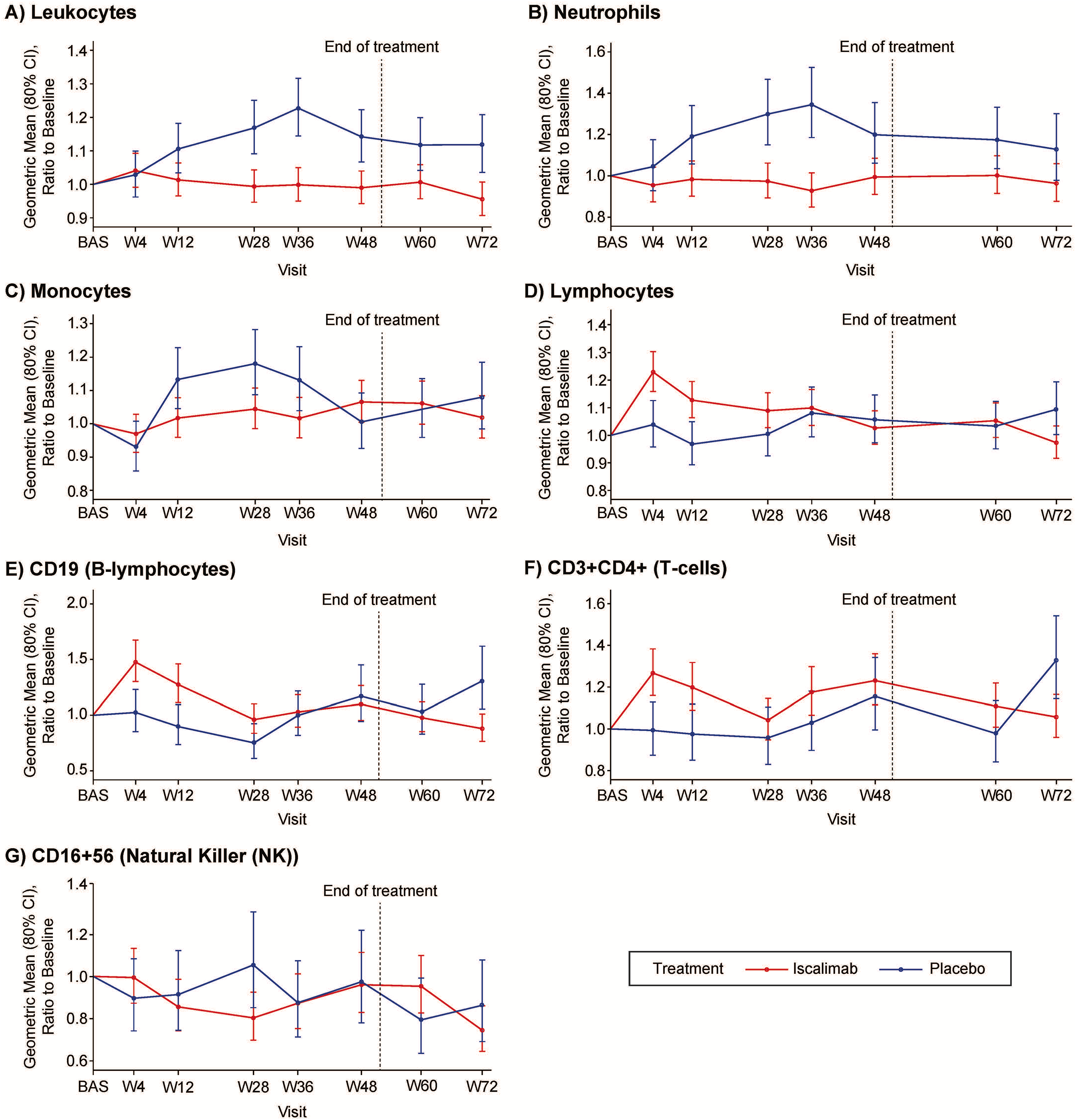
Leukocytes:
Safety and tolerability
There were no deaths or permanent disability. During treatment five serious adverse events (AEs) occurred in four participants under iscalimab (urinary tract infection, diabetic metabolic decompensation, traumatic fracture [unrelated to hypoglycemia], hypoglycemia, and large intestine infection [3.4% each] and one under placebo [mastoiditis, 6.7%]). Four participants had treatment discontinued early (iscalimab n = 4 [13.8%], placebo n = 0 [0%]) due to AEs: one each for transaminase elevations that began before first dose, mild COVID-19, recurrent nasopharyngitis, and neutropenia. Two participants had treatment interrupted due to a proactive trial halt during the COVID-19 pandemic, one in each group. In addition, one participant in placebo withdrew from the trial due to study burden.
