
Abstract
Aims:
The study aimed to determine if the Kindlin-2/Otub1/Slc7a11 cascade could improve cardiac ischemia reperfusion injury by inhibiting ferroptosis.
Results:
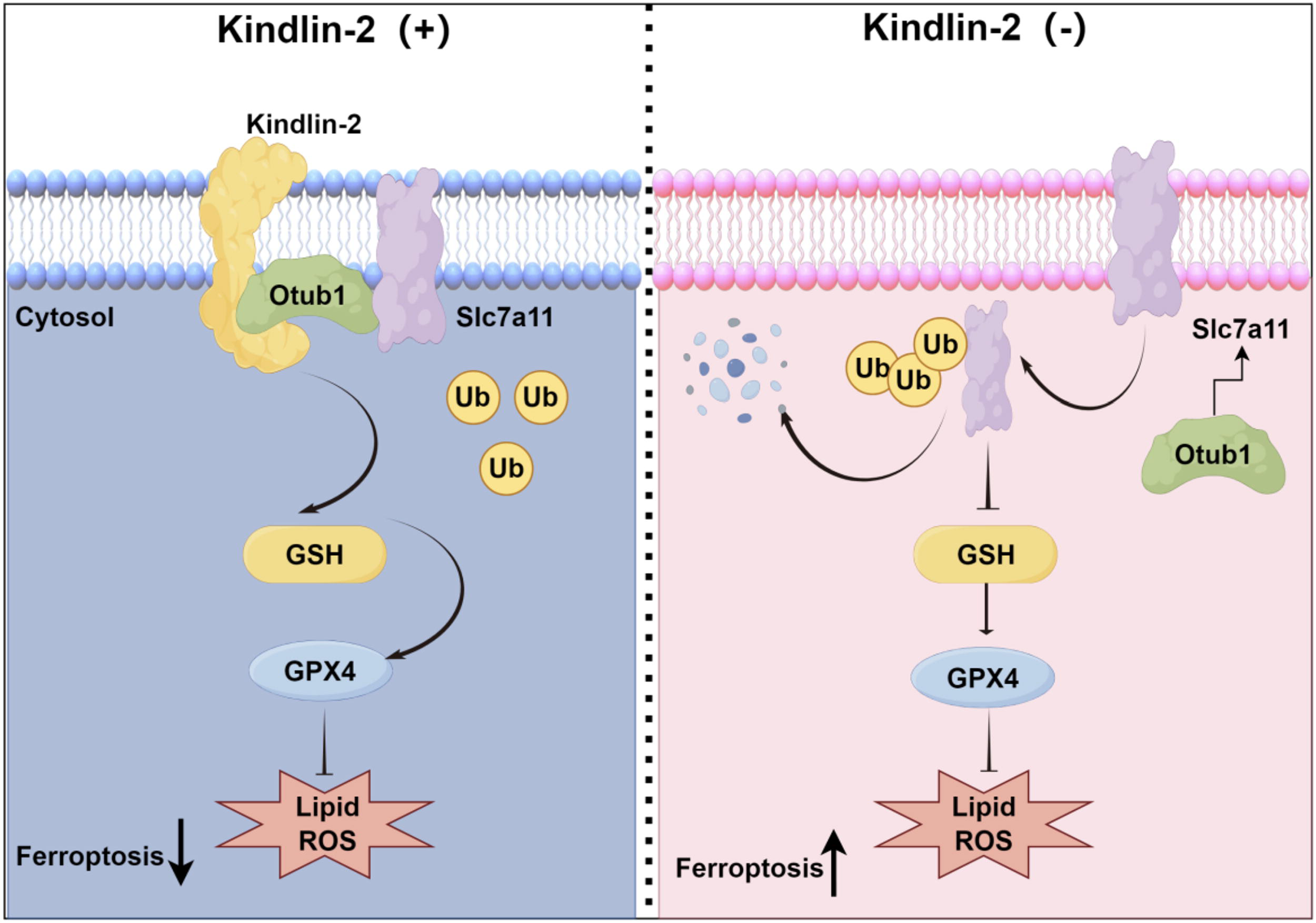
The cardiac tissues of ischemia — reperfusion (I/R) mice, ischemic cardiomyopathy (ICM) patients, and cardiomyocytes underwent hypoxia/reoxygenation stimulation, and the Kindlin-2 levels decreased. Cardiomyocyte-specific Kindlin-2 overexpression alleviated I/R injury by inhibiting cardiomyocyte ferroptosis in vivo while cardiomyocyte-specific low expression of Kindlin-2 impaired cardiac functions, and this was accompanied by cardiomyocyte ferroptosis and reversed by Fer-1. In addition, in vitro experiments verified that Kindlin-2 prevented ferroptosis in cardiomyocytes treated with hypoxia/reoxygenation. An endogenous Kindlin-2 deficiency in cardiomyocytes was subsequently identified to spontaneously induce ferroptosis without exogenous stimulation, which is also prevented by Fer-1. Mechanistically, Kindlin-2 accelerated the interaction between Otub1 and Slc7a11. Consequently, deubiquitinated Slc7a11 contributed to the activation of glutathione (GSH) and Gpx4 to exert the anti-ferroptosis effect. Slc7a11/GSH/Gpx4 cascades strengthened by Kindlin-2 were abolished by Otub1 knock down. Moreover, Otub1 rescued cardiomyocyte ferroptosis and cardiac injury due to the Kindlin-2 deficiency.
Innovation:
Kindlin-2 accelerated the interaction between Otub1 and Slc7a11. Therefore, Slc7a11/GSH/GPX4 cascades were reinforced to improve the deteriorated tissues of I/R hearts by ameliorating ferroptosis.
Conclusions:
Our research revealed that the Kindlin-2/Otub1/Slc7a11 cascade improved cardiac I/R injury by inhibiting ferroptosis; hence, it may be a potential therapeutic target for ICM. Antioxid. Redox Signal. 43, 727–744.
Introduction
Globally, over 17 million people die from cardiovascular diseases every year, and over 50% of the death toll is triggered by myocardial infarction (Laslett et al., 2012; Sacks et al., 2017). Coronary blood reperfusion is extensively applied in clinical treatment to restore the perfusion of ischemic cardiac tissues to restrict the attack of acute myocardial infarction (AMI) (Guo et al., 2023). This is considered the most effective approach to alleviate symptoms and improve the AMI prognosis, but reperfusion can also deteriorate cardiac trauma, and this is termed ischemia — reperfusion (I/R) injury (Zhang et al., 2024). The mitochondrial electron transport chain is disrupted by ischemia stimulation. After the reperfusion of the ischemia area, molecular oxygen brought by fresh blood binds electrons to generate reactive oxygen species (ROS) that further damage cardiac tissue. Moreover, elevated iron was found in the infarct areas that underwent reperfusion therapy (Fang et al., 2019). Based on this, cardiomyocyte ferroptosis is primarily referred to as the significant pathogenesis of I/R injury.
The responses to ischemia stimulation include regulated cell death such as apoptosis, necrosis, and autophagy, which mediate cardiac impairment (Kung et al., 2011; Wu et al., 2021; Yaoita et al., 2000). Artificial ligation of the left coronary artery of mice and apoptosis of cardiomyocytes occur within four hours of reperfusion after ischemia. However, during the late stage of I/R injury (1–7 days), cardiomyocytes primarily undergo ferroptosis, the iron-dependent regulated cell death induced by superfluous lipid peroxidation derived from redox imbalances (Feng et al., 2019). Therefore, ferroptosis of cardiomyocytes is a crucial target for protection against cardiac I/R injury. The solute carrier family 7 member 11 (Slc7a11) is the cystine/glutamate reverse transporter, and it mediates extracellular cystine intake to exchange glutamate. The synthesis of glutathione (GSH), the cofactor of the antioxidant enzyme glutathione peroxidase 4 (GPX4), is then promoted to eliminate the accumulated lipid peroxides and inhibit ferroptosis (Hong et al., 2021; Li et al., 2020; Ye et al., 2022). Otub1 on the cytomembranes is well established to bind to and stabilize Slc7a11 because it is an ovarian tumor (OTU) family member deubiquitinase. Conversely, OTUB1 inactivation induces ferroptosis by reinforcing the ubiquitination of SLC7A11 (Liu et al., 2019).
Innovation
Our results revealed that the deteriorated I/R heart tissues can be prevented by Kindlin-2, which is manifested as ameliorative ferroptosis. Moreover, cardiomyocytic ferroptosis induced by Kindlin-2 deficiency can be mostly rescued by superfluous Otub1. However, the enhancing effect of Kindlin-2 on the Slc7a11/GSH/Gpx4 pathway can be abolished by Otub1 inhibition.
