
Abstract
Aims:
To determine the role of the ubiquitin-activating enzyme UBA1 in macrophage-mediated renal injury during sepsis-associated acute kidney injury (SA-AKI) and to elucidate the underlying molecular mechanism.
Methods and Results:
Using a cecal ligation and puncture mouse model, we evaluated renal function, inflammation, and survival in myeloid-specific Uba1 knockout mice (Uba1M-KO) and littermate controls. Transcriptomic, proteomic, and ubiquitinome analyses were integrated with mechanistic studies in bone marrow–derived macrophages and renal tubular epithelial cell co-cultures. A pharmacologic UBA1 inhibitor (PYR-41) was tested for therapeutic efficacy. UBA1 expression was markedly increased in renal macrophages during SA-AKI. Uba1M-KO mice demonstrated improved survival, preserved renal function, and attenuated inflammatory responses, as evidenced by reduced cytokine production, reactive oxygen species generation, apoptosis, and macrophage infiltration. Mechanistically, UBA1 promoted ubiquitination and degradation of the nuclear pore protein nucleoporin 35 (NUP35), impairing IκBα nuclear import and activating nuclear factor kappa B (NF-κB) signaling. This led to enhanced macrophage inflammatory activation and subsequent renal tubular injury. Pharmacologic inhibition of UBA1 recapitulated the protective effects of genetic deletion in vivo.
Innovation and Conclusions:
This study identifies UBA1-mediated NUP35 ubiquitination as a previously unrecognized checkpoint linking ubiquitin activation to nuclear pore integrity and inflammatory signaling in sepsis. UBA1 drives macrophage-mediated inflammation in SA-AKI by promoting NUP35 degradation and subsequent activation of NF-κB signaling. Targeting UBA1 represents a promising immunomodulatory strategy for the prevention and treatment of SA-AKI. Antioxid. Redox Signal. 44, 859–877.
This is a visual representation of the abstract.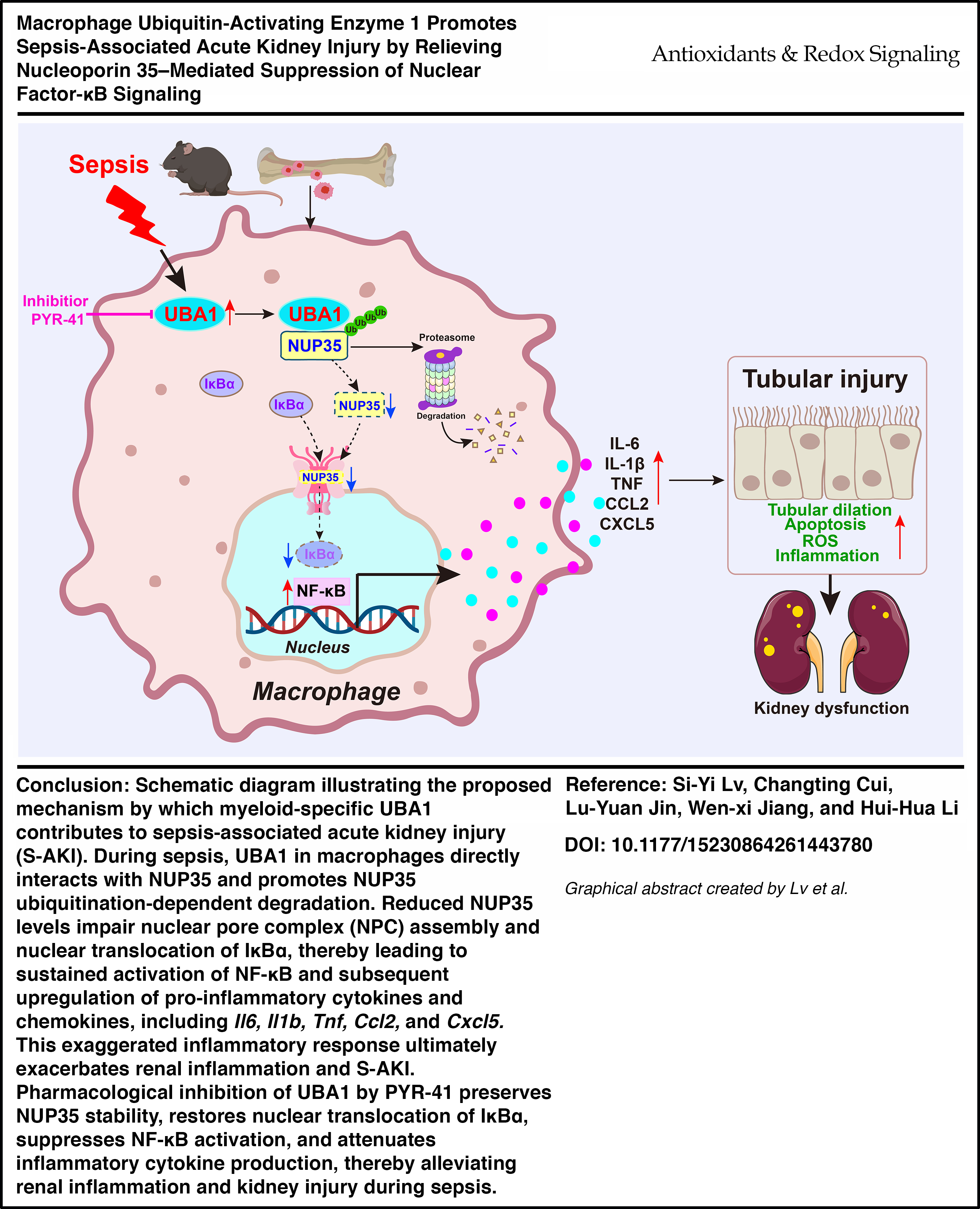
Introduction
Sepsis is a potentially fatal disorder marked by a dysregulated immune reaction to infection, frequently resulting in failure of multiple organs (Rudd et al., 2020). The kidney is particularly vulnerable, with 25% to 75% of patients in Intensive Care Units, who are septic, presenting with sepsis-associated acute kidney injury (SA-AKI) that results in rapid renal dysfunction and significant morbidity and mortality (L Chen et al., 2024; White et al., 2023; Zarbock et al., 2023). Despite extensive research, the molecular and cellular mechanisms underlying SA-AKI are still poorly understood. Several fundamental mechanisms that contribute to the development of SA-AKI have been reported, including disproportionate inflammatory responses, microcirculatory dysfunction, and cellular metabolic disorders. Notably, studies in humans have identified imbalanced inflammation as one pathogenic mechanism of SA-AKI, proposing immune modulation as a promising therapeutic modality (He et al., 2022; Sun et al., 2023). However, evidence from experiments that target the inflammatory cascade in sepsis remains inconclusive. Therefore, elucidating the underlying mechanisms and identifying new treatment strategies are crucial for a better prognosis.
