
Abstract
Background:
We have developed pancreatic beta-cell, antigen-specific, chimeric antigen receptor (CAR) T regulatory cells (Tregs) and explored their therapeutic potential for type 1 diabetes (T1D)/latent autoimmune diabetes of adults (LADA) in human pancreatic tissues ex vivo, and in a spontaneous humanized mouse model (T1D mice) in vivo.
Results:
Using live-cell imaging, we observed these glutamic acid decarboxylase, 65 kD isoform (GAD65)-CAR-Tregs home to human pancreatic islets exvivo and proliferate upon encountering the cognate GAD65 antigen in the islets. Furthermore, human pancreatic-islet activated GAD65-CAR-Tregs also suppressed human T1D cytotoxic T lymphocytes in co-cultures. We confirmed these findings in vivo, in a spontaneous humanized T1D mouse model (T1D mice) by showing that mouse GAD65-CAR-Tregs also suppressed diabetogenic T responsive (Tresp) cells and were superior to normal Tregs. We also show that mouse GAD65-CAR-Tregs homed to mouse pancreatic islets in vivo. Moreover, we conducted a 30-day preclinical trial in T1D mice, and observed normalization of fasting blood glucose, fasting insulin, and glucose tolerance tests in GAD65-CAR-Treg-treated T1D mice. We confirmed by histology, the advancement of Tregs, retreat of T effector cells in GAD65-CAR-Treg-treated mice, that led to the recovery/reconstitution of pancreatic islets.
Discussion:
Taken together, human GAD65-CAR-Tregs homed to human islets, suppressed diabetogenic T cells, and when used to treat T1D mice that mimic the human pathophysiology of T1D, GAD65-CAR-Tregs reversed T1D. Conceivably, the treatment of T1D with GAD65-CAR-Tregs will allow for recovery/reconstitution of beta cells in human patients as well.
Keywords
Article Highlights
We generated glutamic acid decarboxylase, 65 kD isoform (GAD65) chimeric antigen receptor (CAR) regulatory T cells (Tregs) from human and mouse Tregs. Human GAD65-CAR-Tregs homed to ex vivo human pancreatic islets. When cultured with pancreatic-islets, human GAD65-CAR-Tregs were able to suppress human diabetogenic cytotoxic T lymphocytes ex vivo. In a humanized mouse model of T1D, mouse GAD65-CAR-Tregs suppressed T responsive (Tresp) cells and were superior to normal Tregs. We show that mouse GAD65-CAR-Tregs homed to mouse pancreatic islets in vivo as well. We conducted a 30-day preclinical trial in a humanized mouse model of T1D, and observed normalization of fasting blood glucose, fasting insulin, and glucose tolerance tests (GTTs) with histological reversal of T1D.
For human autoimmune diabetes (type 1 diabetes, T1D, and latent autoimmune diabetes of adults, LADA) to develop, two essential elements need to be present: diabetes-susceptibility genes and a targeted autoantigen. 1 Once the immune attack is initiated, autoantigen-specific cytotoxic T cells destroy insulin producing beta cells. Unfortunately, unfit/poor regulatory T cell responses are less capable of suppressing immunity at the site of inflammation in individuals suffering from autoimmune diabetes. 2
The therapeutic application of regulatory T cells (Tregs) for the treatment of autoimmune disorders, although efficacious, has been limited by the scarcity of antigen specific Tregs.3,4 If antigen-specific Tregs could be produced on demand against a desired autoimmune target, antigen-specific immune suppression of autoimmune diseases would be achievable.
One approach to endow T cells with a desired antigen-specific recognition uses chimeric T cell receptors with antibody-type specificity. Adoptive cellular transfer (ACT) therapies using re-directed cytotoxic T cells with antibody-type specificity (chimeric antigen receptor, CAR-T cells) have shown impressive efficacy in the treatment of hematological malignancies. 5 Accordingly, employing such technology to re-direct Tregs to sites of autoimmune attack may be a useful therapeutic approach to alleviate a broad scope of diseases in which uncontrolled autoimmune responses play a major role. As such, the use of CAR-Tregs represents a highly targeted, next-generation approach for immune tolerance in autoimmune diseases, as T1D. This approach is being explored to target disease-relevant peptide/MHC complexes, non-autoantigenic tags co-expressed on transplanted stem cells, or native T cell receptors (TCRs) equipped with chemically inducible signaling complexes to enhance stability and bystander suppression.6–8 However, the choice of antigens is critical, and to achieve site specific immunosuppression, CARs should be directed to a major autoantigen. We therefore designed a strategy to produce CAR-Tregs for the prevention/treatment of T1D, taking advantage of a membrane bound autoantigenic target present in insulin-producing beta cells.
Glutamic acid decarboxylase, 65kD isoform (GAD65) is the enzyme responsible for gamma-aminobutyric acid (GABA) production in pancreatic beta cells.9,10 GAD65 confines itself to a vesicular compartment distinct from insulin-containing granules. 11 In fact, native GAD65 is primarily anchored to Golgi membranes, travels in small (micro) cytoplasmic vesicles, and does not colocalize with insulin carried by large vesicles (granules) in beta cells.11,12 It is well known that GAD65 (+) synaptic-like microvesicles carry GABA (product of GAD65 enzyme) from within pancreatic beta cells to the interstitial space as extracellular vesicles (EVs).11,12 These EVs are also vehicles for disposal of surplus cellular material. Therefore, for GABA paracrine signaling and for maintenance of cellular homeostasis, EVs, carrying membrane bound GAD65, are released from beta cells to the islet interstitial (intra-islet) space.11,12 GAD65 is a well-recognized autoantigen in human T1D/LADA. Most patients generate autoantibodies to GAD65 even years before the onset of hyperglycemia.13,14 Therefore, we chose GAD65 as the target for developing our CAR-Tregs to prevent/treat T1D/LADA.
We report here, the development of pancreatic, beta-cell-specific, GAD65-CAR-Tregs and the exploration of their therapeutic potential against T1D/LADA. We demonstrate their suppressive capacity over diabetogenic T cells from humans with T1D/LADA in vitro, and in a spontaneous T1D model, that closely resembles the human condition in vivo (T1D mouse). 15 We specifically show that GAD65-CAR-Tregs can home to human islets ex vivo and to mouse islets in vivo. We finally report diabetes reversal in a 30-day preclinical trial using GAD65-CAR-Tregs in the T1D mouse model.
Materials and Methods
Development of GAD65 autoantigen-specific CAR-Tregs
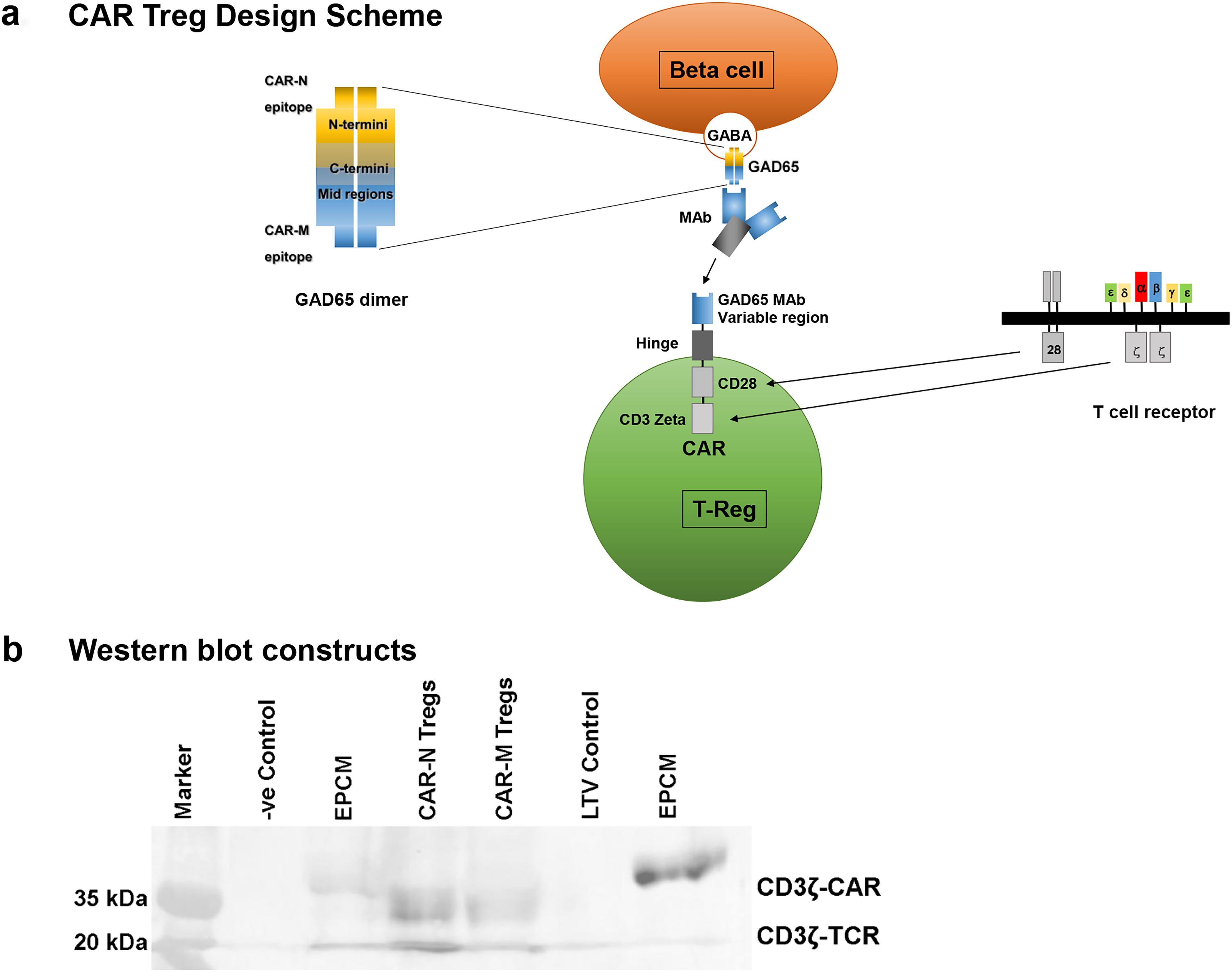
We modified human and mouse Treg cells to enable the expression of CARs and re-direction of specificity to target autoantigen expressed in pancreatic islets. Two GAD65 B cell paratopes (single chain variable fragment, scFv) known to interact with two immunodominant regions in the N-terminal (CAR-N, paratope recognizes AA 39-173) and Middle (CAR-M, paratope recognizes AA 219–243) regions13,14 were selected for assembly onto truncated T cell receptors (via hinge-transmembrane-CD28 and CD3ζ intracellular domains, 2nd generation CAR, Fig. 1a). To generate GAD65-CAR-Tregs, we first isolated regulatory T cells (Tregs) from human blood or mouse spleens using Miltenyi Biotec CD4+ CD25+ Regulatory T Cell Isolation Kits (human, cat # 130-091-301 and mouse, cat #130-091-041). This involved a two-step magnetic separation over MACS® columns to achieve over 90% purity of CD4+CD25+ Tregs. We then introduced GAD65-CAR constructs into these Tregs via lentiviral transduction, with an efficiency of 30%–35%. The transduced cells were expanded 3–5-fold over 4–5 days using IL-2 (2000 ng/mL) and rhGAD65 (4 µg/mL; KRONUS Inc. product ID and lot. # 25FG/RM00-872, 170 South Seneca Springs Way, Suite 105 | Star, ID 83669, USA) and finally re-purified to 99% purity using CD25 microbeads. CAR constructs Western blot for assessment of integrity of CD3ζ in M and N GAD65-CAR constructs, irrelevant construct Epithelial cell adhesion molecule (EPCAM [EPCM]), lentivirus vector alone (LTV), and –ve control (loading dye) is shown in Figure 1b.