The focal adhesion protein, Kindlin-2 (gene name Fermt2), is evolutionarily conserved. It modulates various cellular functions, such as differentiation, migration, and extracellular matrix (ECM) adhesion, under physiological and pathological conditions by functioning on diverse signaling pathways (Cao et al., 2020; Dong et al., 2023b; Gao et al., 2022; He et al., 2020; Wu et al., 2015; Zhu et al., 2020). The roles of Kindlin-2 in regulated cell death have also been extensively studied in different organs. Kindlin-2 improves the apoptosis of nucleus pulposus cells by inhibiting inflammasomes (Chen et al., 2022). Kindlin-2 binds to P53 and affects downstream genes P21 and Sperpin to prevent senescence in chondrocytes (Wu et al., 2022). Kindlin-2 also binds to the cytoplasmic kinase domain of TGFβ and increases the secretion of downstream Colony Stimulating factor 1 (CSF-1). Consequently, the apoptosis of breast cancer can be avoided (Sossey-Alaoui et al., 2017). However, the involvement of Kindlin-2 in ferroptosis has not been studied. Progressive heart failure can be alleviated by the activation effect of Kindlin-2 on Integrin β1 in cardiac tissues (Zhang et al., 2016). Additionally, Kindlin-2 interacts with SUV39H1 and recruits this histone methyltransferase to the promotor of GATA4 (Qi et al., 2019). The marker gene of cardiac hypertrophy transcriptionally decreases, and the symptom is relieved. In summary, the cardiac protection effect of Kindlin-2 has been gradually investigated. However, the improvement of Kindlin-2 during cardiac I/R and the underlying mechanism remain unclear.
We discovered in this study that the mRNA and protein levels of Kindlin-2 in ferroptotic cardiomyocytes were reduced in vivo and in vitro. The anti-ferroptotic effect of Kindlin-2 was potently identified in cardiac I/R injury and H/R-treated cardiomyocytes. Cardiac ferroptosis occurs spontaneously in the absence of Kindlin-2. Mechanistically, Kindlin-2 contributed to the interaction between Otub1 and Slc7a11, and this results in the stabilization of Slc7a11 and activation of GSH and Gpx4 to prevent ferroptosis, as shown in Figure 1. Our conclusions indicated that the Kindlin-2/Otub1/Slc7a11/GSH/Gpx4 axis is a potential approach to cure cardiac I/R injury.
Results
Kindlin-2 is expressed in cardiomyocytes
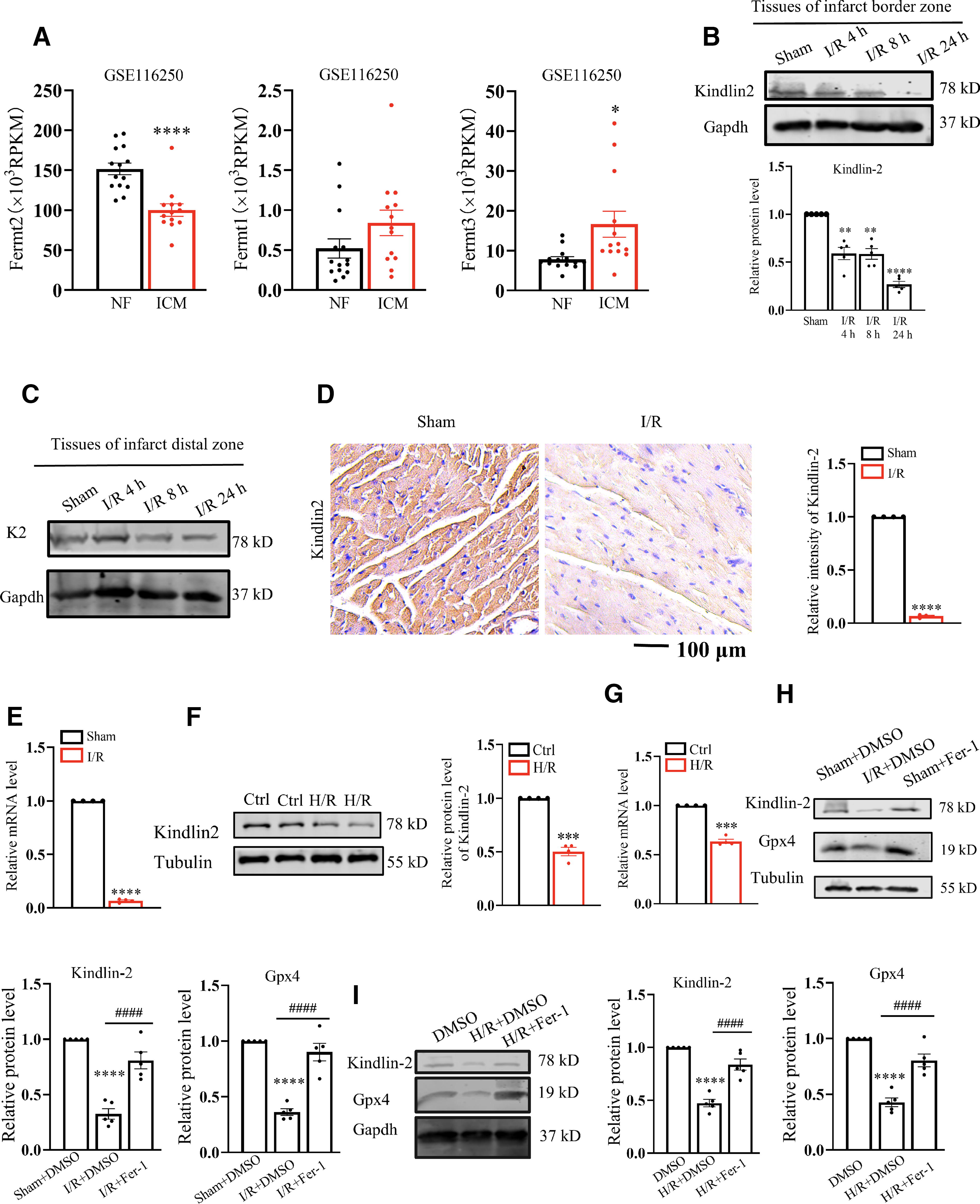
As members of the Kindlin family, Kindlin-1 is primarily expressed in epithelial cells, and hematopoietic cells are rich in Kindlin-3. Kindlin-2 is extensively distributed throughout the body compared to the other members, except in blood cells. Therefore, in most cases, the Kindlin-2 level is much higher than that of Kindlin-1 and Kindlin-3 (Rognoni et al., 2016). We analyzed human ischemic cardiomyopathy (ICM) samples to decipher the cardiac expression profile of the Kindlin (Fermt) family. The expression profiling of GSE116250 dataset was derived from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). As predicted, in healthy hearts, the cardiac Fermt2 level is approximately 20 times that of Fermt3 and 300 times that of Fermt1. Moreover, the cardiac Fermt2 expression, but not those of Fermt1 and Fermt3, is significantly reduced in ICM patients compared with normal individuals (Fig. 2A). Left ventricles of C57BL/6 mice were subjected to ischemia operation for 45 min. The mice were sacrificed at different time points (4 h, 8 h, and 24 h) after the reperfusion of the ischemia hearts. The Kindlin-2 protein levels in the infarct border zone, but not infarct distal zone, of the cardiac I/R mice decreased in a time-dependent manner compared to the left ventricular tissues in the sham group (Fig. 2B and C). The immunohistochemistry (IHC) and quantitative real-time polymerase chain reaction results also reflected the reduced protein and mRNA levels of Kindlin-2 in the infarct border zone of the cardiac I/R mice (reperfusion time was 24 h) (Fig. 2D and E). Next, a lower Kindlin-2 expression was also detected in isolated primary cardiomyocytes treated by hypoxia/reoxygenation (H/R) (Fig. 2F and G). To further decipher the potential effects of Kindlin-2 in cardiomyocytes, protein lysates of primary cardiomyocytes were collected to conduct a mass spectrum analysis. Compared with the IgG group, 1544 proteins were significantly enriched in the Kindlin-2 antibody group and identified to probably interact with Kindlin-2. The Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes pathway were analyzed in https://david.ncifcrf.gov/summary.jsp. We found that cardiac Kindlin-2 was highly correlated with regulated cell death, including ferroptosis (Supplementary Fig. S1A and S1B). Then I/R models were intraperitoneally injected with ferroptotic inhibitor Ferrostatin-1 (Fer-1), and the cardiac Kindlin-2 decline was recovered by Fer-1. The Gpx4 level was used as the positive control to manifest the inhibition of cardiac ferroptosis (Fig. 2H). Similarly, Kindlin-2 restoration also occurred in the in vitro H/R models treated by Fer-1 (Fig. 2I). In summary, cardiac ferroptosis was accompanied by reduced Kindlin-2. However, it remains unclear if cardiac Kindlin-2 prevents ICM by targeting ferroptosis.