Renal macrophages are the most abundant immune cells and are essential for regulating inflammation and tissue repair (Mu et al., 2025; Rayego-Mateos et al., 2022). Upon exposure to pathogen- or damage-associated molecular patterns, macrophages rapidly polarize into a pro-inflammatory phenotype, releasing cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1 beta (IL-1β), and IL-6, which magnify renal inflammation and contribute to tubular epithelial injury (Mu et al., 2025). In contrast, anti-inflammatory or M2 macrophages, which assist in resolving inflammation and facilitating tissue repair, are often insufficient or functionally impaired in SA-AKI (Chen et al., 2019). Thus, this imbalance between pro- and anti-inflammatory phenotypes exacerbates renal injury and delays recovery. Increasing evidence supports the involvement of macrophages in both the onset and progression of SA-AKI (T Li et al., 2022, 2024; Xiang et al., 2023), and therapeutic strategies targeting macrophage activation or pro-inflammatory polarization have shown protective effects in animal models (Fan et al., 2023; Gander-Bui et al., 2023; X Li et al., 2022; Tang et al., 2022; Wang et al., 2021). Nevertheless, the definitive molecular mechanisms that regulate the dynamic phenotypic and functional changes of macrophages, as well as their pathogenic or reparative roles during SA-AKI, remain poorly understood.
The ubiquitin conjugation cascade is required for ubiquitination of protein substrates and degradation by the proteasome or autophagy-lysosome pathways (Barghout and Schimmer, 2021). Ubiquitin-like modifier activating enzyme 1 (UBA1) is the initiating E1 enzyme in the ubiquitin conjugation cascade. Emerging evidence indicates that UBA1 plays a pro-inflammatory role in diverse pathological conditions. A recent study demonstrated that somatic mutations in the UBA1 gene disrupt normal ubiquitination, resulting in a severe systemic inflammatory disorder termed VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome (Beck et al., 2020). Furthermore, UBA1 is involved in the ubiquitination and degradation of multiple key regulatory proteins, such as interferon regulatory factor 3, the autophagy-related LC3 protein, the cell cycle regulatory proteins p53 and p21, as well as the transcription factor c-Jun (Chen et al., 2021; Liao et al., 2020; Qin et al., 2016). Notably, AKI occurs in ∼25% of VEXAS syndrome patients (Kalantari et al., 2025), suggesting that UBA1 dysfunction is closely linked to both inflammation and kidney injury. UBA1 has also been implicated in neurodegenerative (Lambert-Smith et al., 2020) and cardiovascular diseases, including atherosclerosis (Liao et al., 2020), cardiac remodeling (Shu et al., 2018), and aortic dissection (Wang et al., 2025), primarily by enhancing monocyte/macrophage infiltration and sustaining tissue inflammation. In contrast, inhibition of UBA1 using small molecules such as the UBA1 inhibitor 4-(4-(5-nitrofuran-2-ylmethylene)−3,5-dioxopyrazolidin-1-yl) benzoic acid ethyl ester (PYR-41) suppresses inflammatory responses and ameliorates disease progression in preclinical animal models (Liao et al., 2020; Shu et al., 2018; Wang et al., 2025). However, the specific role of UBA1 in macrophage-driven inflammation and its contribution to SA-AKI remain poorly defined and warrant further investigation.
In this study, we determined the functional role of macrophage-specific UBA1 in the development of SA-AKI. Our findings revealed that UBA1 expression in renal macrophages increased progressively during sepsis and correlated with kidney function. By using macrophage-specific Uba1 knockout (Uba1M-KO) mice, we demonstrated that UBA1 deficiency significantly improved survival outcomes, kidney damage and the inflammatory response. We propose that the deletion of UBA1 stabilized NUP35, a component of the nuclear pore complex (NPC), leading to enhanced nuclear translocation of inhibitor of nuclear factor κBα (IκBα) and inhibition of nuclear factor kappa B (NF-κB)–mediated inflammatory reactions. Furthermore, PYR-41 inhibition of UBA1 confirmed the protective effects observed with the Uba1 knockout mice. Therefore, this study provides novel insights into the molecular mechanisms underlying macrophage UBA1-mediated inflammation in SA-AKI and identifies UBA1 as a potential target for therapeutic intervention to protect renal function in sepsis.
Innovation
This study is the first to demonstrate that UBA1 regulates nuclear transport–dependent NF-κB signaling in SA-AKI by targeting NUP35 for ubiquitination and proteasomal degradation. We show that macrophage-specific Uba1 deletion and pharmacologic inhibition of UBA1 significantly improve survival and preserve renal function following sepsis. Mechanistically, UBA1 promotes NUP35 degradation, which impairs nuclear import of IκBα and sustains NF-κB activation in macrophages, thereby driving renal tubular injury. These findings identify a previously unrecognized mechanism linking ubiquitin-dependent 4proteostasis to nuclear pore-regulated inflammatory signaling. Targeting UBA1 may therefore represent a broadly applicable therapeutic strategy for macrophage-driven inflammatory injury in SA-AKI.