Homing of human CAR-Tregs to human pancreatic islets
Blood for CAR-Treg production was drawn 1–2 weeks prior to pancreas surgery from individuals undergoing pancreatectomies. We first isolated Tregs (CD4+CD25+) from peripheral blood (PB) PBMCs (n = 6) of these human subjects and expanded them in vitro. These Treg cells were then transduced with GAD65-CAR-N constructs and later again transduced with a GFP marker. GAD65-CAR-N-Tregs were selectively expanded in the presence of IL-2 and recombinant human rhGAD65 antigen as described above (Fig. 2 top half). Once pancreas tissue became available, it was processed for islet isolation using the collagenase method (with minimal modifications) 16 (Fig. 2 bottom left). Pancreatic islets were then co-cultured with the autologous GAD65-CAR-N-GFP+-Tregs for 7 days. We used IncuCyte S3 Live-Cell System (Sartorius), a real-time system that automatically acquires and analyzes in high definition, phase and fluorescent images of cell cultures, around the clock, while cells remain undisturbed, to monitor the co-culture (Fig. 2 bottom right).
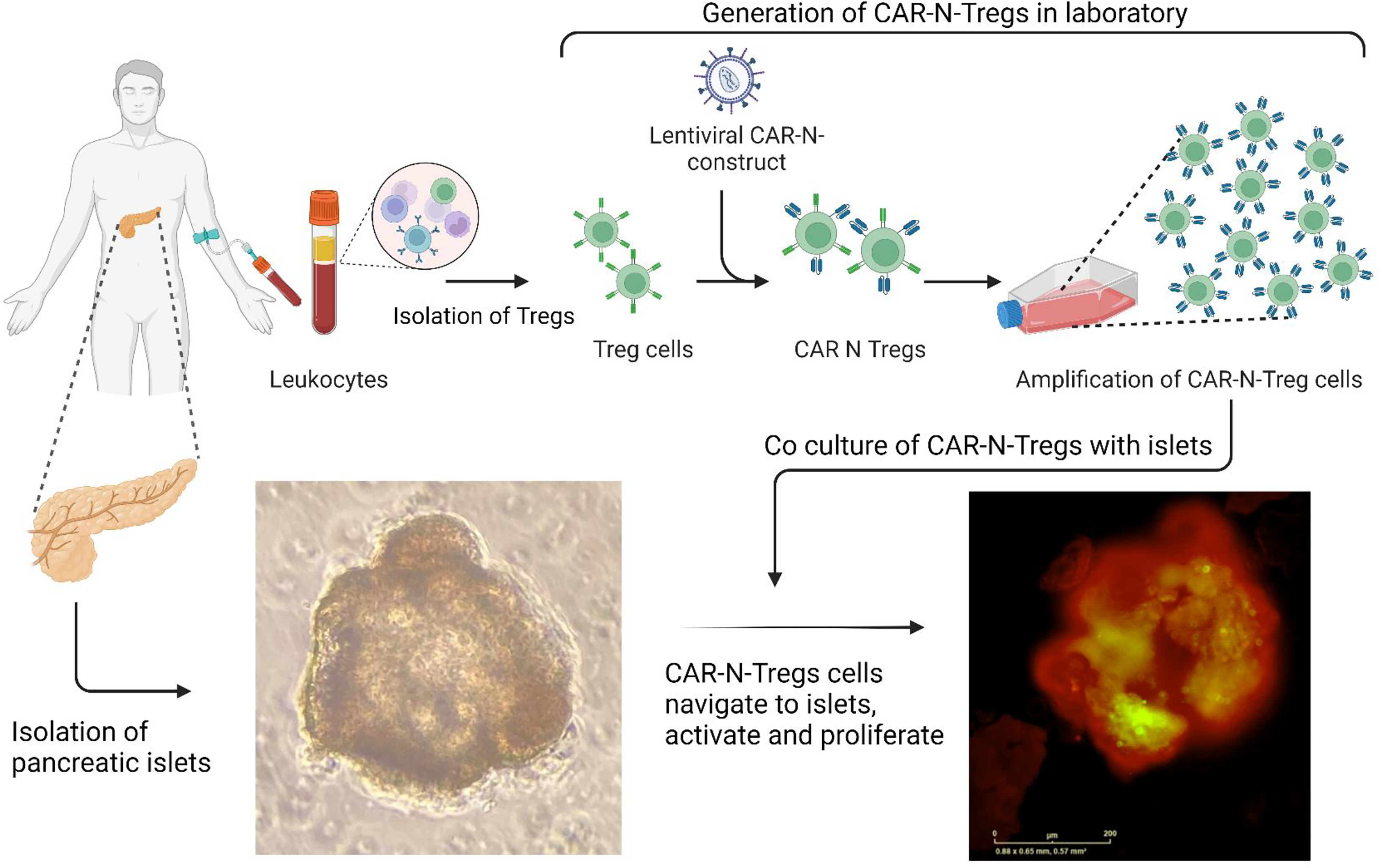
Homing of human CAR-Tregs to human pancreatic islets (ex vivo). This study included recruitment of patients undergoing pancreatic surgery. We collected 10 cc of blood (Fig. 2 top left) 1–2 weeks prior to pancreas surgery from individuals undergoing pancreatectomies; followed by collection of a small piece of pancreas (5 cc wedge) once the pancreas was removed for a specific clinical indication. PBMCs were isolated from blood using FICOL gradient method. CD3 T cells were sorted using human Pan T cell isolation kit (cat # 130-096-535, Miltenyi Biotech.). Tregs (CD4+CD25+) were isolated using human CD4 and Treg isolation kits (cat #s 130-096-533; 130-091-301, respectively). We first isolated Tregs (CD4+CD25+) from peripheral blood PBMCs (n = 6) of these human subjects and expanded them in vitro. These Treg cells were then transduced with GAD65-CAR-N constructs and later again transduced with GFP marker. GAD65-CAR-N-Tregs were selectively expanded in the presence of IL-2 and recombinant human rhGAD65 antigen (Fig. 2 top half). Once pancreas tissue became available, it was processed for islet separation using the collagenase method (Fig. 2 bottom left, unstained islets). All the isolated islets, and cells were washed with sterile RPMI before co-culture. Pancreatic islets were then co-cultured with autologous GAD65-CAR-N-GFP+-Tregs for 7 days. Live-cell immunofluorescence microscopy demonstrated the distinct homing of GAD65-CAR-N-Tregs to functional islets (red-stained for insulin) as early as 24 h of co-culture (Fig. 2 bottom right).
On site suppression of human T1D/LADA cytotoxic T cells by GAD65-CAR-Tregs
Tregs, cytotoxic CD8, and CD4 T cells were isolated from the PB of T1D/LADA patients (n = 3) with Miltenyi Biotec kits using MACS® columns as described above. Next, a batch of Tregs was transduced with GAD65-CAR-N construct using a lentiviral system. To allow for tracking via confocal microscopy, we engineered both GAD65-CAR-N Tregs and a control set of naïve Tregs to express GFP. These cells were then separately co-cultured in the presence of heterologous human pancreatic islets for 76 h (Fig. 3a). Live immunofluorescence imaging showed the enrichment of GFP positive GAD65-CAR-N-Tregs onto insulin red-stained functional pancreatic islet with the progression of the incubation time (visible increase of mid fluorescence intensity by IncuCyte S3 Live-cell System [Sartorius] as above). After 76 h, cells were recovered from cultures and immunofluorescently stained with anti-CD4, anti-CD25, anti-CD8/Interferon gamma (IFNg), and gated for Treg (CD4+CD25+) and cytotoxic T lymphocyte (CTL) (CD8/IFNg) during flow cytometry. They were then analyzed by Flow Jo (BD Biosciences) software (Fig. 3b).