Overexpression of cardiomyocyte-specific kindlin-2 improved I/R-induced cardiac ferroptosis
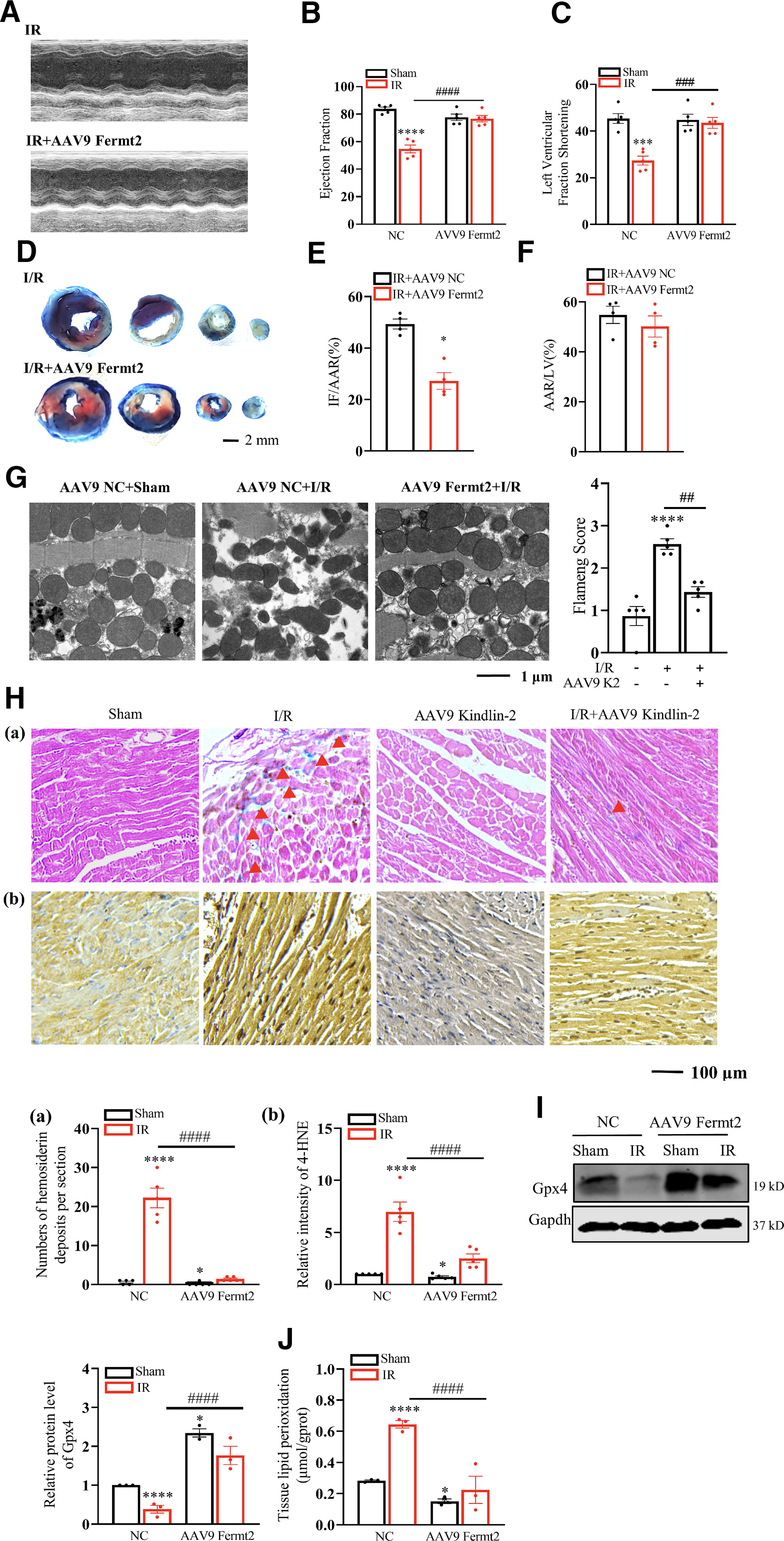
Kindlin-2 regulation on ischemic heart diseases was first examined in mouse models. We injected Adeno-Associated Virus type 9 (AAV9) Fermt2 to establish cardiomyocyte-specific Kindlin-2 highly expressed mice compared with the littermates that received AAV9 negative control (NC). Kindlin-2 reaches the peak at two weeks after the injection of AAV9 NC and AAV9 Fermt2 (Bilal et al., 2021). The protocol above and the AAV9 sequences are concretely described in Supplementary Fig. S2A. We then discovered that the Kindlin-2 and Gpx4 protein levels increased in cardiac tissues injected with AAV9 Fermt2 (Supplementary Fig. S2B). Moreover, as the marker of ferroptosis, 4-HNE in cardiac tissues was decreased by Kindlin-2 (Supplementary Fig. S2C). Therefore, the high expression of cardiac Kindlin-2 likely prevents ferroptosis. The mice were then subjected to cardiac I/R injury and echocardiographic analysis. We discovered that the decline in cardiac functions, such as the ejection fraction (EF%) and fractional shortening (FS%), of the cardiac I/R-injured mice was immensely relieved in the AAV9 Fermt2 mice, which was also manifested in the representative echocardiograph images (Fig. 3A–C). Double Evans Blue (EB) and triphenyl tetrazolium chloride (TTC) staining in the cardiac tissues of the I/R injury mice showed smaller post-I/R infarct sizes in the AAV9 Fermt2 group than in the AAV9 Scramble group (Fig. 3D–F). According to previous findings, an elevated mitochondrial membrane density and lessened mitochondrial size were observed in the cardiac tissues of the I/R model using transmission electron microscopy. Abundant cardiac Kindlin-2 has been found to dramatically mitigate distorted mitochondria after I/R (Fig. 3G). In vivo experiments have also verified that cardiomyocytes primarily undergo ferroptosis during the late stage of I/R injury (1–7 days) (Fang et al., 2019). Due to the previous results that displayed the concurrence between cardiac ferroptosis and Kindlin-2 declination, our purpose became to verify the potential effect of Kindlin-2 in cardiac ferroptosis. The remaining mice were sacrificed to obtain the infarction border zones of the left ventricles, and these were subjected to Prussian blue and 4-HNE staining (Fig. 3H). The results showed abundant Fe2+ deposition and 4-NHE level in the tissues of the cardiac I/R-injured mice that was also visibly abolished by Kindlin-2. Meanwhile, the decreased Gpx4 and increased lipid peroxidation in cardiac tissues of I/R injury can also be recovered using cardiac Kindlin-2 (Fig. 3I and J). In summary, the in vivo results proved that cardiac Kindlin-2 may attenuate I/R injury by repressing ferroptosis.
Kindlin-2 deficiency accelerates cardiac ferroptosis
The objective of this study is to prove the indispensability of Kindlin-2 for cardiac function maintenance. We injected AAV9 ShRNA Fermt2 to knock out Kindlin-2 in the cardiac tissues of mice (Kindlin-2 KO mice). The sequences of AAV9 shRNAs are shown in Supplementary Fig. S3A. Two weeks later, the Kindlin-2 KO mice displayed cardiac dysfunctions that are affirmed by the representative images of the echocardiographs (Supplementary Fig. S3B). There were declines in the EF and FS percentages in the Kindlin-2 KO mice in the absence of external stimuli (Supplementary Fig. S3C and S3D). More cardiac Fe2+ deposition and 4-HNE (Supplementary Fig. S3E) determined that cardiac ferroptosis can be aroused by insufficient Kindlin-2. Next, cardiac ferroptosis in the Kindlin-2 KO mice was indicated by the decreased Gpx4 and increased lipid peroxidation (Supplementary Fig. S3F and 3G). Altogether, the decreased functions and enhanced ferroptosis of mouse hearts can be reversed by Fer-1 administration.