Results
UBA1 expression is upregulated in renal macrophages during SA-AKI
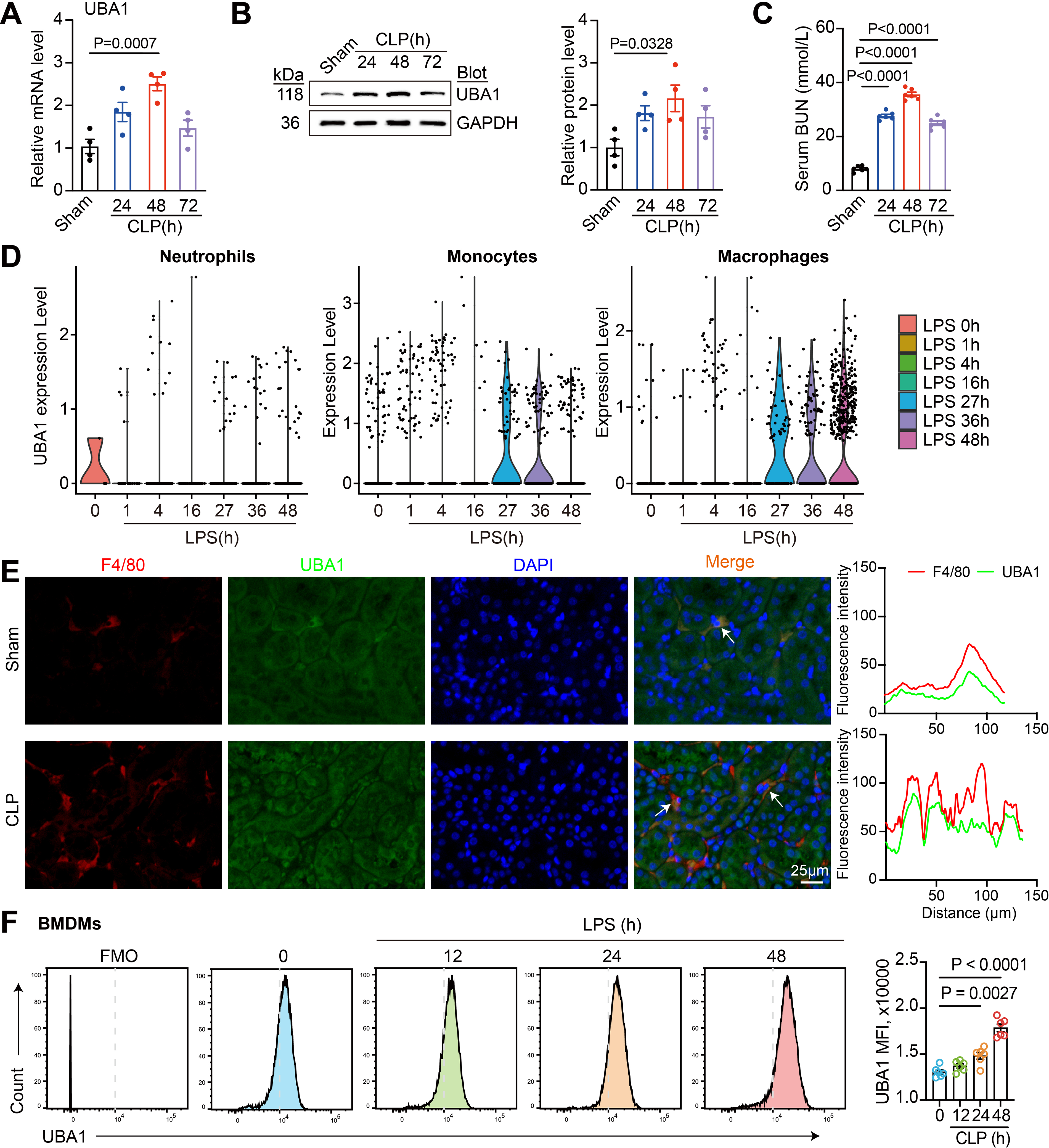
To investigate the expression dynamics of UBA1 during SA-AKI, we used a cecal ligation and puncture (CLP)-induced sepsis mouse model and analyzed UBA1 expression in renal tissues at different time points post-CLP. quantitative PCR (qPCR) analysis revealed a time-dependent increase in renal Uba1 mRNA levels, peaking at 48 h post-CLP (Fig. 1A). Concomitantly, western blotting verified a significant elevation of UBA1 protein levels at 48 h (Fig. 1B), concurring with the severity of kidney injury, as indicated by serum blood urea nitrogen (BUN) levels (Fig. 1C). To determine the cellular source of UBA1 during sepsis, we further analyzed a publicly available single-cell RNA-sequencing dataset (GSE151658) from kidneys of lipopolysaccharide (LPS)-challenged mice. Violin plots revealed that Uba1 expression was markedly increased in monocytes/macrophages over a period of 27–48 h after LPS stimulation (Fig. 1D). To validate this in CLP kidneys, we performed in situ immunofluorescence co-staining of UBA1 and F4/80 (a macrophage-specific antigen). UBA1 was highly elevated in F4/80+ macrophages, while renal parenchymal cells showed minimal change (Fig. 1E), indicating that UBA1 upregulation occurs predominantly in macrophages. In addition, flow cytometry analysis of bone marrow-derived macrophages (BMDMs) stimulated with LPS (200 ng/mL) revealed that UBA1 expression at baseline (0 h, control) was low and increased in a time-dependent manner at 12, 24, and 48 h (Fig. 1F). Taken together, these results confirm that UBA1 is upregulated in macrophages during SA-AKI.
Myeloid-specific UBA1 deletion attenuates SA-AKI
To explore whether UBA1 upregulation in macrophages contributes to the pathogenesis of SA-AKI, myeloid-specific Uba1 knockout mice (Uba1M-KO) were generated by crossing Uba1flox/flox mice with lysozyme M-Cre recombinase (Lyz2-Cre) mice. Uba1flox/flox mice lacking the Cre allele were used as controls (Uba1M-WT) (Fig.S1A). Successful deletion of UBA1 in myeloid cells was confirmed by PCR genotyping and immunoblotting of BMDMs (Fig.S1B). Both Uba1M-KO and Uba1M-WT mice were subjected to CLP surgery and monitored for 48 h to assess the functional role of UBA1 in SA-AKI (Fig.2A).

Survival of both sets of mice was monitored and the results indicated that Uba1M-KO mice had higher survival rates than their Uba1M-WT littermates within 48 h of CLP (Fig. 2B). Serum BUN levels were significantly lower in Uba1M-KO mice, indicating preserved kidney function (Fig. 2C). Functional assessment, including 24-h urine volume, urine creatinine, blood creatinine, urinary protein levels, and urine protein-to-creatinine ratios, demonstrated that Uba1M-KO significantly improved renal function in CLP-treated mice compared with Uba1M-WT controls (Fig. 2D). Histopathological examinations using hematoxylin and eosin (H&E) and periodic acid–Schiff (PAS) staining verified severe tubular damage in control mice. In septic Uba1M-WT controls, we noted renal architecture with increased tubular dilation, cast formation, and loss of the brush border, while these effects were ameliorated in Uba1M-KO mice (Fig. 2E–F). Furthermore, CLP-induced renal apoptosis (as visualized by TUNEL staining) was also reduced in Uba1M-KO mice (Fig. 2G), supporting a protective effect of myeloid-specific UBA1 deficiency toward CLP-induced renal injury.
As oxidative stress and inflammation are key contributors to SA-AKI, we assessed reactive oxygen species (ROS) production, macrophage infiltration and inflammatory cytokine expression. Immunostaining with dihydroethidium (DHE) staining revealed a marked reduction in the ROS levels (Fig. 2H) and number of F4/80+ macrophages (Fig. 2I) in the kidneys of Uba1M-KO mice compared with controls. qPCR analysis further demonstrated that Uba1 deletion significantly decreased the expression levels of Il1β, Il6, Tnf, and C-C motif chemokine ligand 2 (Ccl2) mRNAs following CLP (Fig. 2J). Collectively, these results indicate that myeloid-specific UBA1 deletion protects against SA-AKI by mitigating oxidative stress and inflammation.
UBA1 interacts with NUP35 and promotes its ubiquitination-mediated degradation in LPS-stimulated BMDMs
To identify potential downstream effectors mediating UBA1 function in macrophages, immunoprecipitation (IP) combined with mass spectrometry (MS) was performed on LPS-stimulated BMDMs. Among the candidate interactors, nucleoporin 35 (NUP35), a nucleo-cytoplasmic transporter protein, was highly enriched in anti-UBA1 IP (Fig. 3A). To assess whether UBA1 specifically interacts with NUP35, co-IP assays were conducted with IgG control or anti-UBA1 antibody. Immunoblotting analysis confirmed a direct interaction between UBA1 and NUP35 in BMDMs, and this interaction was enhanced by LPS (Fig. 3B). Immunofluorescence staining further demonstrated nuclear colocalization of UBA1 and NUP35 in BMDMs (Fig. 3C). Thus, these results indicate that NUP35 is a target protein of UBA1.