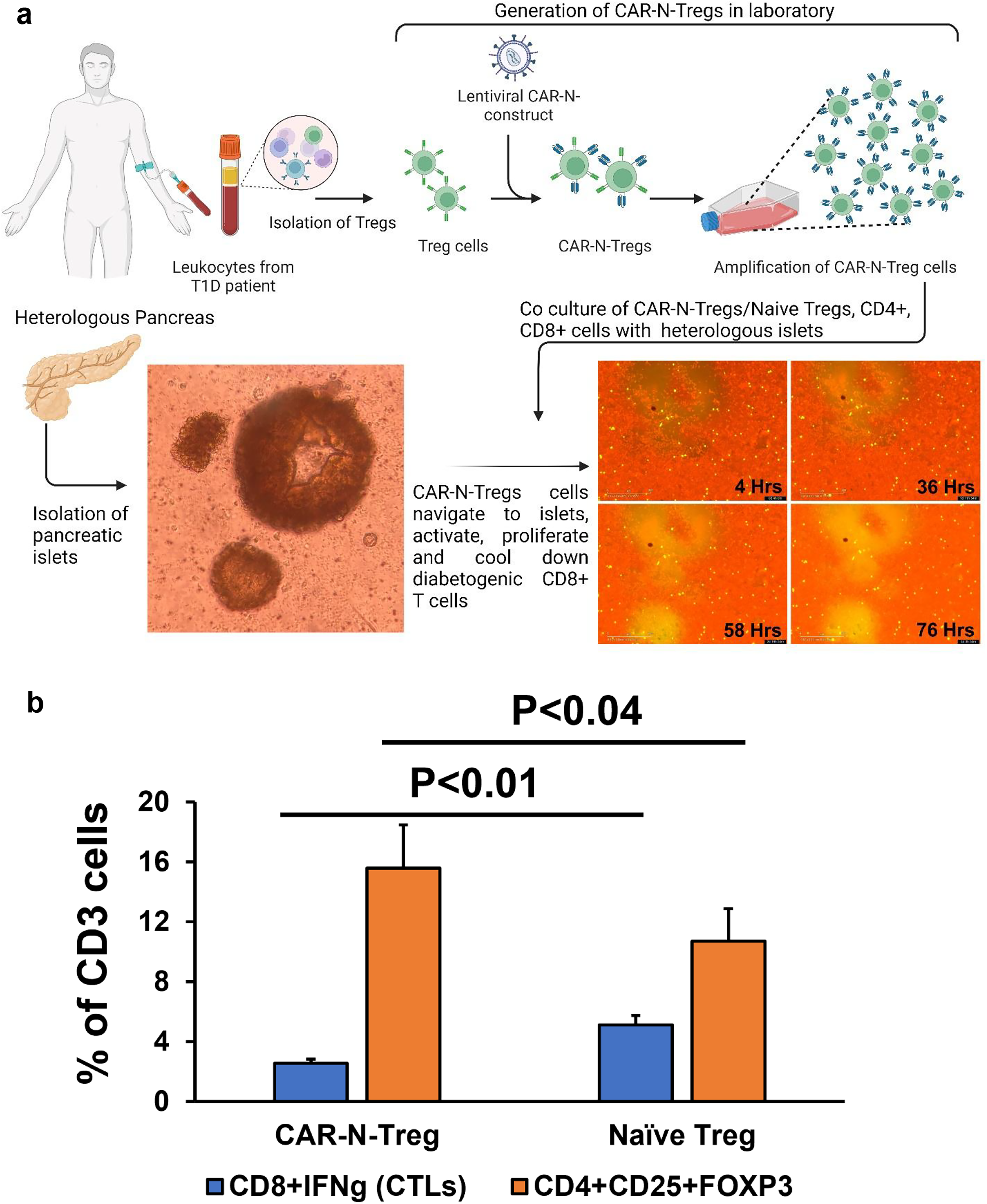
On-site suppression of human T1D/LADA cytotoxic T cells by GAD65-CAR-Tregs in pancreatic islet microenvironment (ex vivo).
17
3 a. Isolation of human Tregs and cytotoxic CD8 and CD4 T cells from peripheral blood of T1D/LADA patients (n = 3) followed by generation of GAD65-CAR-N-Tregs is illustrated on top panel. PBMCs from T1D patients were processed for isolation of Tregs (CD4+CD25+), CD4 as described above (Fig. 2), and CD8 T cells (Miltenyi Biotec, CD8 Cell Isolation Kit, cat #130-096-495). Tregs were transduced with GAD65-CAR-N constructs using lentiviral vector system. A batch of the GAD65-CAR-N-Tregs and naïve Tregs were co-transduced with GFP to monitor for localization/proliferation tracking. GAD65-CAR-N-GFP+-Treg cells were further amplified in the presence of rhGAD65. As before, the amplified GAD65-CAR-N-Treg were further purified using CD25 microbeads (Miltenyi Biotec) to reach 99% purity. A batch of the transduced cells were further analyzed for the expression of CD3+CD4+CD25+FoxP3+. As in previous figure, pancreatic islets were isolated from pancreatic tissue (collagenase method) and cultured ex vivo in DMEM medium under standard conditions overnight (bottom left panel). The GAD65-CAR-N-Treg and naïve Tregs, were co-cultured separately with cytotoxic CD8 and CD4 T cells in presence of heterologous human pancreatic islets (bottom right panel, 4, 36, 58, and 76 h after culture). Live immunofluorescence imaging shows the enrichment of GFP positive GAD65-CAR-N-Tregs onto insulin red-stained functional pancreatic islet with the progression of the incubation time (visible increase of mid fluorescence intensity).
T1D mouse
Among T1D mouse models, the classical nonobese diabetic (NOD) mouse has been instructive in elucidating basic molecular mechanisms involved in autoimmune destruction of pancreatic beta cells. However, it does not carry human diabetes susceptibility genes, and the nature of the primary target antigen is not certain.18–20 Therefore, we worked with HLA-DQA1*0301/DQB1*0302 (DQ8) transgenic, murine MHC-class II molecule-deficient (mII-) C57BL/6-BTBR congenic mice carrying RIP-hGAD65 that were generated to produce a transgenic line that expresses HLA-DQ8 class II in the absence of endogenous murine MHC class II molecules in antigen presenting cells (APCs) and hGAD65 in pancreatic beta-cells. Transgenic mice were selectively in-crossed based on high fasting blood glucose for >30 generations to produce a mouse model that develops diabetes spontaneously. 15 We considered mice to be diabetic when for 2 consecutive days, glucose readings were above 250 mg/dL.
Mouse CAR-Treg in vitro suppression/in vivo proliferation assays
In vitro suppression
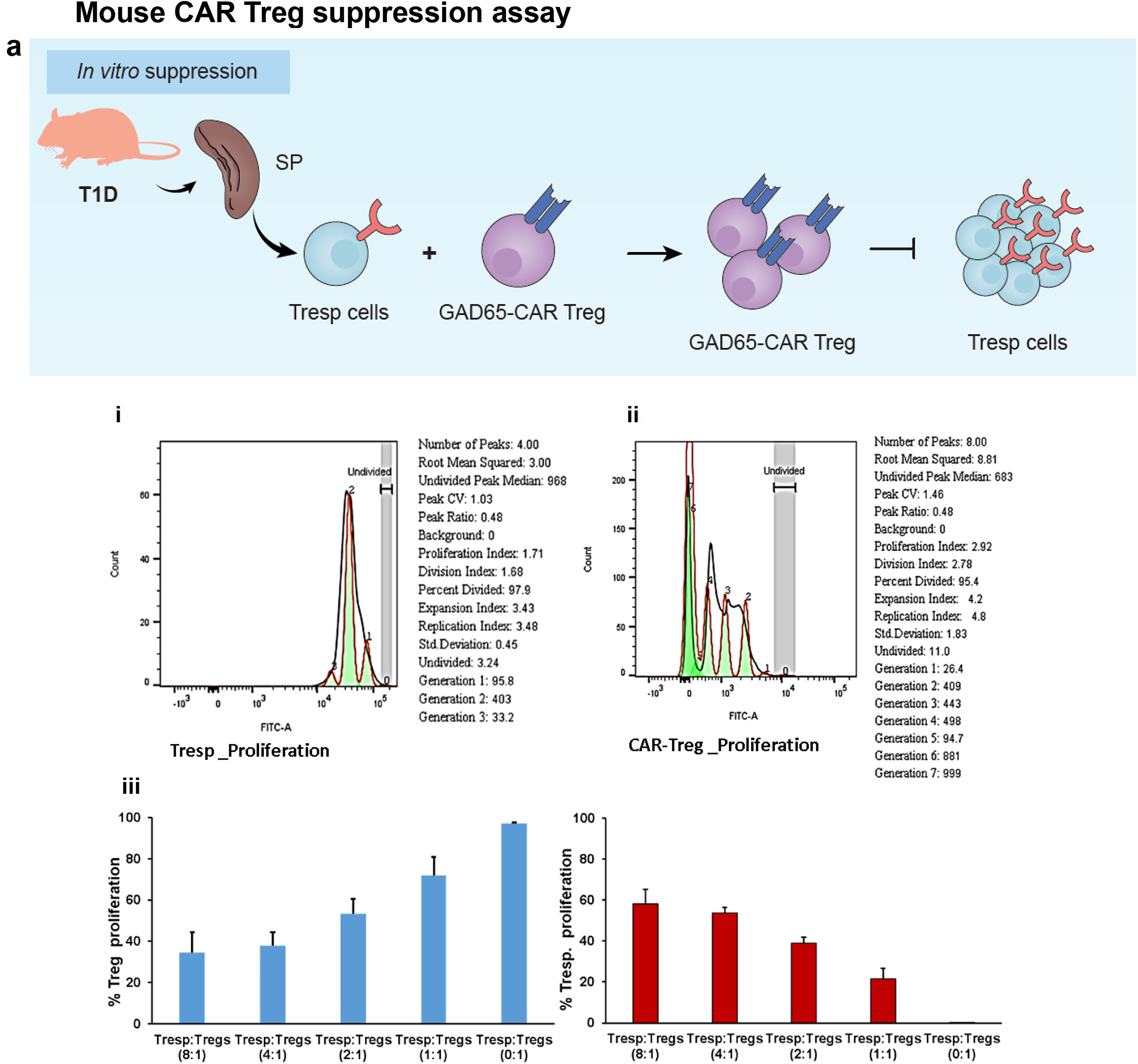
Tregs (CD4+CD25+) and T responsive (Tresp, CD4+CD25−) cells were isolated from T1D mouse spleens using Miltenyi Biotec kits for mouse (catt # 130-091-041) method. Mouse GAD65-CAR Tregs were generated by lentiviral transduction of these isolated Tregs, employing the same methods and constructs as for human cells (see above). Separately, GAD65-CAR Tregs (CD4+CD25+) and Tresp were incubated with IL-2 and rhGAD65 (4 ug/mL), for 4 days. Of note, although enriched, Tresp cells cultures also included APCs in the preparation. Prior to co-culture, both cell populations were labeled with CellTrace (CFSE-FITC), washed and co-cultured at different ratios (0:1; 1:1; 2:1; 4:1; 8:1, respectively). After 5 days, cells were immunofluorescently stained with CD4 and CD25 antibodies, and gated Treg (CD4+CD25+) and Tresp (CD4+CD25−) cell populations were traced by fluorescence (CFSE-FITC, x-axis) for proliferation. Flow cytometry analysis was done with Flow Jo (BD Biosciences) software as described in Miltenyi Biotech in vitro human regulatory T cell suppression assay protocol with little modification (all in Fig. 4a). 17
Continued.