Overall, 41 (93.2%) participants experienced at least one AE (iscalimab: n = 28, 96.6%; placebo: n = 13, 86.7%). AEs attributed to the study drug were proportionally balanced between iscalimab (48.3%) and placebo (46.7%). In particular, AEs in the system–organ–class of infection and infestation were proportionally balanced between iscalimab (82.8%) and placebo (80.0%). Regardless of causality, the most common AEs were hypoglycemia, nasopharyngitis, injection site reaction, COVID-19, and neutropenia. Most AEs were mild-to-moderate in intensity and resolved. Treatment-emergent AEs are provided in Table 3. No difference in change was observed for clinical or laboratory evaluations other than the leukocytes (above), including vital signs, hematology, clinical chemistries, urinalysis, electrocardiogram parameters, or age adjusted linear growth, tanner stage, or bone age.
Treatment-Emergent Adverse Events
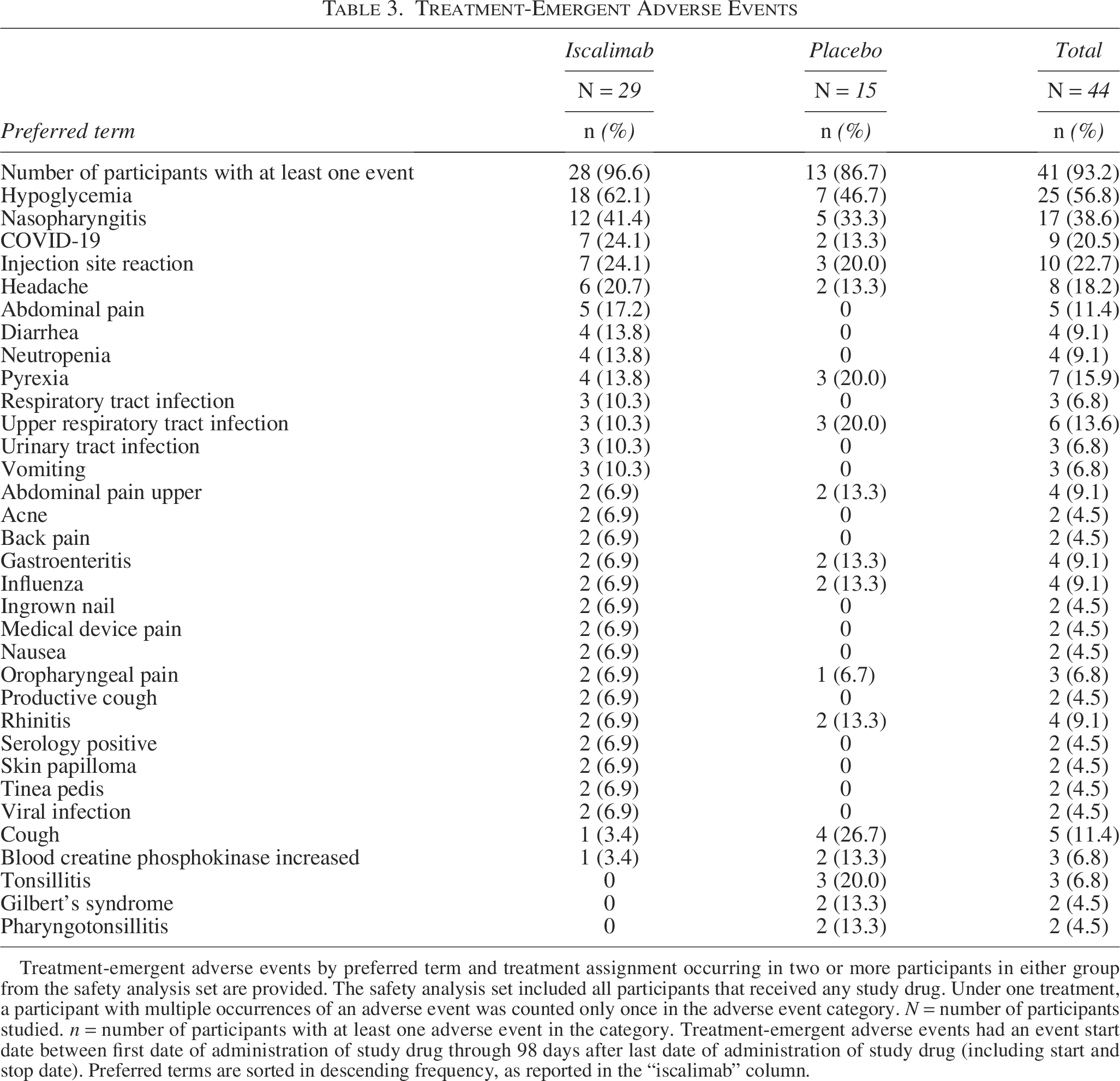
Treatment-emergent adverse events by preferred term and treatment assignment occurring in two or more participants in either group from the safety analysis set are provided. The safety analysis set included all participants that received any study drug. Under one treatment, a participant with multiple occurrences of an adverse event was counted only once in the adverse event category. N = number of participants studied. n = number of participants with at least one adverse event in the category. Treatment-emergent adverse events had an event start date between first date of administration of study drug through 98 days after last date of administration of study drug (including start and stop date). Preferred terms are sorted in descending frequency, as reported in the “iscalimab” column.
Pharmacokinetics of iscalimab in pediatric and young adults with type 1 diabetes
Iscalimab doses were weight based. The Cmax was observed in the samples taken approximately 1.5 h (Tmax) after the start of the initial 30 min loading IV infusion. After that, predose concentrations remained stable during the treatment period and were maintained at comparable concentrations in both weight range groups (Fig. 2, Supplementary Data). There were no preexisting or emergent antidrug antibodies.