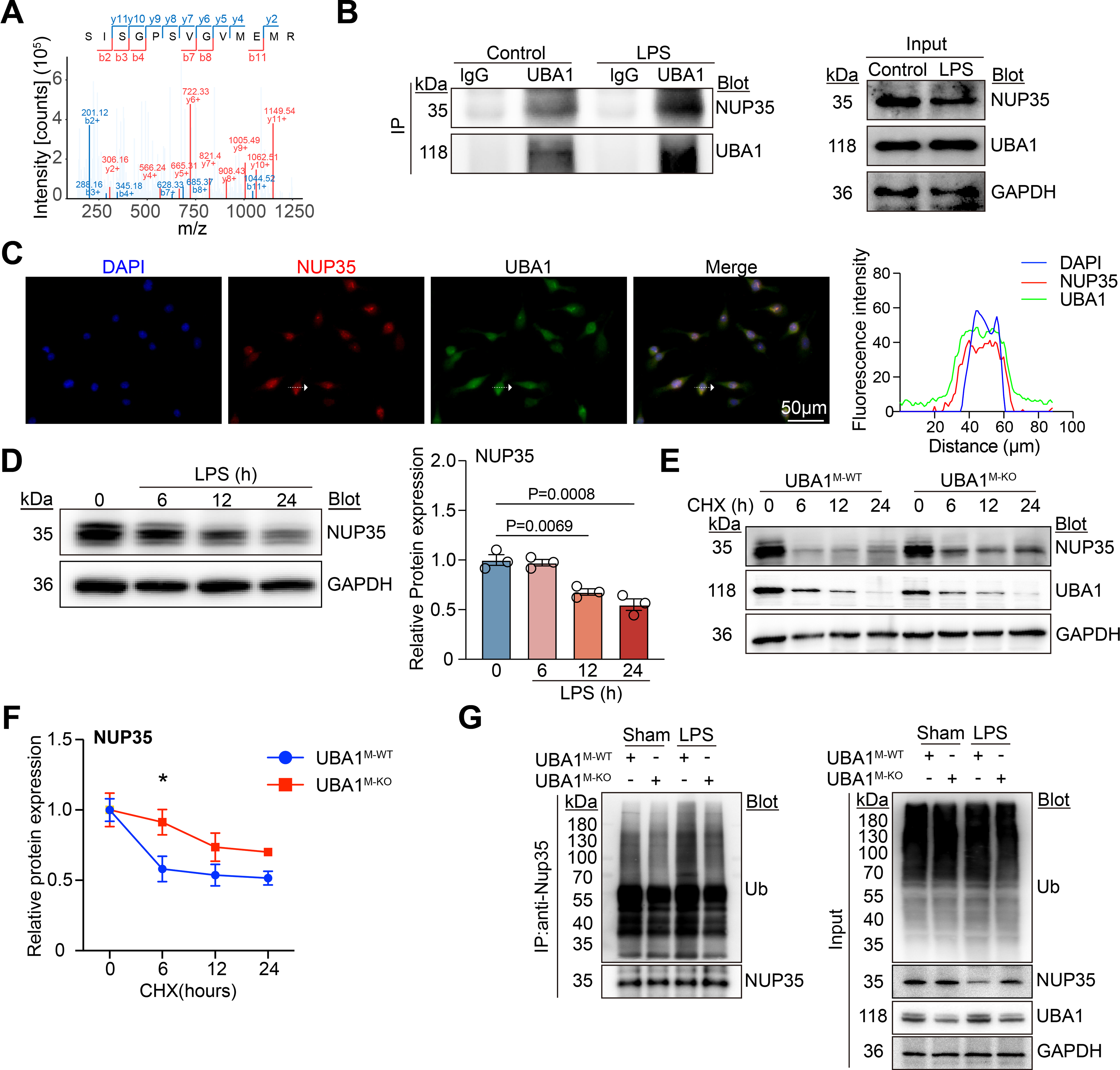
Next, we examined the expression dynamics of NUP35 in BMDMs following LPS stimulation. Immunoblotting analysis revealed a time-dependent decrease in NUP35 protein levels after LPS exposure for 0, 6, 12, and 24 h (Fig. 3D), suggesting that LPS induces NUP35 degradation, which is negatively correlated to UBA1 expression. Interestingly, the degradation of NUP35 was markedly delayed in Uba1M-KO BMDMs, as demonstrated by cycloheximide (CHX) chase assays (Fig. 3E–F), indicating that UBA1 facilitates NUP35 protein turnover. To determine whether UBA1 regulates NUP35 through controlling its ubiquitination, cell lysates were immunoprecipitated with an anti-NUP35 antibody, followed by immunoblotting with an anti-ubiquitin (Ub) antibody. The results showed that LPS-induced ubiquitination of NUP35 in Uba1M-WT BMDMs was markedly attenuated in Uba1M-KO BMDMs (Fig. 3G), suggesting that UBA1 promotes NUP35 ubiquitination and subsequent degradation. Overall, these data demonstrate that UBA1 physically interacts with NUP35 and promotes its ubiquitination-dependent degradation following LPS stimulation.
Ubiquitinome profiling identifies inflammatory and NF-κB-related pathways in Uba1M-WT macrophages
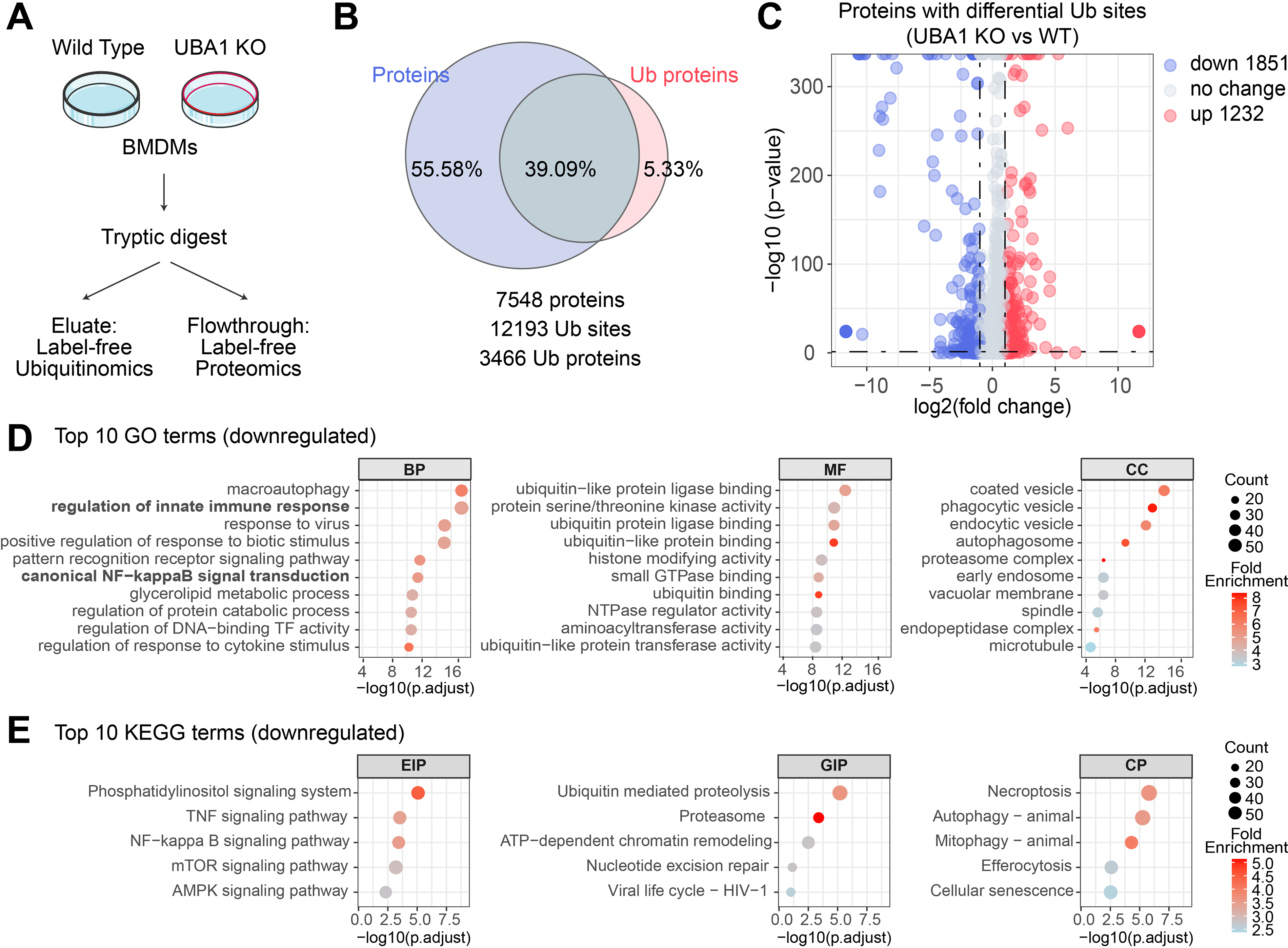
To examine the post-translational mechanisms by which myeloid UBA1 regulates inflammation and renal function, we performed ubiquitination proteomics on BMDMs from Uba1M-KO and Uba1M-WT mice following LPS stimulation (Fig. 4A). In total, 7,548 proteins, 12,193 ubiquitination sites, and 3,466 ubiquitinated proteins were identified, respectively (Fig. 4B). Differential analysis revealed 1,851 downregulated and 1,232 upregulated ubiquitination sites specifically in Uba1M-KO BMDMs (adjusted p < 0.05, |log2FC| > 1) (Fig. 4C), indicating extensive alterations of the macrophage ubiquitinome. Gene ontology (GO) enrichment analysis of downregulated ubiquitination sites revealed that Uba1M-KO predominantly affects proteins involved in macroautophagy, innate immune signaling, inflammatory responses, pattern recognition receptor signaling pathway, and canonical NF-κB signaling transduction. These proteins are associated with molecular function such as ubiquitin-like protein ligase binding and protein kinase activity, and cellular components such as vesicle trafficking and proteasome complexes, highlighting their roles in protein homeostasis and immune regulation (Fig. 4D and Supplementary Table S3). Consistently, Kyoto Encyclopedia of Genes and Genomes pathway analysis validated these findings, displaying enrichment in toll-like receptor signaling, TNF and NF-κB pathways, and ubiquitin-mediated proteolysis as prominently altered pathways in Uba1M-KO macrophages (Fig. 4E and Supplementary Table S4). Comparative changes of gene expression in kidney tissues from Uba1M-KO and Uba1M-WT mice supported these observations, where reduced expression of inflammatory genes