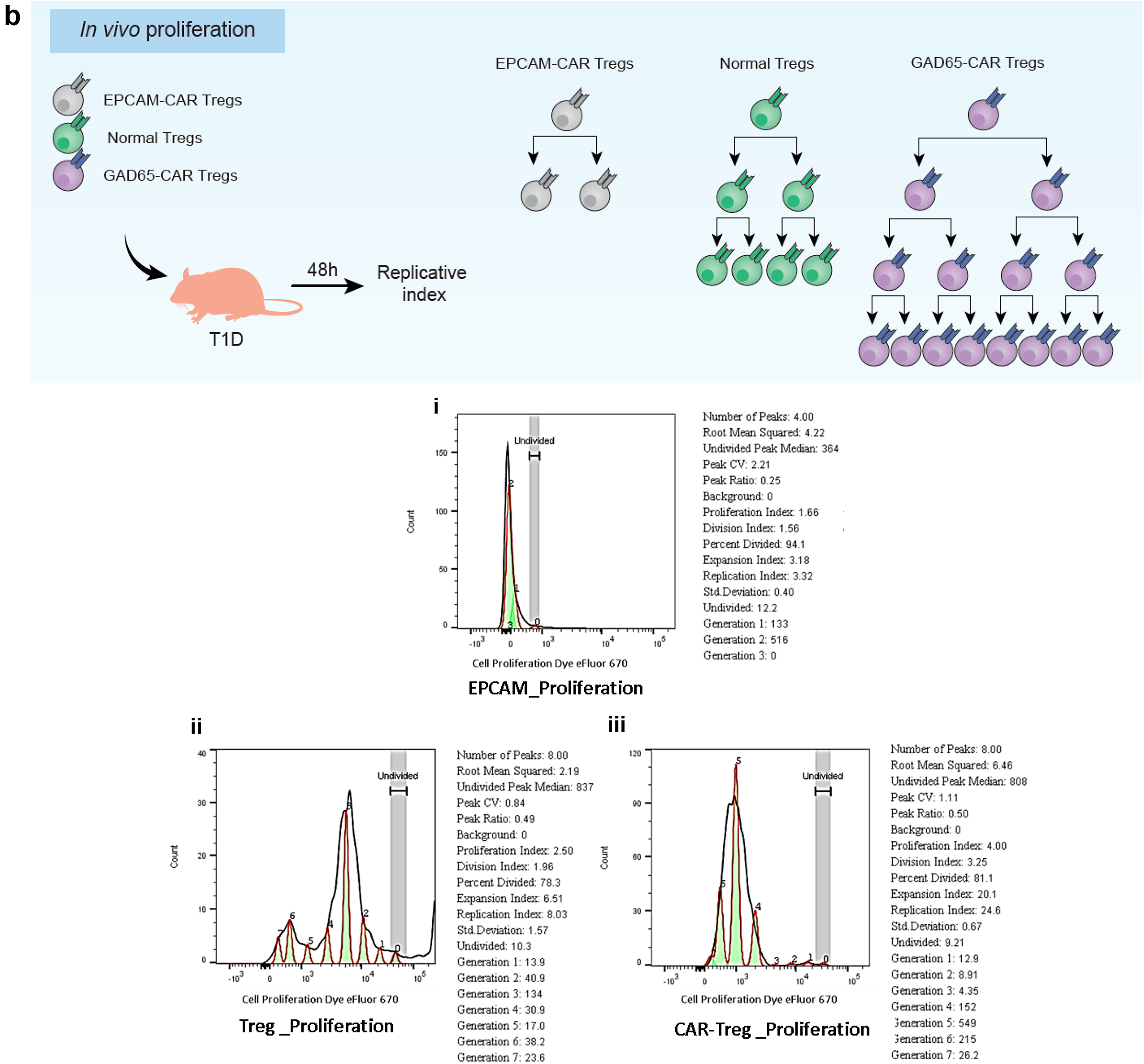
In vivo proliferation
To assess the proliferative capacity of different Treg types in T1D mice, we intravenously infused the T1D mice with GFP-expressing GAD65-CAR Tregs, unmodified (normal) Tregs, or EPCAM-CAR Tregs. Mice were euthanized 48 h after ACT. All Treg (CD4+CD25+) cells were effectively distinguished from non-Treg cells (CD4+CD25−) by flow cytometry (y-axis) based on CD25 Ab recognition. 21 The frequency of proliferating cells (Dye eFluor 670 [x-axis], histograms) was analyzed using Flow Jo (BD Biosciences) proliferation assay software (all in Fig. 4b.).
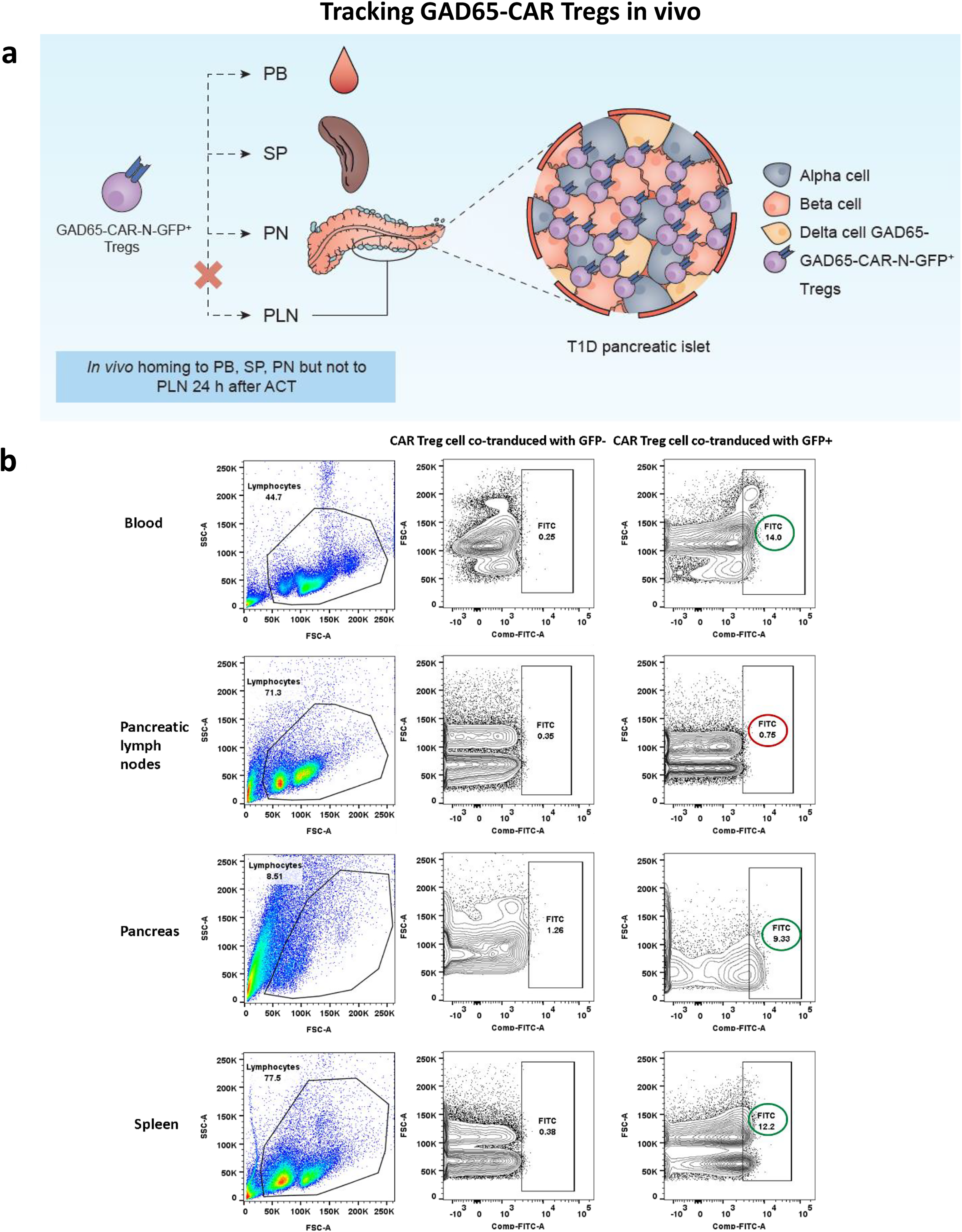
Mouse GAD65-CAR-Tregs also home to the mouse pancreas
A group of T1D mice was intravenously infused with 5 million GAD65-CAR-Treg cells (co-expressing GFP+/−, n = 3), 24 h before euthanasia. PB, pancreatic lymph nodes (PLN), pancreas (PN), and spleen (SP) were processed for single-cell suspensions (Fig. 5a). The single cells were acquired by BD FACSCANTO flow cytometer for detection of GFP in FITC channel (Fig. 5b).
Continued.
GAD65-CAR-N-GFP+-Tregs were infused intravenously in T1D mice and euthanized 24 h after. Samples were prepared as per the standard protocol 22 (Fig. 5c).