Kindlin-2 attenuates cardiomyocyte ferroptosis
It is known that ferroptosis occurs in cardiomyocytes in vitro when exposed to H/R. To further investigate the fundamental roles of Kindlin-2 in cardiomyocyte ferroptosis, primary cardiomyocytes were transfected with the Kindlin-2 plasmid. This was followed by H/R treatment, which is well known to arouse cardiomyocyte ferroptosis in vitro. We found that reduced Gpx4 and increased mRNA level of Ptgs2 in H/R cardiomyocytes can be prevented by Kindlin-2 (Fig. 4A and 4B). In contrast, cardiomyocytes transfected with siRNA Kindlin-2 resulted in less Gpx4 and more Ptgs2 than the siRNA scramble, which can be prevented by Fer-1 (Supplementary Fig. S4A and S4B). The predominant marker of ferroptosis is lipid peroxidation, and it can be triggered by ROS and naturally generate malondialdehyde (MDA) that reflects the lipid peroxidation extent (Tang et al., 2021). Therefore, lipid ROS, cytosolic ROS, mitochondrial ROS, and MDA levels were measured. Moreover, on account of eliminating the accumulated lipid peroxides and inhibiting ferroptosis, GSH level was also tested. Both elevated inducement and termination products of lipid peroxidation triggered by H/R stimulation can be immensely restrained by Kindlin-2 (Fig. 4C–4G). Undoubtedly, cardiomyocytes lacking in Kindin-2 exhibited higher levels of lipid ROS, cytosolic ROS, mitochondrial ROS, and MDA and lower levels of GSH, which can also be recovered by Fer-1 (Supplementary Fig. S4C–S4G). In addition, attenuated cell viability in response to H/R can be alleviated by Kindlin-2 (Fig. 4H). The in vitro results demonstrated that the decisive effect of cardiac Kindlin-2 on anti-ferroptosis was clearly defined in this study.

Kindlin-2 alters Slc7a11 stability by modulating its ubiquitination
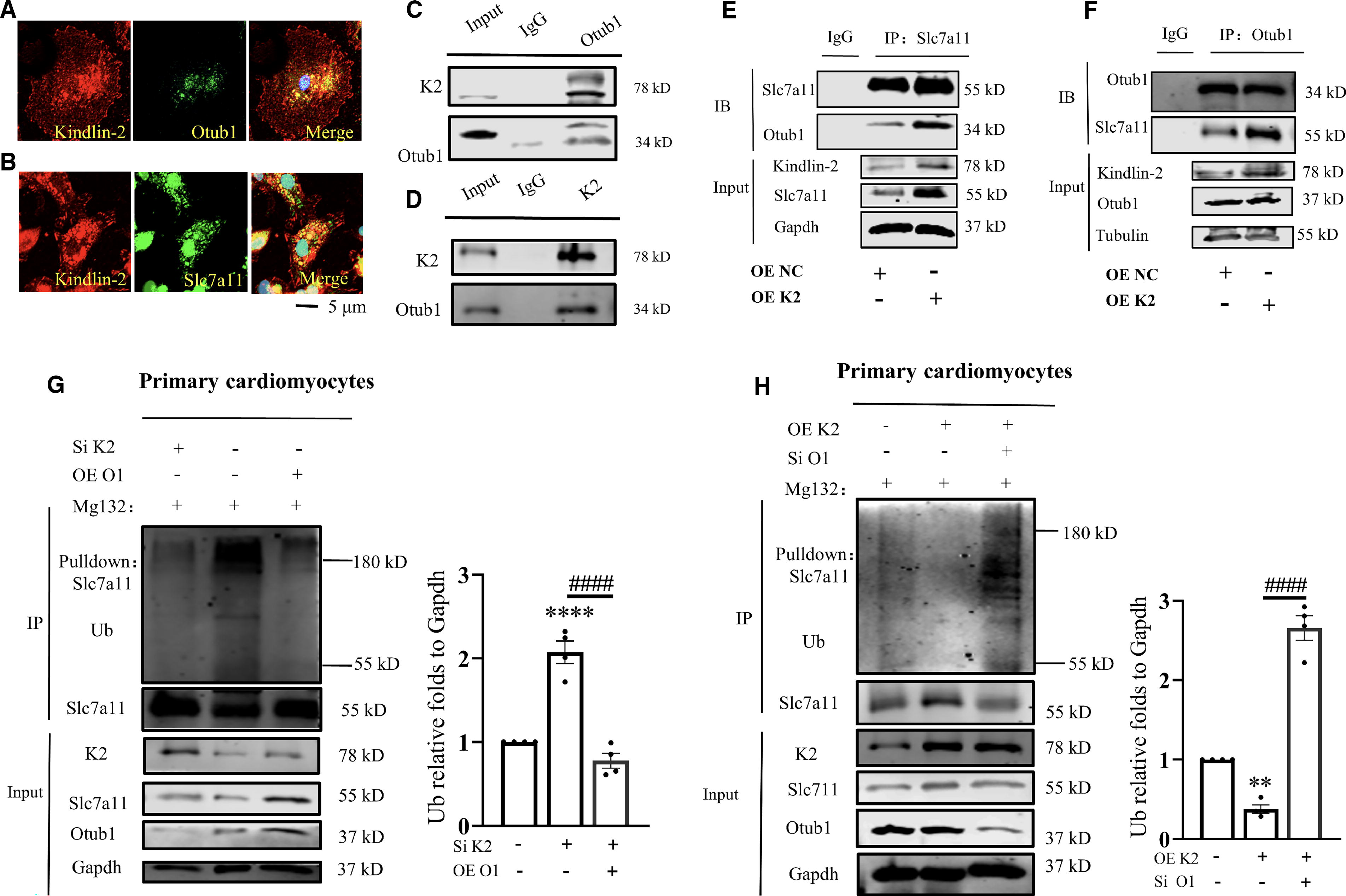
The ferroptotic inhibition capacity of cardiac Kindlin-2 was well established according to the preceding results. However, the underlying mechanism of the protective response above still requires further exploration. As the classical amino acid transporter, Slc7a11 is highly involved in I/R-induced cardiac ferroptosis (Ding et al., 2023; Fang et al., 2020). The results of previous publications and our result demonstrated that Slc7a11 and Kindlin-2 decreased in the cardiac tissues of I/R mice in parallel. Based on these observations, we determined whether Kindlin-2 modulated Slc7a11 expression in cardiomyocytes. The interaction between Kindlin-2 and Slc7a11 was predicted using AlphaFold. (Fig. 5A). A Co-Immunoprecipitation (Co-IP) assay further demonstrated the interaction between Kindlin-2 and Slc7a11 (Fig. 5B and 5C). Furthermore, Kindlin-2 KO drastically decreased the Slc7a11 protein level without affecting the Slc7a11 mRNA expression in cardiomyocytes and cardiac tissues (Fig. 5D–G). We then performed cycloheximide (CHX) experiments and found that Kindlin-2 markedly decreased the Slc7a11 protein stability in cardiomyocytes (Fig. 5H). Cardiomyocytes were then treated with MG132 (proteasome inhibitor) to verify the pathway that determined the Slc7a11 degradation.

Results showed that MG132 increased the Slc7a11 protein level that was knocked down, although Kindlin-2 was knocked out (Fig. 5I). Therefore, Kindlin-2 KO-induced Slc7a11 may rely on the proteasomal pathway. Slc7a11 protein polyubiquitination was analyzed to further verify if Kindlin-2 modulated Slc7a11 degradation through proteasome degradation. Results showed that Kindlin-2 KO increased the Slc7a11 polyubiquitination levels in cardiomyocytes (Fig. 5J). In contrast, Kindlin2 overexpression (OE) reduced the polyubiquitination in cardiomyocytes (Fig. 5K).
Kindlin-2 inhibits Slc7a11 ubiquitination through Otub1
The GO analysis showed that Kindlin-2 may function in a ubiquitin-dependent pathway. As with deubiquitinase, Otub1 binds to and stabilizes Slc7a11 on the cell membrane to inhibit ferroptosis (Liu et al., 2019). We next investigated if Otub1 was involved in Kindlin-2-inhibited Slc7a11 polyubiquitination. Immunofluorescence assays showed the colocalization between Kindlin-2 and Otub1/Slc7a11 in cardiomyocytes (Fig. 6A–B). The interaction between Kndlin-2 and Otub1 was then confirmed using Co-IP assays (Fig. 6C–D). However, the protein and mRNA level of Otub1 were not affected by the Kindlin-2 deficiency (Supplementary Fig. S5A and B). We then discovered that Kindlin-2 OE increased the interaction between Otub1 and Slc7a11 (Fig. 6E–F). In contrast, Kindlin-2 KO reduced the binding capacity of Otub1 and Slc7a11 (Supplementary Fig. S5C and S6D). Cardiomyocytes treated with the Otub1 SiRNA manifested decreased Slc7a11 and Gpx4 protein levels without altering Kindlin-2 (Supplementary Fig. S5E). Therefore, Slc7a11 stability regulated by Kindlin-2 may be determined by the interaction between Otub1 and Slc7a11. Furthermore, Kindlin-2 KO dramatically elevated Slc7a11 ubiquitination that was blunted by Kindlin-2 (Fig. 6G). Conversely, Kindlin-2 could not prevent Slc7a11 ubiquitination if Otub1 was knocked down (Fig. 6H). Altogether, the Otub1/Slc7a11 cascade may be the downstream outcome of Kindlin-2, which should be verified in a following study.