and NF-κB targets provided complementary evidence toward the functional impact of UBA1-dependent ubiquitination (Supplementary Fig. S2). Transcriptomic analysis of kidney tissues from Uba1M-KO and Uba1M-WT mice provided complementary evidence by showing reduced expression of inflammatory genes and NF-κB target genes (Supplementary Fig. S2). Taken together, these data demonstrate that UBA1 deficiency selectively reduces the ubiquitination of proteins involved in inflammatory signaling and NF-κB-associated pathways.
UBA1 deficiency suppresses NF-κB signaling in macrophages
Next, we sought to assess whether Uba1M-KO alters NF-κB signaling activity in macrophages. The key components of the canonical NF-κB pathway, including NF-κB p65 and IκBα, were examined in LPS-stimulated BMDMs. Immunoblotting analysis of whole-cell lysates showed that LPS robustly induced phosphorylation of p65 and IκBα in Uba1M-WT BMDMs; however, this response was markedly attenuated in Uba1M-KO BMDMs (Fig. 5B), suggesting that Uba1M-KO suppresses canonical NF-κB signaling via increasing IκBα in BMDMs.
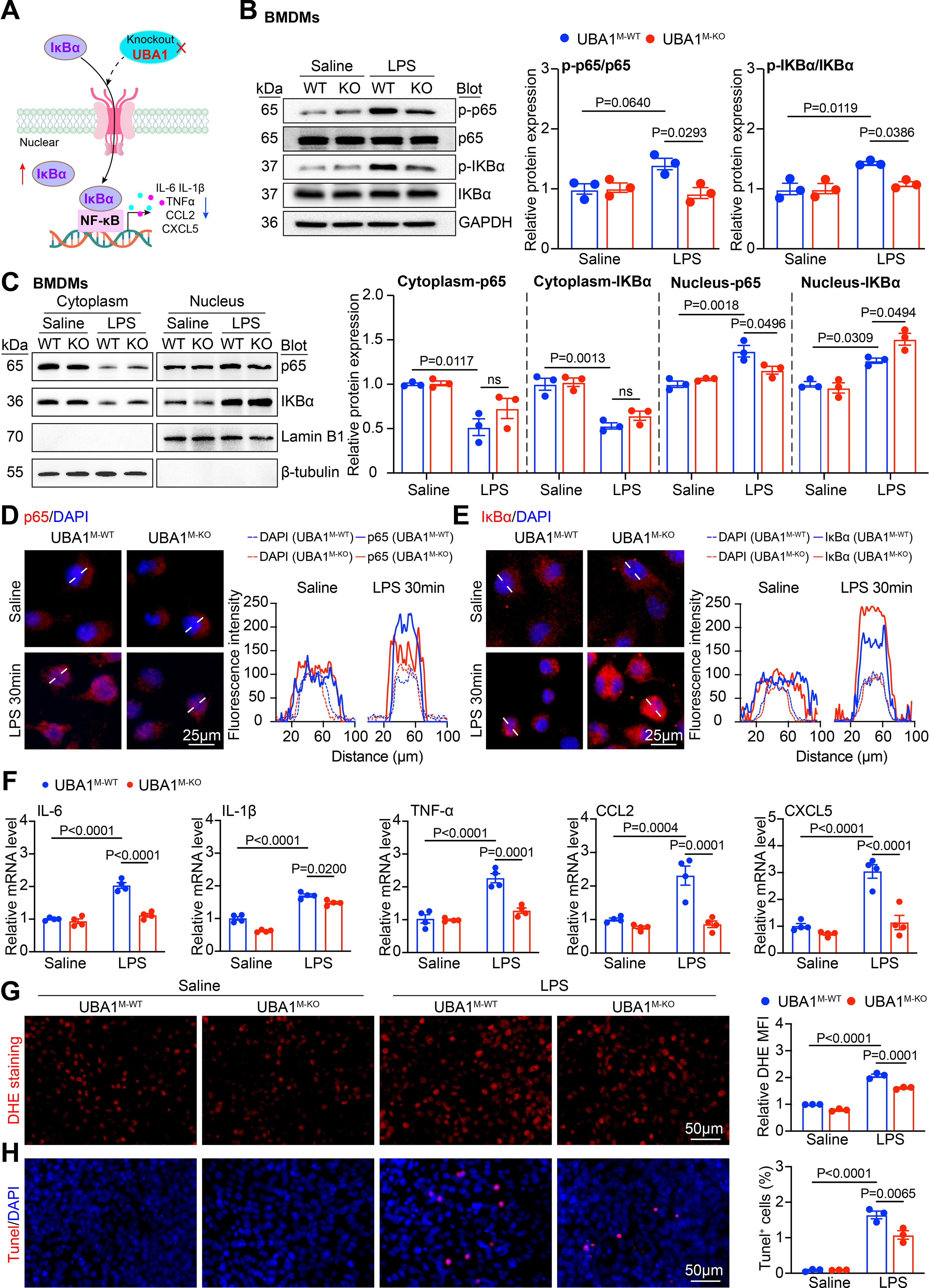
As activation of NF-κB is driven by the subcellular distribution of p65 and IκBα, we then analyzed cytoplasmic and nuclear localization of both these components. Subcellular fractionation followed by immunoblotting revealed that LPS stimulation led to pronounced nuclear accumulation of p65 in Uba1M-WT BMDMs, but this effect was substantially reduced in Uba1M-KO cells. Notably, Uba1M-KO resulted in a marked increase in nuclear accumulation of IκBα compared with Uba1M-WT cells, indicating enhanced nuclear retention of this inhibitory protein under inflammatory stimulation (Fig. 5C). Immunofluorescence staining consistently confirmed that increased nuclear p65 levels and decreased nuclear IκBα proteins in Uba1M-WT BMDMs following 30 min of LPS exposure; however, these changes were significantly reversed in Uba1 BMDMs (Fig. 5D–E), mirroring the observation from immunoblots. To assess the functional consequence of these localization changes, expression levels of canonical NF-κB target genes were measured. qPCR analysis revealed that LPS-induced transcription of proinflammatory cytokine and chemokine genes, including Il6, Il1β, Tnf, Ccl2, and C-X-C motif chemokine ligand 5 (Cxcl5), was significantly reduced in Uba1M-KO BMDMs compared with Uba1M-WT controls (Fig. 5F), indicating diminished NF-κB transcriptional activity. In addition, Uba1M-KO BMDMs exhibited lower intracellular ROS levels (assessed by DHE staining) (Fig. 5G), and the percentage of apoptotic cells (TUNEL staining) (Fig. 5H), demonstrating enhanced cellular resilience to inflammatory stress. Collectively, these results support a model in which UBA1 deficiency restrains NF-κB activation by enhancing nuclear accumulation of IκBα and thereby suppressing inflammatory gene transcription, oxidative stress, and apoptosis in macrophages (Fig. 5A).