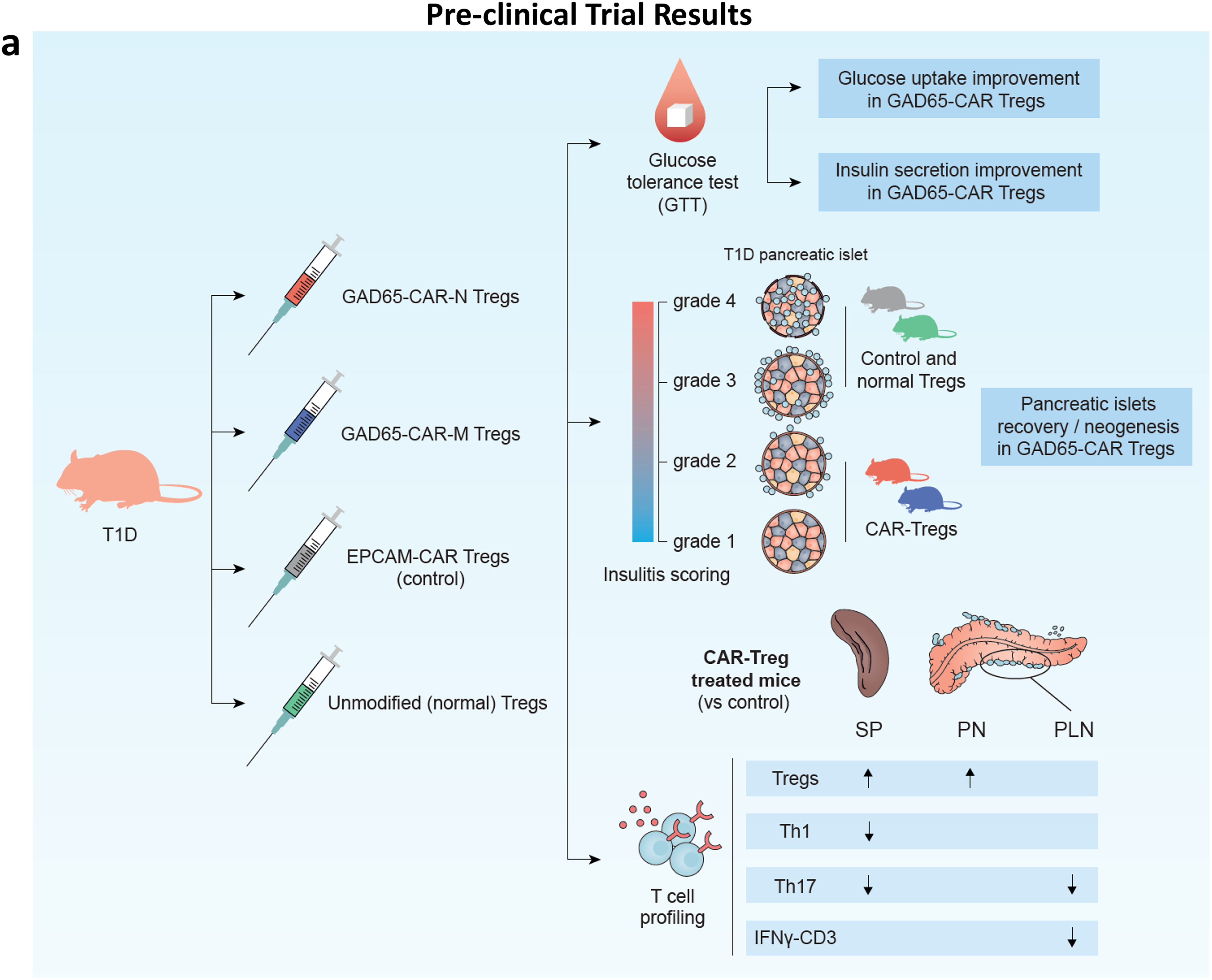
Preclinical trial (30 days)
(Fig. 6) Humanized mice (T1D model) were separately treated with 5 million GAD65-CAR-M-Treg, GAD65-CAR-N-Treg cells, EPCAM-CAR-Treg, and unmodified (normal) Treg cells. The mice were euthanized 30 days post-ACT.
Fasting blood glucose, GTT, and plasma insulin test
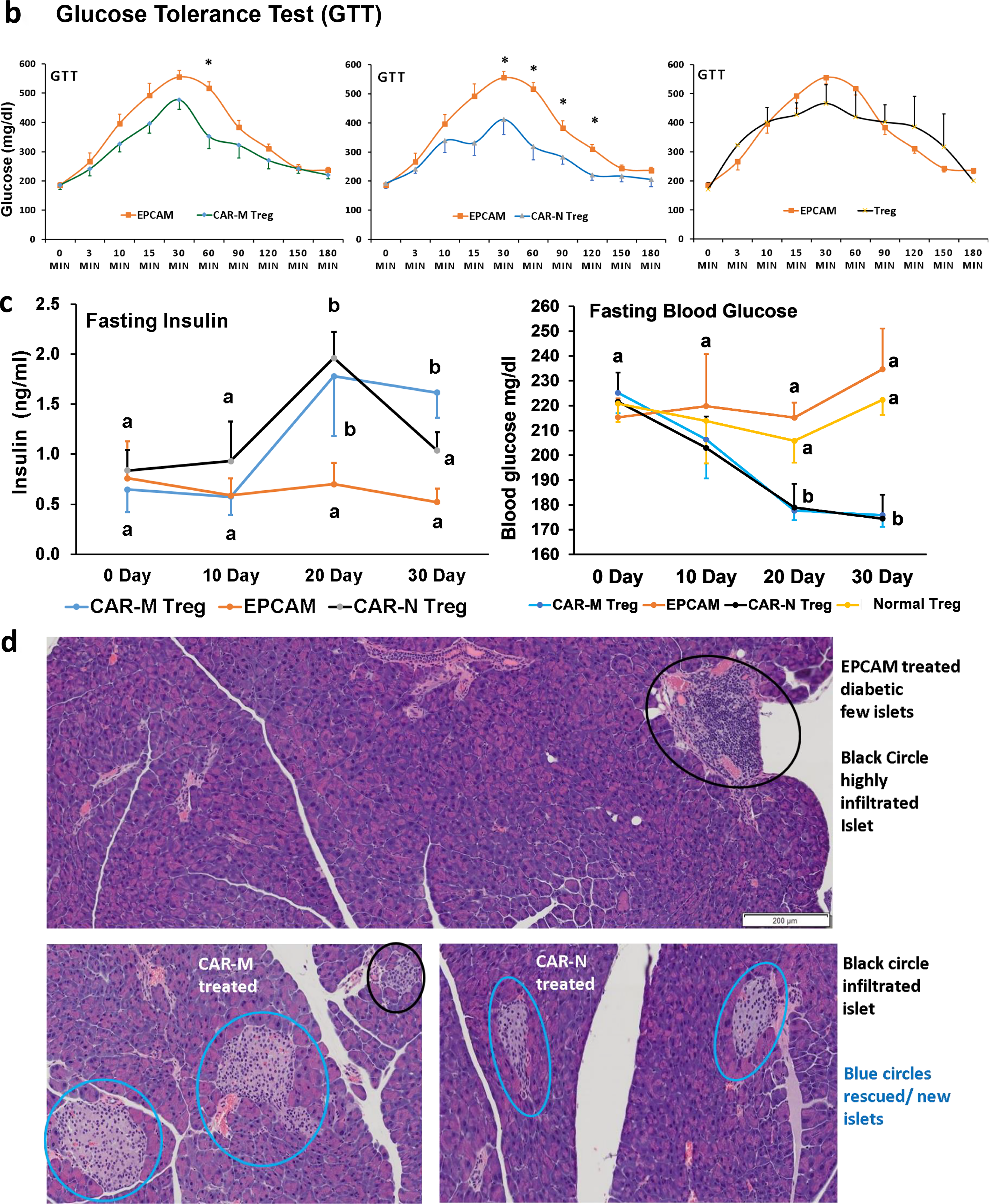
Fasting blood glucose levels were monitored weekly for 30 days, both before and after ACT. A GTT was conducted pretrial and at least twice thereafter. For these tests, mice were fasted for 10 h, then given an intraperitoneal glucose injection (2 g/kg). Blood glucose was measured at different intervals (as depicted in Fig. 6b) using an Ascensia Breeze Glucometer (Bayer). Plasma insulin concentrations were measured at fasting at 0, 10, 20, and 30 days after adoptive cell transfer using a mouse ultrasensitive insulin ELISA kit (Crystal Chem, Inc.) (Fig. 6c).
Continued.
Continued.