Anti-ferroptosis effect of cardiac kindlin-2 was abolished by Otub1 deficiency
Previous study results have demonstrated the known interaction and protection effect of Otub1 on Slc7a11 that relies on Kindlin-2. We then investigated if Otub1 was the downstream target of Kindlin-2. Tail vein injections of AAV9 Fermt2 and AAV9 Sh RNA Otub1 in the C57BL/6 mice were performed. After two weeks of treatment, I/R models were established, and representative images of the echocardiograph, EF%, and FS% of the left ventricle implied no recovery effect of Kindlin-2 on declining cardiac functions caused by I/R operation if Otub1 was inhibited (Fig. 7A–C). Increased infarct areas, distorted mitochondria, Fe2+ deposition, 4-HNE level, and lipid peroxidation in I/R mice lacking Otub1 were also not improved by Kindlin-2 (Fig. 7D–I).
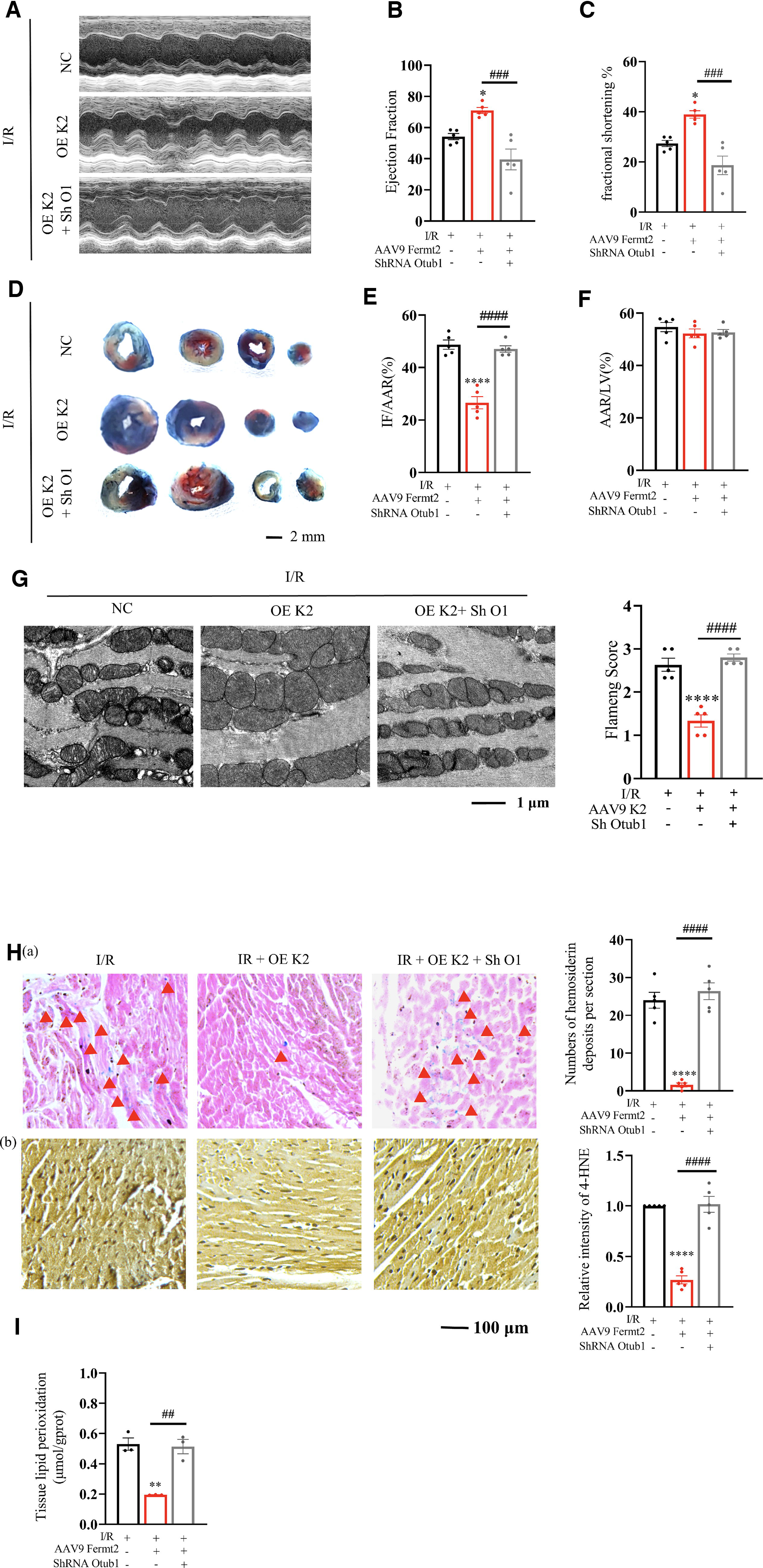
The in vitro experiments showed that due to the advantage of reinforced Slc7a11/GSH/Gpx4, cardiomyocytes that overexpressed Kindlin-2 exhibited strong resistance to H/R-induced ferroptosis, such as attenuated mRNA level of Ptgs2, lipid ROS, cytosolic ROS, mitochondrial ROS, and MDA. However, there was almost no inhibitory effect of Kindlin-2 in ferroptosis when Otub1 was disrupted (Supplementary Fig. S6A-G). We conclude that the protective effect of Kindlin-2 on cardiac ferroptosis and weakened functions highly depends on the existence of Otub1.
Increased Otub1 level reversed cardiac ferroptosis caused by kindlin-2 deficiency
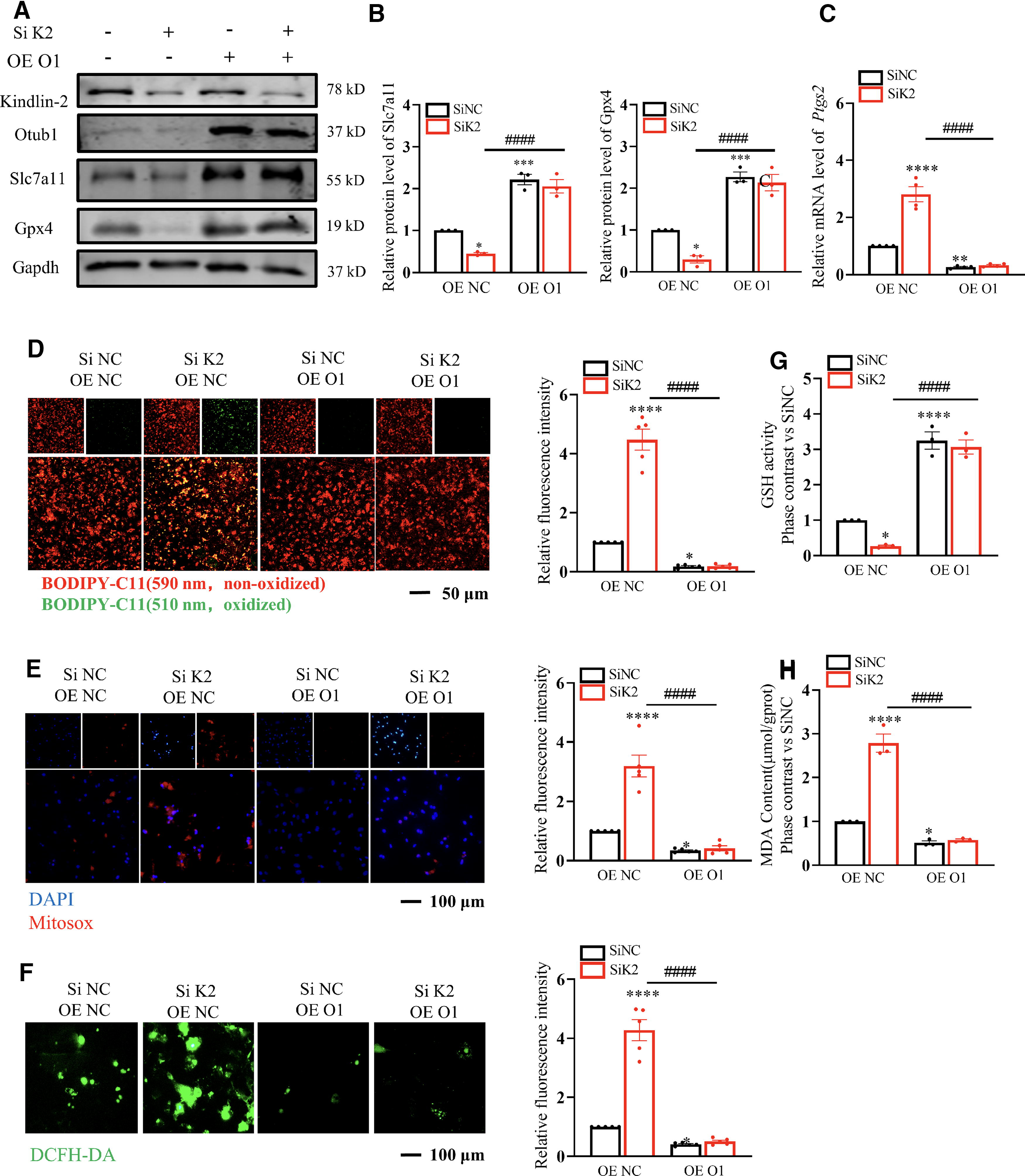
The anti-ferroptotic effect of cardiac Kindlin-2 was forcefully confirmed by intensifying Otub1/Slc7a11 cascades. Next, we examined if excessive Otub1 could reverse ferroptosis preceded by Kindlin-2 deficiency. First, we simultaneously transfected the Otub1 plasmid and Kindlin-2 siRNA into cardiomyocytes for 48 h. Results showed that the declination of Slc7a11 and Gpx4 and the increased mRNA level of Ptgs2 caused by Kindlin-2 knockdown were reversed by cardiac Otub1 (Fig. 8A–C). Subsequently, we also demonstrated the superabundant MDA, lipid ROS, cytosolic ROS, mitochondrial ROS, and decreased GSH level produced by the absence of Kindlin-2 could also be eliminated by Otub1 OE (Fig. 8D–H). Collectively, the in vitro results indicated that elevated Otub1 partially rescued the ferroptosis caused by Kindlin-2 deficiency, while the artificially reduced Kindlin-2 could not be recovered by the involvement of Otub1. The following tasks were focused on in the in vivo experiments. We injected both AAV9 Kindlin-2 shRNA and AAV9 Otub1 (Supplementary Fig. S7A) into adult mice. Representative images of the echocardiographs, EF%, and FS% of the left ventricles denoted the remarkable recovery effect of Otub1 on cardiac impairment induced by scarce Kindlin-2 (Supplementary Fig. S7B-7D). Moreover, in the Kindlin-2 KO group, the increased 4-HNE, Fe2+ deposition, lipid peroxidation, and decreased cardiac Gpx4 in cardiac tissues can also be mitigated by Otub1 (Supplementary Fig. S7E-G). In summary, Kindlin-2 prevented cardiac ferroptosis by consolidating the Otub1/Slc7a11/GSH/Gpx4 axis.
Kindlin-2 interacts with HIF-1 α in cardiomyocytes
Promoted stiffness and collagen biogenesis in invasive breast cancer benefit from abundant Kindlin-2 and Hypoxia-inducible factor 1α (HIF-1α). Furthermore, HIF-1α was demonstrated to interact and positively regulate Kindlin-2 in MCF (human breast cancer cells) (Xue et al., 2020). We also observed the interaction between HIF-1α and Kindlin-2 in cardiomyocytes (Supplementary Fig. S8A and B). In addition, under H/R stimulation, protein level of HIF-1α and Kindlin-2 in cardiomyocytes were decreased (Supplementary Fig. S8C). Combining our job with previous studies, deficiency of HIF-1α and Kindlin-2 is probably involved in cardiomyocyte ferroptosis under H/R stimulation.
Discussion
Kindlin-2 functions in an integrin-dependent manner as the focal adhesion protein, and this has been confirmed in cardiomyocytes (Zhang et al., 2016). In addition, the integrin-independent effects of Kindlin-2 also positively regulate cell behaviors by stabilizing target proteins and the involved cascades (Dong et al., 2023b; Tang et al., 2023). There are known effects of Kindlin-2 on heart failure, and we then planned to explore the correlation between the Kindlin family and ICM. As the major member in cardiac tissues, decreased Kindlin-2 was discovered in cardiomyocytes undergoing anoxic stimulation and in ischemic cardiac tissues. We first discovered that the deteriorated tissues of I/R hearts could be prevented by Kindlin-2 that was manifested as ameliorative ferroptosis. There was a small amount of Kindlin-2 in ferroptotic cardiomyocytes, so we aimed to reveal the anti-ferroptotic effect of cardiac Kindlin-2, and these findings were further authenticated in in vivo and in vitro experiments. In contrast, we also discovered that cardiomyocytes that lacked Kindlin-2 gave rise to ferroptosis. Although several publications have highlighted the protective roles of Kindlin-2 in regulated cell death of various cell types, the anti-ferroptotic ability of Kindlin-2 has never been reported. In this study, our systematic research regarding the effects of Kindlin-2 on cardiac ferroptosis has been proven for the first time.