Discussion
The CD40-CD154 costimulatory pathway is essential for T cell-dependent immune responses, antigen-presenting cell function, and development of humoral memory. 18 These immune functions have been implicated in the pathogenesis of multiple autoimmune diseases, including T1DM. Given the multiple preclinical studies in genetic and transgenic models of T1DM showing a role of CD40:CD154 costimulatory activation contributing to insulitis and diabetes,8–10,12,14 human data demonstrating the presence of CD40 in human pancreatic β cells, and higher soluble and cellular CD40 in new-onset stage 3 T1DM,16,17 we proposed that blocking CD40:CD154 costimulation would slow progression of pancreatic β cell destruction in new-onset stage 3 T1DM.
In this Phase 2 (2a) trial evaluating iscalimab, a fully human anti-CD40 monoclonal antibody, in adolescent and young adults with new-onset stage 3 T1DM, no difference in β cell function assessed by C-peptide during the MMTT is observed at week 52, although the size of the study limits the interpretation of the efficacy results. Despite randomization, the iscalimab had lower stimulated C-peptide at baseline than placebo. Interestingly almost no further decline in stimulated C-peptide in the iscalimab treatment group is observed, whereas the placebo group had additional loss of β cell function that is similar in magnitude than the average rate of decline in other epidemiological studies.25,30 In contrast to the normalized stimulated C-peptide(AUC) at 52 weeks, the annualized rate of change of stimulated C-peptide suggests a slower decline of β cell function with iscalimab compared with placebo, although this efficacy result must also be interpreted with caution due to small sample size and as this analysis was not prespecified, and no correction for multiple testing has been applied. As participants were not on a unified insulin regimen, and insulin doses were adjusted according to individual and local targets, the lower HbA1c in the iscalimab group compared with placebo must also be interpreted cautiously.