NUP35 mediates UBA1-dependent macrophage-driven tubular epithelial injury
Based on the observation that UBA1 deficiency suppresses NF-κB activation in macrophages, we next examined whether NUP35 mediates this effect. To address this, small interfering RNA (siRNA) was applied to knock down NUP35 in BMDMs. Immunoblotting indicated that Uba1M-KO BMDMs showed a marked reduction in phosphorylation of p65 and IκBα proteins compared with scramble-transfected Uba1M-WT BMDMs following LPS stimulation. In contrast, knockdown of NUP35 abrogated this suppressive effect (Fig. 6B). Furthermore, Uba1M-KO BMDMs showed a significant decrease in nuclear p65 protein levels and an increase in nuclear IκBα protein levels, whereas these effects were eliminated by si-NUP35 knockdown (Fig. 6C). Consistent with these signaling changes, NUP35 knockdown also abolished the Uba1-KO–mediated reduction in the mRNA levels of pro-inflammatory cytokines (Il6, Il1β, Tnf, Ccl2, and Cxcl5; Fig. 6D). Together, these results indicate that NUP35 is required for the inhibitory effect of UBA1 deficiency on NF-κB–driven inflammatory responses in macrophages.
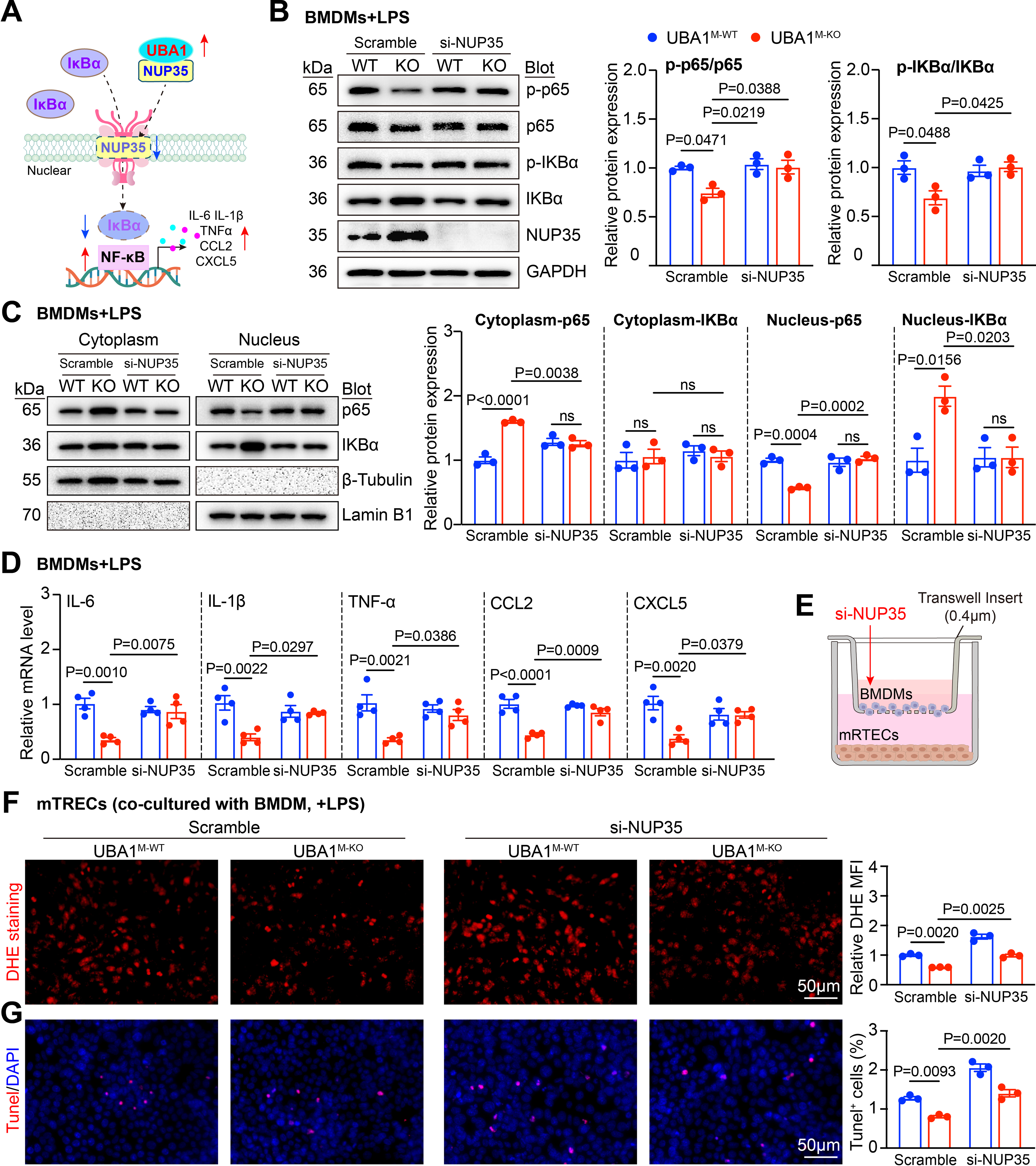
To test whether Uba1M-KO BMDMs directly protect renal epithelial cell injury, a transwell coculture system of BMDMs and renal tubular epithelial cells (RTECs) was established (Fig. 6E). Upon LPS stimulation, Uba1M-KO BMDMs exhibited a reduced capacity to induce ROS production (DHE staining) and apoptosis (TUNEL staining) in RTECs compared with Uba1M-WT BMDMs. Conversely, the Uba1M-KO-mediated protective effect was lost upon NUP35 knockdown (Fig. 6F–G). Collectively, these results support a model in which NUP35 mediates the suppressive effect of UBA1 deficiency on NF-κB activation by regulating nuclear accumulation of IκBα, thereby attenuating inflammatory gene transcription and renal epithelial cell injury induced by LPS (Fig. 6A).