Blood reperfusion of the ischemic area is an important clinical ICM treatment (Zhou et al., 2021). However, reperfusion probably is accompanied by more serious injuries. A recent study found that cardiac ferroptosis and not apoptosis mediates long-term I/R injury (Fang et al., 2019). Our conclusions emphasized the possibility of Kindlin-2 as a potential target for the treatment of cardiac I/R injury. We unprecedently confirmed that Kindlin-2 intensified the interaction between Otub1 and Slc7a11 to reinforce Slc7a11/GSH/Gpx4, the classical anti-ferroptosis pathway (Ning et al., 2023). Physiologically, to avoid error responses, improperly folded proteins are degraded by the ubiquitin proteasome system (UPS), which also mediates pathological processes. Ubiquitins have the advantage of the three-step enzymatic cascade that can transfer them to substrates through the E1, E2, and E3 enzymes in sequence to submit the polyubiquitinated protein to proteasomes for proteolysis (Dong et al., 2023a). As the well-known protein modification at the post-translational level, ubiquitination can be reversed by deubiquitinases. In terms of mechanism, Otub1 can stabilize Slc7a11 by deactivating E2 conjugating enzymes indispensable for polyubiquitination. As anticipated, our study confirmed that the regulation of Kndlin-2 on Slc7a11 was UPS-dependent (Liu et al., 2019).
Our results revealed that cardiomyocytic ferroptosis induced by Kindlin-2 deficiency can be primarily rescued by superfluous Otub1. However, the enhancement effect of Kindlin-2 on the Slc7a11/GSH/Gpx4 pathway can be abolished by Otub1 inhibition. As a result, the major role of the Otub1/Slc7a11/GSH/Gpx4 axis played by Kindlin-2 was validated. Our findings suggested that the Kindlin-2/Otub1/Slc7a11 axis is a new orientation for treating and preventing cardiac I/R injury. Furthermore, our findings demonstrate the interaction between HIF-1α and Kindlin-2 in cardiomyocytes. Through upregulating Slc7a11/GPX4 axis, the ferroptosis resistance of HIF-1α in cardiomyocytes has been well established. (Ge et al., 2023). HIF-1α also enhances Kindlin-2 expression in MCF7 (Xue et al., 2020). Therefore, Kindlin-2 in cardiomyocytes is probably involved in the HIF-1α/Slc7a11/Gpx4, the anti-ferroptotic cascades. In response to H/R stimulation, decreased HIF-1α may abolish Kindlin-2/Slc7a11/Gpx4 axis to aggravate ferroptosis.
Ferroptosis has no specific markers, even though it was discovered years ago (Dixon and Olzmann, 2024). Ferroptosis is distinguished from other cell death types by increased membrane density and reduced cristae of shrinking mitochondria (Liu et al., 2023). Much research has been dedicated to the detection of the vital ferroptosis functions during the occurrence and development of diseases such as tumors, ischemia-reperfusion injury, kidney injury, blood diseases, lung diseases, and gastrointestinal diseases (Li et al., 2020; Qiu et al., 2020; Yan et al., 2021). Different manners of cell death, such as apoptosis, ferroptosis and autophagy, have intersections, and whether or not the certain regulatory network can be integrated by these dead types remains controversial (Shen et al., 2023). In addition, the antagonistic and synergistic effects among different cell deaths have not yet been illustrated (Bai et al., 2023).
Our study does contain limitations, even though the anti-ferroptotic roles of Kindlin-2 have been clearly illuminated. First, it is still unknown if cardiac Kindlin-2 can regulate other forms of cell death. Second, the cardiac specificity of AAV9 injection is not so focused as in cre-loxp mice (Zacchigna et al., 2014). The volume of AAV injection is also an obstruction to success (Tsuji et al., 2023). Even so, the synthesis of our current work and previous achievements shows that Kindlin-2 has the potential to inhibit multiple cell death modes concurrently and is conducive to solving the above problems. Therefore, further investigations should be conducted.