Hematoxylin/eosin staining of pancreas/islet scoring
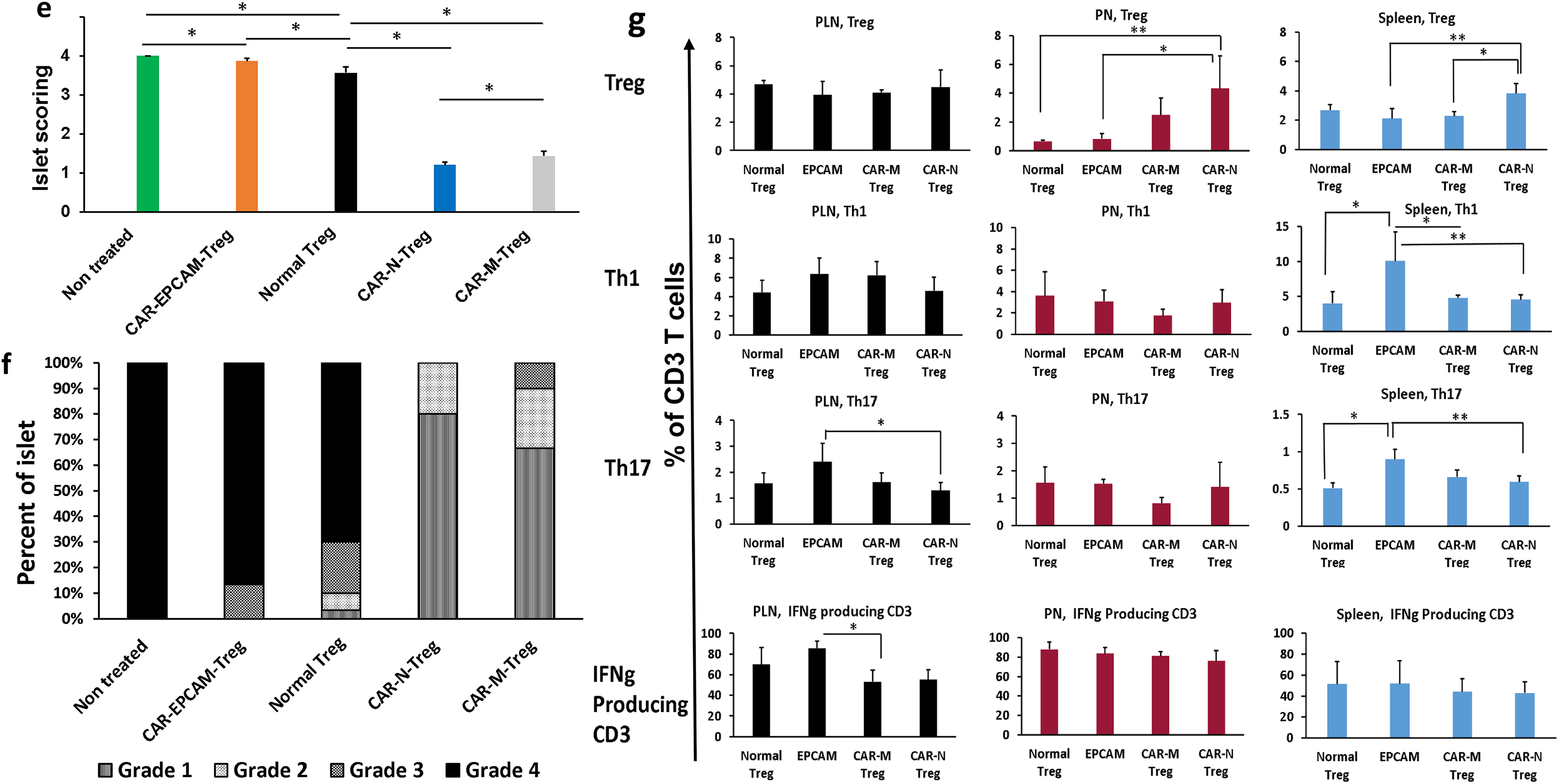
Pancreatic sections were stained with hematoxylin/eosin (H&E) and slides were analyzed by optical microscope for histological identification, localization of lymphocytic infiltration, and for classification of islets’ architecture/insulitis scoring (as previously described) 23 (Fig. 6d.; e. and f. more samples provided in Supplementary Fig. S2).
Immune cell profiling
Intra and extracellular staining of immune cells was performed as previously described.24–26 (Fig. 6g. please see Fig. legend for details).
Statistics
Data for fasting glucose, insulin, GTT, and flow-cytometry were analyzed for normality using the Kolmogorov–Smirnov test and transformed to natural logarithms or ranks as appropriate when not normally distributed. Otherwise, flow-cytometry of various cell phenotypes was statistically analyzed using the SAS MIXED procedure (version 9.3, SAS Institute, Inc.). The significant effects were further tested to locate the difference in means using the least significant difference test (for difference among time points in GTT for example). The statistical significance threshold was at P ≤ 0.05. Data are presented as the mean ± SEM.
Study approval
All human subject experimentation was approved by the University of Toledo IRB.
All animal protocols were approved by the University of Toledo Animal Research committee.
Results
Development of beta-cell, antigen-specific CAR-Tregs
Genetic modification of Treg cells to enable expression of CARs and redirection of specificity toward antigens expressed by pancreatic beta cells was accomplished by selecting human autoantibody paratopes against GAD65. We generated CAR-N-Tregs that recognize a human GAD65 immunodominant epitope in the N-terminus of human GAD65 protein. We also generated CAR-M-Tregs that recognize another human GAD65 immunodominant epitope in the Middle region of the human GAD65 protein (Fig. 1a. CAR-Treg Design Scheme). Both, N and M GAD65 paratopes were designed to mimic naturally occurring human autoantibodies from diabetic patients. CAR-N paratope specifically recognizes a GAD65 area close to the membrane-anchoring domain (therefore, solely recognizes membrane bound GAD65) while CAR-M paratope specifically recognizes a GAD65 hydrophilic area also exposed in the monomeric form of the autoantigen (therefore, also recognizes soluble GAD65). The presence of GAD65-CAR-N/M-CD3ζ and endogenously expressed TCR-CD3ζ confirmed the integrity of CAR construct in Tregs (Fig. 1b).
Human GAD65-CAR-Tregs home to human pancreatic islets
We wanted to determine if these human GAD65-CAR-Tregs could recognize the endogenous human GAD65 protein and home to human pancreatic islets. This study involved drawing 10 cc of blood (Fig. 2 top left) 1–2 weeks prior to pancreas surgery from individuals undergoing pancreatectomies; followed by collection of a small piece of pancreas (5 cc wedge) once the pancreas was removed for a specific clinical indication. We first isolated Tregs from PB (n = 6) of these human subjects and expanded them in vitro. These Treg cells were then transduced with GAD65-CAR-N and GFP lentiviral constructs. GAD65-CAR-N-Tregs were selectively expanded in the presence of rhGAD65 antigen (Fig. 2 top half). Once pancreas tissues became available, they were processed for islet separation using the collagenase method (with modifications) 16 (Fig. 2 bottom left). Pancreatic islets were then co-cultured with autologous GAD65-CAR-N-GFP+-Tregs for 7 days. We used IncuCyte S3 Live-Cell Imaging System (Sartorius), a real-time imaging system that automatically acquires and analyzes in high definition, phase and fluorescent images of cell cultures, around the clock, while cells remain undisturbed, to monitor the co-cultures. Live-cell immunofluorescence microscopy demonstrated the distinct homing of GAD65-CAR-N-GFP+-Tregs to islets as early as 24 h of co-culture (Fig. 2 bottom right). Importantly, while in contact with functional islets (red stained for insulin), proliferation of GAD65-CAR-N-GFP+-Tregs was clearly noticeable within 48–72 h. Thus, it is clear that GAD65-CAR-N-Tregs recognize endogenous GAD65 on isolated islet in an MHC-independent manner, 27 activate themselves, and proliferate.
GAD65-CAR-Tregs suppress human T1D/LADA cytotoxic T cells in pancreatic islet co-culture
Having shown that GAD65-CAR-Tregs were able to find their cognate antigen in pancreatic islets and proliferate, we designed the following experiment to see if their anti-inflammatory potential could suppress cytotoxic T cells in situ. To demonstrate suppression of T1D/LADA cytotoxic T cells by GAD65-CAR-Tregs in situ, we first isolated Tregs and cytotoxic CD8 and CD4 T cells from PB of T1D/LADA patients (n = 3). A batch of Tregs was used to generate GAD65-CAR-N-Tregs. GAD65-CAR-N-Treg and Naïve Tregs were also made to express GFP for tracking and were co-cultured with activated cytotoxic CD8 and CD4 T cells (Fig. 3a. top half) in presence of human pancreatic islets (Fig. 3a. bottom left). GAD65-CAR-N-Tregs homed to islets, got activated, proliferated (Fig. 3a. bottom right, clockwise, 4, 36, 58, and 76 h postculture immunofluorescence images) and suppressed autologous cytotoxic (expressing IFNg) CD8 and CD4 T cells (CTLs, Fig. 3b). Flow cytometry demonstrated the superiority of GAD65-CAR-N-Tregs over naïve Tregs in suppressing CTLs (GAD65-CAR-N-Treg/Naïve Treg:CTL ratio 1:1) in the pancreatic islet microenvironment (Fig. 3b). GAD65-CAR-N-Tregs had significantly higher (P < 0.04) proliferative capacity compared with naïve Tregs (in terms of CD3 T cell percentage-expansion; Fig. 3b orange bars) while in co-culture. Also note, the CTL suppression was significantly higher (P < 0.01, Fig. 3b blue bars) with GAD65-CAR-N-Treg than with Naïve Treg cells.
Mouse GAD65-CAR-Tregs also suppress activated T responsive cells in vitro in a dose-dependent manner
As done for human samples, mouse Tregs were engineered using the same constructs described above for human experiments. Tregs were isolated from spleen (as done for human PBMCs) of T1D mice (hGAD65/hDQ8 transgenics) 15 and initially incubated with IL-2 and rhGAD65 (4 µg/mL) for 4 days. Mouse Tregs were transduced with lentiviral construct for the making of GAD65-CAR-N-Tregs. At the same time, mouse T responsive (Tresp, CD4+CD25−) cells, also isolated from T1D mouse spleen, were also initially incubated with IL-2 and rhGAD65 (4 ug/mL) for 4 days (Fig. 4a. cartoon provides a summary of this experiment). After 4 days, both cell populations were incubated in CFSE. For determining the proliferative capacity, GAD65-CAR-N-Tregs and Tresps, cells were first assessed for proliferation (Fig. 4a. i: Tresp proliferation; ii: GAD65-CAR-N-Treg proliferation). Cell aliquots of Tresp (CD4+CD25−) and GAD65-CAR-N-Treg (CD4+CD25+) were then co-cultured for 5 days (n = 4) under different co-culture ratios. After 5 days, cells were immunofluorescently stained with CD4 and CD25 antibodies to differentiate GAD65-CAR-N-Tregs from Tresps. Flow-cytometry analysis was used to quantify suppressive/proliferative responses. The results were similar to what was observed with human cells. For example, at a 1:1 ratio, the proliferation of GAD65-CAR-N-Tregs tripled that of Tresps (Fig. 4a. iii graph summarizes data of the in vitro suppression/proliferation assay described). Noticeably, mouse GAD65-CAR-N-Treg suppression of mouse Tresp was observed even at the 1:8 ratio.