Pharmacological inhibition of UBA1 ameliorates sepsis-associated kidney injury in mice
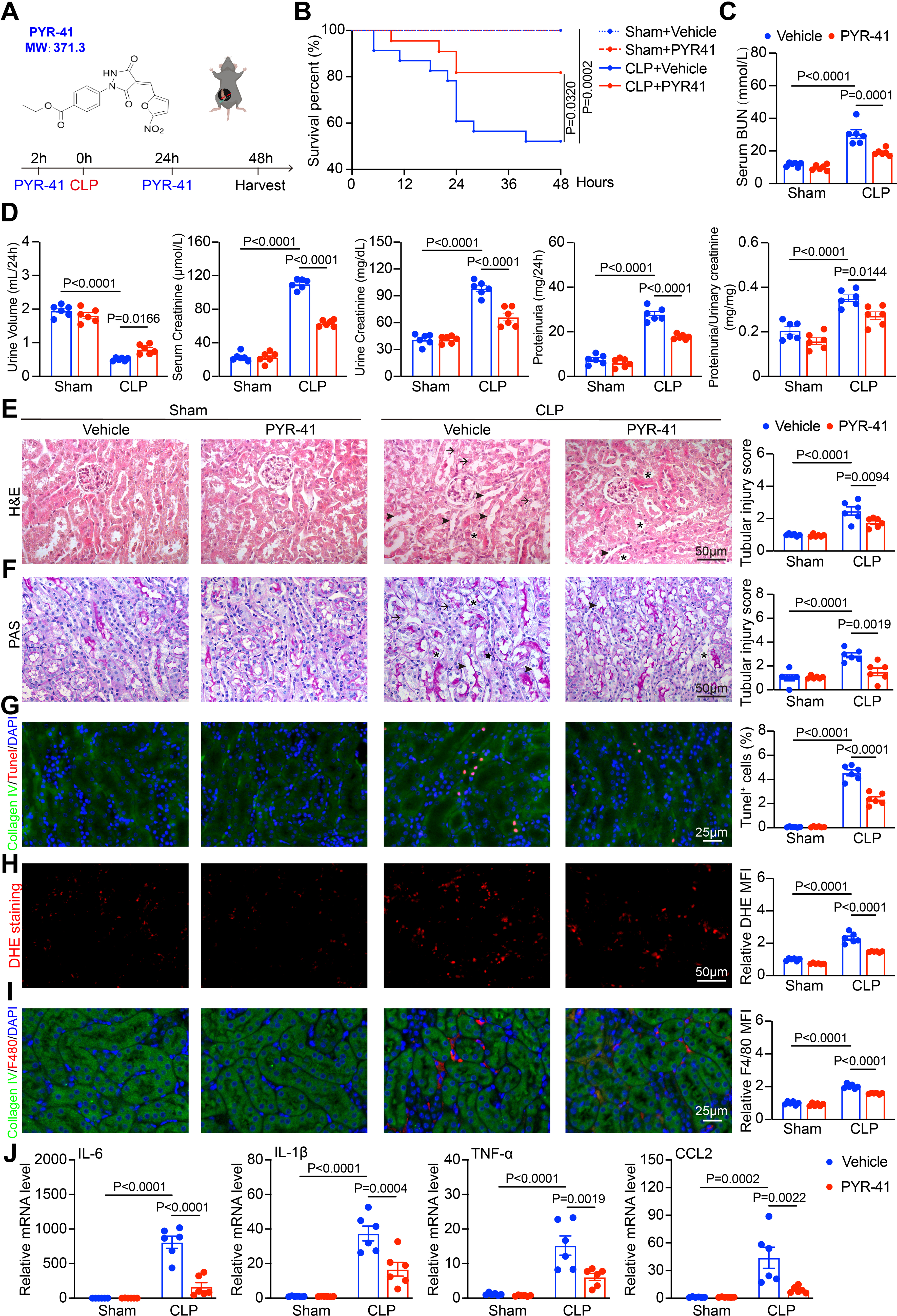
As UBA1 deficiency ameliorated macrophage-mediated inflammation, and thereby inhibited the development of SA-AKI, we therefore investigated whether selective inhibition of UBA1 could exert protective effects in SA-AKI. The UBA1 inhibitor PYR-41 was administered intravenously into WT mice which were then subjected to CLP surgery and observed over 48 h (Fig. 7A). PYR-41 treatment significantly improved the 48-h survival rate compared with vehicle treatment (Fig. 7B) and markedly reduced serum BUN levels (Fig. 7C). Functional assessments, as reflected by 24-h urine volume, urine creatinine, blood creatinine, urinary protein, and urine protein/creatinine ratio, further confirmed that PYR-41 treatment attenuated renal function induced by CLP (Fig. 7D). Accordingly, H&E staining of the renal cortex (Fig. 7E) and PAS staining of the renal medulla (Fig. 7F) both demonstrated that PYR-41 treatment alleviated CLP-induced tubular injury, as visualized by reduced tubular damage, preservation of the brush border in cortical tubules, and decreased cast formation in medullary tubules. Furthermore, TUNEL (Fig. 7G) and DHE staining (Fig. 7H) showed lower apoptotic cells and ROS levels in renal cells of PYR-41-treated mice post-CLP. Immunofluorescence staining of F4/80 demonstrated that CLP-induced macrophage infiltration in the kidneys of vehicle-treated mice was markedly attenuated by PYR-41 treatment (Fig. 7I). Consistent with this, qPCR analysis confirmed a significant downregulation of Il1β, Il6, Tnf, and Ccl2 in PYR-41–treated mice (Fig. 7J), supporting the anti-inflammatory effect of UBA1 inhibition. Overall, these findings validate that inhibition of UBA1 can alleviate the development of SA-AKI, and underscore its promise as a potential therapeutic strategy.
Discussion
In this study, we identify UBA1 as a key regulator of macrophage-mediated inflammation and kidney injury in sepsis. UBA1 was upregulated in renal macrophages during SA-AKI. Myeloid-specific deletion of Uba1 suppressed macrophage infiltration, reduced inflammatory cytokine production, and attenuated oxidative stress; collectively, these changes preserved tubular architecture and improved renal function. Mechanistically, UBA1 interacted with NUP35 and promoted its ubiquitination-dependent degradation. This process restricted the nuclear retention of IκBα and enabled NF-κB activation, which resulted in the upregulation of downstream inflammatory gene expression. Notably, pharmacological inhibition of UBA1 significantly ameliorated CLP-induced renal dysfunction (Graphical Abstract). Collectively, these findings reveal a previously unrecognized role of the UBA1–NUP35–NF-κB axis in the pathogenesis of SA-AKI and provide mechanistic insight into how UBA1 regulates macrophage-driven inflammation.