Material and Methods
Animals
Eight-week-old male C57BL/6J mice weighing 20–25 g were raised by Changsheng Bio-Technology, Liaoning, China. A room temperature (RT) of 20°C–25°C and humidity of 55 ± 5% were provided for the facility that housed the mice. The total number of mice is 150. The fresh air exchange frequency was 10–15 times/h. A 12-h light and 12-h dark cycle was maintained every day. The studies did not discriminate sex, and both male and females were used. All animal operations strictly abided by the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health and were approved by the Animal Ethical Committee of Harbin Medical University (No. IRB3007822).
Mouse model of cardiac I/R injury
The I/R injuries in male mice were induced using a 45-min ischemia followed by 24-h reperfusion. A 2% avertin solution (0.1 mL/10 g body weight; Sigma-Aldrich Corporation, United States) was intraperitoneally injected into the mice to induce deep anesthesia. The mice were then subjected to endotracheal intubation. Blunt dissection of the intercostal muscles exposed the hearts to ligate the left anterior descending coronary arteries with 7–0 nylon wire. A total of 45 min later, the wire was dismantled to arouse reperfusion of the left ventricle for 24 h. The mice were anesthetized again to measure their cardiac functions. The mice were then sacrificed to collect the cardiac tissues and serum for future experiments.
Primary cardiomyocyte preparation and culture
Neonatal C57BL/6 mice (P1–P3) were sterilized using 75% alcohol. The chests were scissored without injuring the enterocoeles to isolate the hearts. After being rinsed by phosphate buffer solution (PBS), the heart tissues were cut into small pieces and digested in trypsin buffer. (Trypsin 0.25%, ethylenediaminetetraacetic acid 0.53 mM, Beijing Solarbio Science & Technology Co., Ltd., China). Dulbecco’s Modified Eagle Medium (DMEM, Gibco, USA) supplied with 10% fetal bovine serum (Lonsera, South America) and 1% double antibiotics (10 KU/mL Penicillin G Sodium salt, 10 mg/mL Streptomycin Sulfate, meilunbio, Liaoning, China) was applied to culture cells in an incubator (37°C, 5% CO2) for 90 min. Suspended cells were added into six-well plates at a density of 1 × 106 cells/well. Cells are available for experiments after a 48-h culture. The primary cardiomyocytes were incubated under hypoxic conditions (37°C with a humid atmosphere of 5% CO2 and 1% O2) for 12 h, followed by a 24-h reoxygenation to establish an in vitro H/R model.
Infection of the adeno-associated virus 9 carrying Fermt2, Otub1, and ShRNA Fermt2
The construction of an adeno-associated serotype 9 (AAV9) virus vector using the interference sequence of the Fermt2 gene (AAV9 shFermt2) and Otub1 gene (AAV9 sh Otub1) was established by the Shanghai Hanbio Biotechnology Co., Ltd. (Shanghai, China). The construction of the adeno-associated virus vector 9 that overexpressed the Fermt2 gene (AAV9 Fermt2) and the Otub1 gene (AAV9 Otub1) was accomplished by Genechem (Shanghai, China). AAV9 of 1.5 × 1011 titers diluted by 0.2 mL normal saline was injected into the tail vein of one mouse. Two weeks later, the gene expression in the mouse reached the peak, and following experiments were conducted.
Echocardiographic measurements
The M-mode of Vevo2100 echocardiographic system (Visual Sonics, Toronto, ON, Canada) with a 10-MHz probe frequency was used to measure the functions of the left ventricles. The anesthetized mice were depilated at the fore breast using depilatory cream. The mice’s body temperatures and body positions were maintained using heating pads at 37°C. The M-mode recording tests were conducted using the left ventricular end diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV), left ventricular end-diastolic diameter (LVIDd), and left ventricular systolic diameter (LVIDs) for three cycles to obtain the average values for statistical analysis. The EF = (LVEDV−LVESV)/LVEDV × 100%, and the FS = (LVID−dLVIDs)/LVIDd × 100%, respectively.
Transfection of plasmids and siRNAs
The siRNA of Kindlin-2 and a NC was designed by Ribobio (Guangzhou, China). The siRNA of Otub1 was from Sigma. The Kindlin-2 and Otub1 plasmids were constructed by Genechem (Shanghai, China). The plasmids and siRNAs above were transfected into cardiomyocytes via Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) and X-treme GENE siRNA Transfection Reagent (Roche, Mannheim, Germany) according to the directions. The siRNA sequences for the mice are listed in Supplementary Table S1.
Inhibitor treatment
Ferrostatin-1 (Fer-1, HY-151563, MedchemExpress, USA) powder was evenly dissolved into the clarified dimethyl sulfoxide (DMSO) reserve liquid. The terminal concentration and stimulation time were 10 μM and 24 h. The clarified DMSO reserve liquid of 100 μL at 25.0 mg/mL was added to 400 μL PEG300 and mixed evenly. A total of 50 μL Tween-80 was added to the system and mixed evenly. We then continued to add 450 μL of normal saline volume to 1 mL. According to the above system, 8 mL solvent was prepared, and 2 mg Fer-1 powder was dissolved into this. The mice were administered intraperitoneal injections of Fer-1 at 1 mg/kg/day the day before and after the I/R surgery. The mice were administered intraperitoneal injections of Fer-1 at 1 mg/kg/day the day before the shRNA tail vein injection. Fer-1 administration was also started 24 h before ShRNA injection and lasted for 14 days. MG-132 (HYHY-13259, MedchemExpress, USA) powder was dissolved into the clarified DMSO reserve liquid. The terminal concentration and stimulation time were 10 μM and 6 h, respectively. The terminal concentration and stimulation time of the H/R were 200 nM and 12 h, respectively.
Cycloheximide assay
The primary cardiomyocytes were seeded in 12-well plates and transfected with siRNA scramble and siRNA Fermt2. Twenty-four hours after the transfection, the cells were incubated with 100 μg/mL CHX dissolved using DMSO. The total protein lysates were harvested at the indicated time points, followed by immunoblotting.
Evans blue (EB)/triphenyl tetrazolium chloride (TTC) double staining
After the establishment of the cardiac I/R mice, the risk area (AAR) and infarct area (IF) of the mice were visually identified using EB/TTC double staining. The mice were anesthetized again, and the chest cavities were reopened. The original ligation sites loosed 24 h previously were ligated again to reocclude the LAD. A total of 0.5 mL of 1.5% Evans blue (Solarbio, China) was injected into the left ventricles through the aortas. Instantly, the perfused hearts were detached and rinsed using cold PBS. After the hearts were frozen in a −80°C fridge for 5 min, the sections between the ligature and apex were cut into four pieces of nearly uniform thicknesses. The slices were placed in a 1% TTC (2,3,5-triphenyltetrazolium chloride; Solarbio, China) solution and incubated at 37°C for 5 min. After this, the samples were fixed in 4% (w/v) paraformaldehyde (PFA) for 30 min and available for photographing. The blue-stained areas are the nonischemic areas, the red-stained areas are the viable myocardium of the AAR, and the white-stained areas are the infarct areas of the AAR. Computer plane measurements were used to calculate the staining area of the continuous sections that were further analyzed using ImageJ software.
Transmission electron microscopy
Several apical pieces approximately 1 mm3 in size were acquired from fresh samples and immediately fixed in 2.5% glutaraldehyde and placed in a 4°C refrigerator overnight. The samples were rinsed with PBS for 3 × 15 min, fixed with 1% osmic acid solution for 1–2 h, rinsed with PBS for 3 × 15 min, then rinsed with water for 3 × 15 min. The tissues were subsequently dehydrated with 30%–50%–70%–80%–95%–100% ethanol for 15 min each time and 100% propylene oxide for 3 × 15 min each time. The samples were then subjected to infiltration embedding in the sequence below: propylene oxide: EMBed 812 = 1:1 (2 h, RT); propylene oxide: EMBed 812 = 1:3 (overnight, RT); and pure EMBed 812 for (4 h, RT). The pure EMBed 812 was then poured into the embedding models, and the samples were inserted into the pure EMBed 812 and maintained at 37°C for 48 h and 45°C for 12 h. The embedding models with resin and samples were placed in a 60°C oven to polymerize for at least 48 h. The resin blocks were trimmed into 60–70 nm thick sections that were transferred onto copper grids and stained with uranium acetate (25 min) and lead citrate (5 min). The dry samples were examined under TEM at a voltage of 80 kV for image acquisition and analysis.