Mouse GAD65-CAR-Tregs proliferate in vivo and are superior to normal mouse Tregs
Although the human islet co-culture experiment clearly showed antigen-specific suppression of Tresps by GAD65-CAR-Tregs, it was performed ex vivo. Therefore, to investigate antigen-specific suppression/proliferation in vivo, EPCAM-CAR-Treg (irrelevant CAR-Treg), unmodified (normal) Tregs (GAD65-specific), and GAD65-CAR-N-Treg cells all co-expressing GFP were infused into T1D mice (n = 4) (Fig. 4b cartoon summary). Mice were euthanized 48 h after infusion of either EPCAM-CAR-GFP+-Tregs, normal-GFP+-Tregs, or GAD65-CAR-N-GFP+-Tregs. All Treg cell types were then identified based on GFP co-expression by flow cytometry. Histograms in Figure 4b show the proliferation and replication peaks. The replication indexes for EPCAM-CAR-Tregs was 3.32 (Fig. 4b. i), for GAD65-specific-normal Tregs was 8.03 (Fig. 4b. ii), and for GAD65-CAR-N-Tregs was 24.6 (Fig. 4b. iii). Therefore, the in vivo replication of GAD65-CAR-N-Tregs was 3 times that of normal Tregs and 8 times that of irrelevant, EPCAM-CAR-Tregs. This shows that GAD65-CAR-N-Tregs are capable not just of localizing antigen (pancreas; only site of GAD65 expression in T1D mice, see next) but also proliferate in an antigen-specific manner.
Mouse GAD65-CAR-Tregs also home to the mouse pancreas
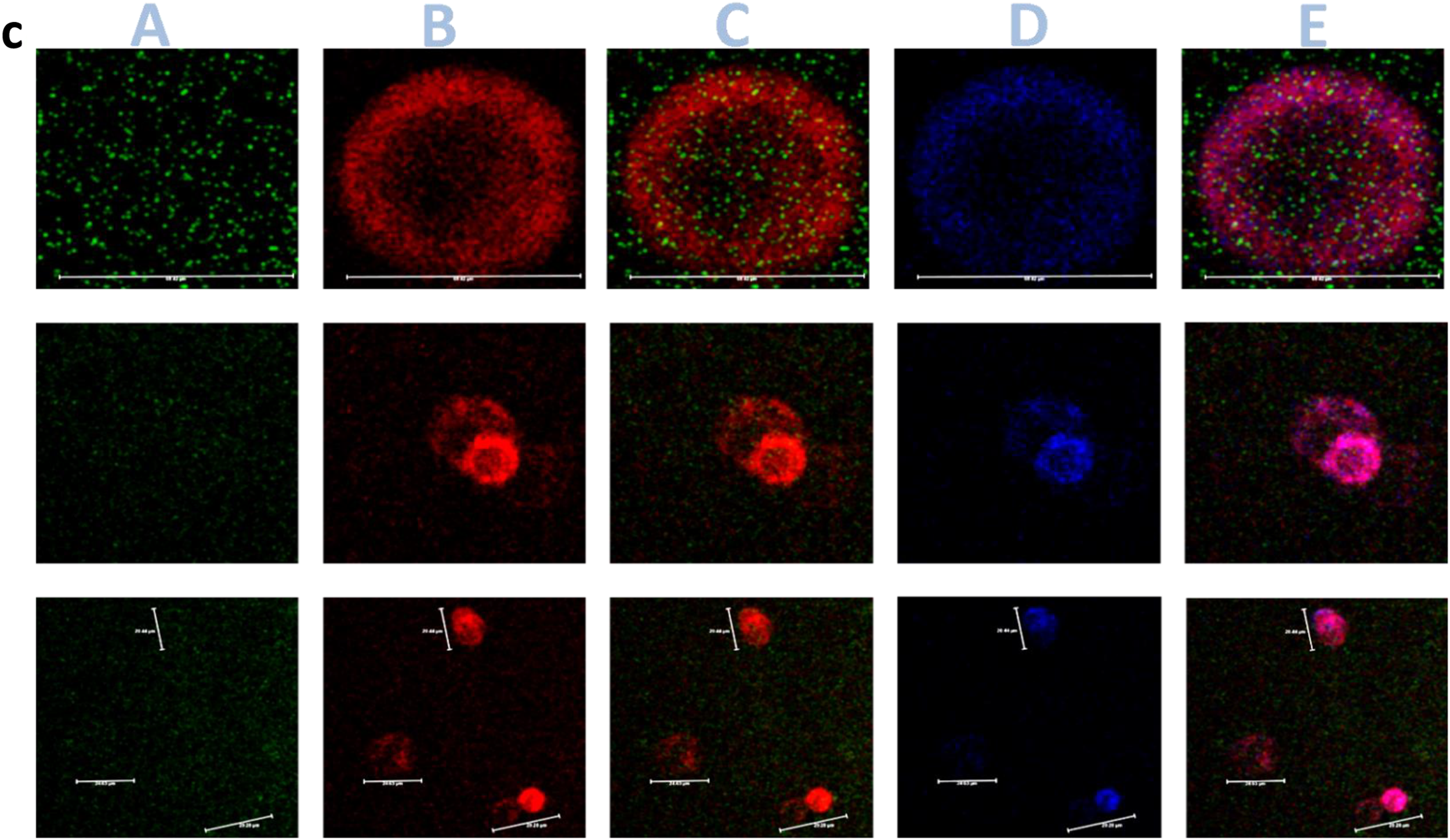
In order to assess for GAD65-CAR-Tregs homing to mouse pancreas in vivo (direct observation), a group of T1D mice were euthanized 24 h post GAD65-CAR-N-GFP+-Treg infusion. Flow-cytometry and confocal-image experiments demonstrated homing of GAD65-CAR-N-GFP+-Tregs to pancreas peri-insular areas 24 h after intravenous infusion (Fig. 5). Tracking of GAD65-CAR-N-GFP+-Treg cells in PB, PLN, PN and spleen (SP) by flow cytometry (Fig. 5a. cartoon summary; 5 b. raw data), demonstrated the selective presence of the GAD65-CAR-N-GFP+-Tregs in the pancreas (PN, Fig. 5b., Row 3). Although GFP labeled GAD65-CAR-N-Tregs were also detectable in the periphery (Blood, Fig. 5b, Row 1 and Spleen, Row 4), they did not accumulate in peri-PLN (PLN Fig. 5b, Row 2) at 24 h post-infusion as expected (GAD65 is not expressed in lymph nodes). Figure 5c shows morphology of pancreatic islets and localization of GAD65-CAR-N-GFP+-Treg cells in peri-islet areas on direct confocal immunofluorescence imaging (3 D iDISCO imaging). The homing of GAD65-CAR-N-GFP+-Tregs to the islets of Langerhans 24 h after intravenous injection demonstrates the redirectionality that the specific CAR, GAD65 paratope, provides to GAD65-CAR-N-Tregs toward GAD65 autoantigen localization (i.e., pancreatic islets). An enlarged image of an islet (Supplementary Fig. S1) demonstrates the overlay of GAD65 and insulin expressing beta cells (purple) interacting with GAD65-CAR-N-GFP+-Tregs (green).
Reversal of glucose intolerance and diabetes after GAD65-CAR-Treg treatment (preclinical trial)
GAD65-CAR-Tregs were next assessed along with unmodified (normal) Tregs or Tregs expressing irrelevant CAR control (EPCAM) for efficacy/superiority in preventing/treating T1D in T1D mice. Four groups of 6–8 week-old T1D mice (n = 7–8 each) were first tested for glucose tolerance (GTT) and glucose-mediated insulin secretion. The results (pre-CAR Treg infusion) were similar for all groups and abnormal as expected for T1D mice. All animals showed signs of glucose intolerance and there were non-significant variations among the groups (not shown).
The four groups of T1D mice were subsequently infused with 5 million Treg cells as follows:
Group 1: GAD65-CAR-N-Tregs,
Group 2: GAD65-CAR-M-Tregs,
Group 3: EPCAM-CAR-Tregs (irrelevant CAR control),
Group 4: Unmodified (normal) Tregs.
GTTs were also performed 15 and 30 days post-CAR Treg/Treg cell infusion. In addition, fasting glucose and insulin secretion tests were specifically performed at the 0, 10th, 20th, and 30th day of all Treg treatments.
The cartoon illustration in Figure 6a summarizes the results of the 30-day GAD65-CAR-Tregs/normal (unmodified) Tregs/EPCAM-CAR-Treg preclinical trial in T1D mice. GTT significantly worsened in control (EPCAM-CAR, Group 3) and unmodified Treg (normal Treg, Group 4)-treated Groups as compared with GAD65-CAR-N/M-Treg-treated Groups 1 and 2 (Fig. 6b). Specifically, GTT results for GAD65-CAR-M versus EPCAM-CAR-Treg-treated groups, showed significantly lower blood glucoses at 60 min after glucose challenge, while GTT in GAD65-CAR-N versus EPCAM-CAR-Treg-treated groups showed significantly lower blood glucoses at 30-, 60-, 90- and 120-min postglucose challenge (Fig. 6b). Of note, unmodified (normal, Group 4) Tregs were ineffective on reversing glucose intolerance (also on Fig. 6b). Fasting blood glucoses were already significantly lower in GAD65-CAR-N/M-Treg treated groups at 20- and 30-days postinfusion (Fig. 6c, right panel). In addition, fasting insulin secretion tests were significantly worse for Group 3 as compared with Groups 1 and 2. Fasting insulin secretion significantly improved as early as 20 days post GAD65-CAR-N/M-Treg (Groups 1 and 2) treatment as compared with Group 3 EPCAM-CAR control (Fig. 6c left panel).
Pancreatic islets are rescued/regenerated with GAD65-CAR-Treg treatment (preclinical trial)
At 30 days, all mice were euthanized, and half of the pancreases were processed for morphological analyses. Unmodified (normal) Treg as well as control EPCAM-CAR-Treg treated pancreases showed few, if any, healthy islets while GAD65-CAR-Treg treated pancreases appeared almost completely recovered. As shown in Figure 6d and Supplementary Fig. S2, healthy islets of Langerhans were only present in GAD65-CAR-Treg-treated mice (blue circles). GAD65-CAR-M-Treg-treated pancreases still showed some lymphocyte-infiltrated islets at 30 days (black circles). Importantly, rescued/regenerated pancreatic islets in GAD65-CAR-Treg-treated mice explained the normalization of glucose homeostasis demonstrated by the GTT and fasting insulin tests’ recovery at 30 days. Pancreatic sections were next subjected to morphological analysis of insulitis and score-graded (1-best, 4-worst). Untreated controls as well as EPCAM-CAR-Treg and unmodified (normal) Treg treated animals had insulitis scores graded at or near 4. GAD65-CAR-Treg treatment mostly reversed the insulitis (Fig. 6e and f).