The iscalimab dosing regimen in this trial included an initial intravenous loading dose to fully saturate the CD40 binding sites, followed by a body weight adjusted subcutaneous maintenance dose administered weekly for 51 weeks. The recommended body weight-adjusted dosing was to provide consistent plasma exposures for all treated participants in this study. Concentrations of iscalimab achieved in plasma are considered therapeutic, as levels higher than approximatively 40 μg/mL completely suppress recall TDAR in nonhuman primates. 18 High systemic predose concentrations of iscalimab are maintained during the treatment period reflecting the weekly dosing schedule. Lack of efficacy is unlikely to result from limited pathway blockage. Studies in the DRBB rat model of T1DM demonstrate that CD40-CD154 blockade is more effective when administered early compared with later in the autoimmune process. 15 It is possible that the more advanced disease, as manifest by the lower baseline stimulated C-peptide, would have attenuated potential efficacy findings.
Treatment with iscalimab administered subcutaneously once-weekly was generally safe and well tolerated. Lower circulating leukocytes, neutrophils, and monocytes can be attributed to attenuation of the increase seen in the placebo participants, rather than a fall in circulating concentrations with iscalimab. While the differences in circulating leukocytes may be due to the inhibition of CD40:CD154 signaling, increased neutrophil to lymphocyte ratios are seen with poor glycemic control and may reflect the lower HbA1c with iscalimab. 31 AEs in the system–organ–class of infection and infestation were not more common with iscalimab treatment than placebo.
The strength of this study was its broad exploration of the CD40:CD154 pathway and design based on the INNODIA Master Protocol, with highly standardized procedures and data capture, and introduction of CGM glucometrics. The main limitation of this Phase 2 (2a) study is the small sample size due to premature stopping of the trial in the context of company policy decision, thereby requiring caution in interpretation of mixed efficacy findings, especially at the primary outcome with no improvement of C-peptide during MMTT at 52 weeks, and improvement in other glycemic measures, including annual rate of C-peptide decline and HbA1c. Insulin doses could not be adequately captured as many participants did not maintain insulin dosing records despite being provided electronic or paper diaries for this purpose and were vague on actual amounts administered. Conduct of the trial in pediatric participants with immunomodulatory therapy during the worldwide COVID-19 pandemic was challenging and led to two participants discontinuing. Protocol amendment for strategic reasons to limit enrollment reduced power. Although T1DM is more common in Caucasians, all participants were Caucasian which could limit generalizability. It is not possible to determine if subgroups of patients with T1DM might respond or if there would be greater β cell protection with treatment initiated at stage 2 rather than stage 3 disease.
Conclusions
Disease-modifying treatments are needed for individuals with early stages of T1DM. This trial assessed pharmacokinetics, safety, tolerability, and preservation of pancreatic β cell function at 52 weeks by measurement of C-peptide during MMTT in trial in adolescents and young adults with new-onset T1DM randomized to iscalimab or placebo. Pharmacokinetic profiles were similar across the range of body weights of study participants, and concentrations were stable with no emergent antidrug antibodies. Iscalimab has an acceptable safety and tolerability profile. Although the primary end point of this trial was not met at 52 weeks, two additional glycemic end points (annual rate in decline in C-peptide and control of diabetes [ HbA1c]) provide supportive evidence of potential beneficial treatment effects. Further clinical investigation into the role of the CD40:CD154 pathway in the pathophysiology of T1DM and as a potential treatment modality is warranted.
Footnotes
Acknowledgments
The authors acknowledge the contribution of the patients and their families, and the site investigators and their study teams. Novartis thanks the multiple employees from Novartis who participated in trial conduct and oversight, biomarkers, data management, and data analysis.