Quantitative real-time PCR
The total RNA was extracted from adherent cells or cardiac tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, United States). The quality of the total RNA was measured using NANODrop2000c (Thermo Scientific, Carlsbad, USA), and 500 ng RNA was reversely transcribed into cDNA using ReverTra Ace qPCR RT Kit (Toyobio Co., Ltd., Japan). The ABI 7500 Fast Real-Time PCR system (Applied Biosystems, Foster City, USA) was used to test cDNA levels by using SYBR Green (Roche, Basel, Basel-City, Swiss). The expression levels of the target genes were normalized to the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expressions. The relative quantitative expressions were calculated using the 2-ΔΔCT method. All primers used in this study are listed in Supplementary Table S2.
Western blotting
A radioimmunoprecipitation lysis buffer (Beyotime, Shanghai, China) was used to extract proteins from the cardiomyocytes and cardiac tissues. Then samples underwent ultrasonication, and surplus debris was removed using a super centrifugal process. The total protein concentration was measured using the bicinchoninic acid Protein Assay Kit (Beyotime, Shanghai, China). Protein samples were run in 12.5% (vol/vol) sodium dodecyl sulfate (SDS)–polyacrylamide gels. The separated proteins were transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA) that were subsequently blocked by 5% skim milk and incubated with primary antibodies at 4°C overnight. After eluting the primary antibodies, the secondary antiantibodies were cross-linked with the membrane for 60 min at RT and under lucifugal conditions. An Odyssey infrared imaging system (LI-COR, Lincoln, NE, United States) was used to scan the membranes and show the target bands. The relative protein levels were measured and analyzed using Image Studio software. The primary antibodies of this study are summarized in Supplementary Table S3.
Co-Immunoprecipitation
The cells were lysed using IP lysis buffer (Beyotime, Shanghai, China) that contained protease inhibitors (Sigma-Aldrich Corporation, United States). The cell lysates were swirled for 30 s and stood on ice for 5 min. The process above was repeated three times, and samples were centrifuged for 15 min at 4°C, 12,000 × g. We acquired the supernatant and precleared the samples using protein G Magnetic Beads (MedChemExpress, United States) at 4°C overnight. Then 0.5 mg of cell lysate was subjected to the indicated antibodies (Kindlin-2, Proteintech, Cat#: 11453-1-AP; Slc7a11, Abmart, Cat#: T57046; Otub1, Abcam, Cat#: ab175200; HIF-1, Cat#: RT1278), and the mixture was rotated at 4°C overnight. The samples were then placed on a magnet rack to adsorb and sedimentate the beads. The supernatant was removed, and the beads were rinsed with cold PBS three times. The loading buffer that contained the beads was boiled at 100°C for 10 min. The input and IP samples were detected using an immunoblot analysis. Antibodies of the immunoblot analysis are listed below. (Ubiquitin, Biodragon, Cat#: BD-PM33045; Otub1, Invitrogen, Cat#: MA5-38255; Slc7a11, Invitrogen, Cat#: MA5-50633; Kindlin-2, Proteintech, Cat#: 11453-1-AP; HIF-1, Cat#: HA721997). The primary antibodies of this study are summarized in Supplementary Table S3.
Cardiomyocytes ROS assay
The lipid-ROS levels of living cells were measured using the Image-iT® Lipid Peroxidation Kit (C10445, Thermo Fisher Scientific, USA) according to the protocols of specification. Primary cardiomyocytes were subconfluently inoculated in 24-well plates and incubated with 5 μm C11-BODIPY at 37°C for 20 min. The images were obtained using a fluorescence microscope (Olympus). A green fluorescence indicated oxidized cell membranes, and a red fluorescence indicated non-oxidized cell membranes. Cytosolic ROS of living cells was measured by the Kit (S0033M, Beyotime, China), according to the protocols of specification. Subconfluent cardiomyocytes were incubated with 10 μM DCFH-DA probe at 37°C for 20 min. Mitochondrial ROS of living cells was measured by Kit (S0061M, Beyotime, China), according to the protocols of specification. Subconfluent cardiomyocytes were incubated with 5 μM MitoSOX dye and Hoechst 33342 (C1027, Beyotime, China) at 37°C for 30 min. The images were obtained by fluorescence microscope (Olympus).
Immunofluorescence staining for cells
The sub-confluent primary cardiomyocytes were fixed using 4% (w/v) PFA for 30 min at RT. After being washed with PBS to thoroughly remove the PFA, samples were treated with a permeable solution (Beyotime) for 15 min at RT and then a blocking solution (Beyotime) for 1 h at RT. The primary antibodies were incubated with the cells for 1 h under RT and washed with PBS three times. The cells were then incubated with secondary antibodies (Anti Mouse, Invitrogen, Carlsbad, CA, USA, Cat#: A11008; Anti-Rabbit, Invitrogen, Carlsbad, CA, USA, Cat#: A11036) for 1 h at RT. The coverslips were fixed on microscope slides with ProLong Gold Antifade reagent that contained 4′,6-diamidino-2-phenylindole (DAPI) (Abcam, Cambridge, MA, USA, Cat#: ab104139). The images were obtained using a fluorescence microscope (Olympus). The antibodies used in this study are summarized in Supplementary Table S3.
Immunohistochemistry (IHC) staining and Prussian blue staining
The hearts of the euthanized mice were perfused with saline through the left ventricles to remove blood. After being washed by PBS, cardiac tissues were fixed with 4% (w/v) PFA for 24 h and embedded in paraffin. The paraffin samples were cut continuously at a thickness of 5 μm, and the sections were roasted at 60°C for 2 h. The samples were then dewaxed and incubated in a 10-mM citrate buffer for antigen extraction. A 3% hydrogen peroxide solution was added at RT for 10 min to block the endogenous peroxidase. The heart sections were then incubated with a primary antibody against Kindlin-2 (Abclonal, Cat#: A8709) or 4-HNE (bs6313R, Bioss, China) at 4°C overnight. Goat anti-rabbit IgG/HRP (PV6001, GoldenBridge) was then incubated at RT for 20 min. Then, 3,3-diaminobenzidine (DAB) staining and hematoxylin reverse staining were conducted. Finally, the neutral rubber seals of the cardiac sections were obtained, and images were captured using a fluorescence microscope (Olympus).
Moreover, 5-μm cardiac paraffin sections were also stained using a Prussian blue iron stain kit (Cat: RS3980, G-clone Bio-technology Co., Ltd., Beijing, China). All the operations were performed according to the manufacturer’s instructions.
MDA and GSH analysis
MDA was measured using an MDA kit (Cat: A003-1-2, Nanjing Jiancheng, Jiangsu, China) in the cell lysates. GSH was measured using a GSH kit (Cat: A006-2-1, Nanjing Jiancheng, Jiangsu, China) in the cell lysates. All of the operations strictly followed the manufacturer’s instructions and were quantified with absorbance at 450 nm on a microplate reader (Bio-Tek, Richmond, VA, USA).
Lipid-peroxidation
Tissues of similar size were added to 0.9% normal saline at 1:9 (g/L) and ground into homogenate in ice bath. After 30-min standing, according to the manufacturer’s instructions of Kit (Cat: A106-1-1, Nanjing Jiancheng, Jiangsu, China), the samples were centrifuged to acquire the supernatant for lipid peroxidation assessment and quantified with absorbance at 586 nm on a microplate reader (Bio Tek, Richmond, VA, USA).
Statistical analysis
Each experiment was repeated at least three times. The data are expressed as the mean ± SDs calculated using GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA). For multiple comparisons, a two-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was performed. An unpaired two-sided Student’s t-test was used for the comparison between the two groups. A p < 0.05 was considered statistically significant.
Authors’ Contributions
Y.D., F.W., and K.L. equally to this study. Y.D., F.W., and K.L., contributed equally to this work. W.D. and Y.D., designed research. F.W., Y.D., Y.Y., K.L., S.Y., and Z.Q., performed experiments. Y.D., X.S., W.D., Y.Y., X.P., Y.L., and Y.Z. analyzed the data. Y.D. and W.D., wrote the article.
Footnotes
Acknowledgment
The authors acknowledge the assistance of the State Key Laboratory of Frigid Zone Cardiovascular Diseases, Department of Pharmacology.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by grants from the Natural Science Foundation of China [82273928, U21A20339]; Youth Project of Scientific Research Institution of Heilongjiang Province [CZKYF2023-1-C047]; CAMS Innovation Fund for Medical Sciences (CIFMS) 2019-I2M-5-078. Postdoctoral Fellowship Program of China Postdoctoral Science Foundation [GZC20240354].
Data Collection
Electronic laboratory notebook was not used.
Ethics Statement
All animal experiments were approved by the Harbin Medical University Animal Ethical committee (No. IRB3007822).
Data Availability
All data that support the findings of this study are available from the corresponding author upon reasonable request.
Consent for Publication
All the authors consent for publication.
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.