Spleen, peri-PLN, and pancreas lymphocytic profile confirms therapeutic response (preclinical trial)
As mentioned above, 30-days post-trial, animals were euthanized and immune organs as well as half of the pancreases were processed into single cell suspensions. Single cells were stained with fluorochrome-conjugated antibodies for immune cell profiling. Lymphocytes from all euthanized mice were characterized for their functional status. Specifically, using flow-cytometry defined markers of cytotoxicity and regulation, we characterized SP, PLN, and pancreas (PN) infiltrating lymphocytes. This immune-cell profiling revealed that T1D mice treatment with beta-cell specific, GAD65-CAR-Tregs led to enrichment of Treg cells at the pancreas level (Fig. 6g, Row 1). This Treg enrichment was also statistically significantly higher in the spleen (SP, GAD65-CAR-N-Treg-treated Group 1, Fig. 6g, Row 1). Furthermore, the consequent downregulation of T helper (Th) 1 (Fig. 6g, Row 2), and Th17 (Fig. 6g, Row 3) responses at SP, PLN (site of T cell activation), and PN level (site of cytotoxic execution in case of T1D) correlated with disease recovery post GAD65-CAR-N/M-Treg treatment. Of note, IFNg producing CD3 population was significantly reduced in PLN of the GAD65-CAR-M-Treg-treated group as well (Fig. 6g, Row 4).
Discussion
The successful use of CAR technology for the generation of antigen-specific CTLs for the treatment of cancer 5 suggests that a similar approach could be used to generate autoantigen-specific CAR-Tregs for the treatment of autoimmune diseases and transplants’ tolerance. Moreover, CAR-Tregs should be superior to naïve Tregs since CAR-Treg activation is MHC-independent, i.e., CAR-Tregs directly recognize autoantigens, activate themselves, and robustly proliferate without the need of APCs.
The creation of HLA-A2-specific CAR (A2-CAR)-Tregs and their application in the generation of antigen-specific human CAR-Tregs was first reported by MacDonald et al., just few years ago. 28 In vitro, A2-CAR-expressing Tregs maintained their expected phenotype and suppressive function before, during, and after A2 stimulation. In mouse models, A2-CAR-Tregs were superior to Tregs expressing an irrelevant CAR at preventing xenogeneic graft-versus-host disease caused by HLA-A2+ Tresp cells. On the other hand, Tenspole et al. generated insulin-specific CAR-Tregs using phage-display technology. 29 However, the insulin-specific CAR-Tregs were unable to prevent/cure spontaneous diabetes in NOD/Ltj females despite their presence reported after 4 months of adoptive transfer. 29
Ours is the first report on GAD65-specific CAR-Tregs for T1D prevention/treatment. For treating T1D/LADA, we selected GAD65 paratopes mostly because GAD65 autoantibodies are present in the majority of patients years before the onset of disease (reviewed). 1 We reasoned that if human autoantibodies are capable of reaching the target autoantigen in beta cells, CAR constructs that mimic those autoantibodies would do the same. We purposely chose for the selection of paratopes, immunodominant areas of the antigen that are either close to the membrane anchoring domain (GAD65-CAR-N-Treg) or in a hydrophilic region of the monomeric form of the autoantigen (GAD65-CAR-M-Treg). The immunodominance of these two GAD65 regions has been previously documented.13,14 Although GAD65 autoantibodies are a hallmark biomarker in T1D, their prevalence is not universal, ranging from 60% to 80% in most cohorts when assessed with other autoantibodies like IA-2A, IAA, ICA, or ZnT8A. However, leveraging the phenomenon of bystander suppression where CAR-Tregs inhibit immune responses via cytokines like IL-10, IL35, or cell modulation, helps justify GAD65 targeting as a viable option even for GAD65 autoantibodies negative patients.
Engineered human GAD65-CAR-Tregs efficiently homed to human pancreatic islets and proliferated after GAD65 antigen recognition. The homing of the GAD65-CAR-N-Tregs to the islets of Langerhans provided evidence for antigen specific re-directionality. Live immunofluorescence microscopy demonstrated the distinct homing of GAD65-CAR-N-human-Tregs to human pancreatic islets as early as 24 h of co-culture. More importantly, proliferation of human GAD65-CAR-N-Tregs was clearly demonstrated within 76 h of co-culture. Since the activation of GAD65-CAR-N-Tregs was not MHC-restricted, their immunosuppressive efficiency was therefore superior to that of naïve Tregs. GAD65-CAR-N-Treg cytokine profile from the co-culture supernatant was characteristic of activated Tregs (not shown).
These same GAD65-CAR-N-Tregs were able to immunosuppress diabetogenic cytotoxic T cells in situ when co-cultured with human islets. GAD65-CAR-N-Tregs seem to first recognize GAD65 protein present in the islets; activated themselves; robustly proliferate and suppress autologous IFNg-producing CD8 T cells (CTLs), which are considered killer T cells in T1D/LADA. Just with this human experimental data, we could speculate that GAD65-CAR-Treg may induce tolerance against diabetogenic T cells and reverse T1D/LADA. Moreover, this strategy could also be adopted for human islet transplantation to reduce rejection and maintain the longevity of transplanted islets. Therefore, we wanted to address these questions in an animal model as well.
In vitro suppression assays using mouse GAD65-CAR-N-Treg and mouse Tresp (T responsive) cells confirmed the powerful suppressive effect of mouse GAD65-CAR-N-Tregs over activated mouse Tresp cells in a dose-responsive manner. As shown, mouse GAD65-CAR-N-Treg suppression of Tresp was observed even at a 1:8 ratio (1 mouse GAD65-CAR-Treg to 8 mouse Tresps). Moreover, in vivo proliferation of GAD65-CAR/GFP+-mouse-Tregs in comparison with unmodified (normal) Treg and irrelevant EPCAM-CAR/GFP+-mouse-Tregs, demonstrated that the replication index of GAD65-CAR/GFP+-Tregs was 3–6 times higher in just 48 h. This illustrates the specificity of GAD65-CAR-Tregs and their proliferative capacity in an antigen-specific manner in vivo as well. In addition, the functional superiority of GAD65-CAR-Tregs over normal Tregs was demonstrated in vivo. Although normal Tregs isolated from spleen of T1D mice were responsive to GAD65, their replication index was about 1/3rd that of genetically engineered GAD65-CAR-Tregs.
Antigen-specific GAD65-CAR-Tregs were used for T1D prevention/treatment trial in the mentioned T1D mouse model that closely resembles the human disease (preclinical trial). 15 The recovery from insulitis/diabetes in GAD65-CAR-Treg-treated animals was indicative of cytotoxic T cell suppression. This could be due to either bystander effect or direct interaction between the GAD65-CAR-Tregs and T effector cells. As shown, the antigen-specific proliferative capacity of GAD65-CAR-Tregs was 4–5 times higher than that of unmodified (normal) Tregs. This provides “in vivo” evidence for functional superiority of GAD65-CAR-Tregs over normal Tregs on their diabetes reversal capacity observed in the GAD65-CAR-N/M-Treg-treated groups. Differential enrichment of Tregs (4–5 folds) at the site of immune activation (PN) in the GAD65-CAR-N/M-treated groups represents local GAD65-CAR-N/M-Tregs activation and proliferation. This also validates the functional stability of GAD65-CAR-N/M-Tregs at 30-day post-treatment. The effectiveness of GAD65-CAR-N/M-Tregs therapy is reflected in the rescuing of the islets from cytotoxic immune attack, which led to improved insulin release, GTT, and reversal to normal fasting blood glucose levels. No adverse events were observed along the preclinical trial although none of the cytokines produced by Tregs (i.e., short-lived adenosine; potent anti-inflammatory IL10 and IL35; and tumor suppressor-TGF-beta) are known to be toxic (as opposed to the known CAR T cytotoxic cell-induced cytokine release syndrome when nonregulatory T cells are used for cancer treatment). 5 Of note, since GAD65 is expressed in GABAergic neurons, we monitored for neurological complications which we did not observe. However, since we were working with beta-cell-specific transgenics, limited off-target effects in this model system were expected.
This is the first time a human CAR-Treg was constructed against GAD65 autoantigen from T1D/LADA patients, which could successfully recognize the target antigen and home to pancreatic islets in exvivo condition. 30 GAD65-CAR-Treg immunotherapy was successful in suppressing the diabetogenic T cells from T1D/LADA patients in ex vivo condition as well as abrogating the diabetes phenotype in an animal model of T1D. 31 Likely, Treg re-direction using antigen-specific GAD65-CAR-Tregs capable of recognizing immunodominant regions of this clinically relevant autoantigen and consequent CTL downregulation, will allow for recovery and reconstitution of beta cells in human patients as well. Next step would be to extend these observations beyond 30 days. Therefore, a 6-month preclinical trial for safety and long-term efficacy ought to follow.
Authors’ Contributions
S.I. contributed to the design of the work, the acquisition, analysis, interpretation of data and drafting the article. P.D. contributed to the design of the work, the acquisition of data. S.I.R. contributed to the acquisition and analysis of data and drafting the article. M.A.A.J. contributed to the design of the work and interpretation of data. A.A.-K. contributed to acquisition and analysis of data. A.J. contributed to acquisition and analysis of data. K.A.S. contributed to subject recruitment and procuring human pancreatic tissues. T.A.T.-R. contributed to subject recruitment and procuring human pancreatic tissues. N.S. contributed as a clinical research coordinator in recruitment and procuring human blood and pancreatic tissues. J.C.J. conceived the design of the work, contributed to the acquisition, analysis, interpretation of data, drafted and finalized the article. J.C.J. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Acknowledgments
Dr. Andrea L. Kalinoski, PhD., Technical Director Integrated Core Facilities at the University of Toledo for confocal microscopy.
Author Disclosure Statement
No potential conflicts of interest relevant to this article were reported.
Funding Information
This work was funded by unrestricted funds from the University of Toledo (J.C.J. and S.I.) and Helmsley Charitable Trust (J.C.J.). The Leona M. & Harry B. Helmsley Charitable Trust Grant #2206-05333.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.