Author Disclosure Statement
The study was sponsored by Novartis. A.B.G., N.H., M.H., M.F.H., J.K., C.J.D., M.Q., and K.W. are employees of Novartis. Site investigators received compensation proportional to effort on the trial. Bosi serves or has served on the advisory panel or on the Speakers bureau for Abbott, Medtronic, Sanofi, and Roche. Cardona-Hernandez has received speaker’s fees from NovoNordisk, Boehringer, Novartis, Bayer, Sanofi-Aventis, and Menarini and advisory board fees in the past from: Eli Lilly, Ascensis, Novo Nordisk, and Sanofi-Aventis. C.M. serves or has served on the advisory panel for Novo Nordisk, Sanofi, Eli Lilly and Company, Novartis, Dexcom, Boehringer Ingelheim, Bayer, Roche, Abbott, Medtronic, Insulet, Biomea Fusion, SAB Bio, and Vertex. Financial compensation for these activities has been received by KU Leuven; KU Leuven has received research support for C.M. from Medtronic, Novo Nordisk, and Sanofi; C.M. serves or has served on the Speakers bureau for Novo Nordisk, Sanofi, Eli Lilly and Company, Medtronic, Dexcom, Insulet, Abbott, Vertex, and Boehringer Ingelheim. Financial compensation for these activities has been received by KU Leuven. C.M. is president of EASD. All external support of EASD is to be found on ![]() .
.
Funding Information
This project received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No. 945268 (INNODIA HARVEST). This Joint Undertaking receives support from the Union’s Horizon 2020 research and innovation program, “EFPIA,” “JDRF” (now called Breakthrough T1D), and “The Leona M. and Harry B. Helmsley Charitable Trust.” Conclusions solely reflect the author’s view, and the Joint Undertaking is not responsible for any use that may be made of the information it contains.
Supplemental Material
Appendix
Marieke den Brinker, MD, PhD
UZ Antwerpen
Drie Eikenstraat 655
2650 Edegem
Belgium
Robert Hilbrands, MD, PhD
UZ Brussel
Av. du Laerbeek 101
Jette 1090
Belgium
Chantal Mathieu, MD, PhD
UZ Leuven
Herestraat 49
3000 Leuven
Belgium
Nicole Seret, MD
CHC Mont Légia
Boulevard Patience et Beaujonc 2
4000 Liège
Belgium
Alfonso Soto Gonzalez, MD
Complejo University Hospital
Lugar Xubias De Arriba 84
15006 A Coruña
Spain
Roque Cardona-Hernandez, MD
Hospital Sant Joan de Déu
Passeig Sant Joan de Déu, 2
08950 Esplugues de Llobregat, Barcelona
Spain
Francisco Jose Tinahones, MD
Hospital Virgen de la Victoria
Campus de Teatinos S/N
29010 Málaga
Spain
Tabitha Randell, MD
Consultant in Paediatric Endocrinology and Diabetes
Head of Service PGHAN
Nottingham Children’s Hospital
Derby Rd, Lenton
Nottingham NG7 2UH
United Kingdom
Emanuele Bosi, MD
San Raffaele Hospital and Vita Salute San Raffaele University
Via Olgettina, 60
20132 Milano, MI
Italy
Riccardo Schiaffini, MD
IRCCS Ospedale Pediatrico Bambino Gesu
Piazza Sant’Onofrio 4
00165 Roma, RM
Italy
Sonia Toni, MD
Meyer Children’s Hospital IRCCS
Viale Gaetano Pieraccini, 24
50139 Firenze, FI
Italy
Tadej Battelino, MD, PhD
University Medical Centre Ljubljana
University of Ljublijana
Bohoričeva ulica 20
1000 Ljubljana
Slovenia
Thomas Danne, MD
Work conducted at:
Kinderkrankenhaus auf der Bult
Janusz-Korczak-Allee 12
30173 Hannover
Germany
Now located at:
Comprehensive Health Research Centre (CHRC)
NOVA Medical School, Universidade Nova de Lisboa
Rua Rodrigues Cabrilho 5 6dto
1400–321 Lisboa
Portugal
Desiree Dunstheimer, MD
Universitaetsklinikum Augsburg
Stenglinstraße 2
86156 Augsburg
Germany
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.