UBA1, the primary E1 enzyme in the ubiquitin–proteasome system (UPS), is central to protein homeostasis, cellular stress responses, and immune signaling (Barghout and Schimmer, 2021). Aberrant UBA1 activity has been implicated in a variety of pathological conditions, including atherosclerosis (Liao et al., 2020), cardiac remodeling (Shu et al., 2018), and aortic dissection (Wang et al., 2025). Elevated UBA1 expression has been reported to promote monocyte/macrophage recruitment and long-term tissue inflammation, whereas UBA1 inhibition attenuates sepsis-associated lung and liver injury (Matsuo et al., 2018). Despite this evidence, the specific contribution of myeloid UBA1 to the development of SA-AKI remained unclear. Here, using Uba1M-KO mice and BMDMs, our study demonstrates that deletion or inhibition of UBA1 significantly improves CLP-induced renal dysfunction and attenuates tubular epithelial injury, apoptosis, ROS production, and inflammatory responses (Fig. 1, 7). These findings uncover a novel role of UBA1 as a critical regulator of SA-AKI through its influence on macrophage-dependent inflammation.
Accumulating evidence indicates that monocyte/macrophage are central drivers of immune responses and subsequent sepsis (Kuwabara et al., 2022; Privratsky et al., 2023). Upon microbial insult, these cells are activated and produce numerous pro-inflammatory mediators, which lead to the cytokine storm contributing to multiorgan damage (Carcillo and Shakoory, 2024). The NF-κB pathway is a key signaling hub in this process by orchestrating transcription of inflammatory mediators, chemokines, and adhesion molecules (Karki and Kanneganti, 2021; Liu et al., 2024). Under quiescent conditions, NF-κB is retained in the cytoplasm by the inhibitor of κB (IκB). Upon activation by the presence of microbes or their antigens, IκBα undergoes phosphorylation, ubiquitination and proteasomal degradation, enabling the release and nuclear translocation of NF-κB subunits (e.g., p65), which in turn initiate transcription of pro-inflammation-associated genes (Yu et al., 2020). The regulation of NF-κB nuclear translocation is tightly controlled by the UPS. For example, E3 ligases such as tumor necrosis factor receptor associated factor 6 and HECT, C2, and WW domain-containing E3 ubiquitin protein ligase 2 have been shown to modulate NF-κB nuclear dynamics (S Chen et al.,2024; Zhao et al., 2023). However, the role of E1 enzymes, particularly UBA1, in this regulatory framework has remained poorly understood. Here, ubiquitination proteomic analysis of BMDMs combined with transcriptomic analysis of kidney tissues showed that deletion of UBA1 specifically suppressed the ubiquitination of proteins that regulate NF-κB target genes involved in inflammatory responses. Furthermore, we elucidated the underlying mechanism and found that UBA1 deficiency increased nuclear retention of IκBα, thereby suppressing NF-κB activation and downstream cytokine production.
To further explore how UBA1 regulates IκBα-NF-κB signaling in macrophages during sepsis, we performed co-IP followed by MS in BMDMs to identify potential targets of UBA1. Among the UBA1-interacting protein candidates, NUP35 (NUP35), a key nucleocytoplasmic transport protein, attracted our attention and was selected for further investigation. NUP35 is an important subunit of the NPC, which facilitates bidirectional transport between the nucleus and the cytoplasm and plays critical roles in maintaining NPC structure, gene expression and immune regulation (Y Li et al., 2024). Studies indicate that NUPs modulate NF-κB signaling by affecting nucleocytoplasmic translocation, including NUP153 (Liu et al., 2020), and NUP358/RanBP2 (Liu et al., 2020). NUP93 (Neely et al., 2023), NUP62 (Makiyama et al., 2022), POM121 (Ge et al., 2019), and NUP160 (Xie et al., 2021). These studies highlight the NPC as a critical regulatory hub for NF-κB dynamics. However, the role of NUP35 in regulating NF-kB signaling and macrophage inflammatory response has remained unexplored. Here, we identified NUP35 as a downstream target of UBA1-mediated ubiquitination in macrophages. Inflammatory stimulation induces UBA1-dependent ubiquitination and proteasomal degradation of NUP35 (Fig. 3), whereas Uba1 deficiency stabilizes NUP35 and promotes nuclear retention of IκBα, thereby suppressing NF-κB activation. Given the essential role of NUP35 in NPC assembly, these results suggest that UBA1 regulates NF-κB signaling, at least in part, through modulation of IκBα transport. Collectively, our results defined UBA1–NUP35–IκBα/NF-κB axis as a new mechanism to regulate macrophage-mediated inflammation and SA-AKI development, providing novel mechanistic insight into how ubiquitin signaling intersects with nuclear pore dynamics to control innate immune responses during sepsis.
Given the critical roles of UBA1 in various inflammatory diseases, inhibition of UBA1 represents a promising therapeutic strategy for the treatment of these conditions. UBA1 inhibitors such as TAK-243 (MLN-7243) have shown in efficacy in preclinical models of acute myeloid leukemia (Barghout et al., 2019) and small cell lung cancer cells (Majeed et al., 2022), without accompanying toxicity. PYR-41, an irreversible and cell-permeable UBA1 inhibitor, suppressed angiotensin II-induced cardiac remodeling (Shu et al., 2018), mitigated sepsis-related hepatic injury (Jin et al., 2026), attenuated sepsis-associated lung injury (Matsuo et al., 2018), and reduced atherosclerosis development (Liao et al., 2020) by inhibiting macrophage pro-inflammatory responses and oxidative stress. Our findings extend these observations by demonstrating that UBA1 inhibition by PYR-41 ameliorates SA-AKI, suggesting that targeting UBA1 may offer a broad immunomodulatory strategy applicable to multiple sepsis-associated organ injuries.
Several limitations of this study should be acknowledged. First, the molecular mechanisms linking UBA1-mediated ubiquitination to NUP35 remain incompletely defined, including the identification of the specific E3 ligases responsible. Second, whether UBA1 inhibition confers similar protection to other organs affected by sepsis, such as the cardiovascular system, requires further investigation. Third, UBA1 may interact with antioxidant pathways such as Nrf2 signaling, which could modulate the balance between pro-inflammatory and anti-inflammatory macrophage responses (Calabrese et al., 2010). Finally, the pharmacokinetics and safety profile of UBA1 inhibitors such as PYR-41 in clinical settings require thorough evaluation. Addressing these issues will help clarify the therapeutic advantages of targeting the UBA1–NUP35 axis.
In summary, our results underscore the pivotal role of UBA1 in macrophage-mediated inflammation during SA-AKI and uncover a novel molecular pathway linking UBA1 to NF-κB regulation through NUP35-mediated IκBα nuclear trafficking. These findings highlight UBA1 as a central regulator of inflammation in SA-AKI and a promising therapeutic target. Future studies should explore tissue-specific roles of UBA1, evaluate additional selective UBA1 inhibitors such as TAK-243 in vivo, and investigate potential crosstalk with antioxidant pathways to fully realize the translational potential of UBA1-targeted therapies in sepsis.
Materials and Methods
Animals
C57BL/6J male mice (8–10 weeks old) were obtained from Vital River Laboratory Animal Technology Company (Beijing, China) and housed under specific pathogen-free conditions with a 12-h light/dark cycle at a controlled ambient temperature of 20°C and ad libitum access to food and water. The animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Beijing Chaoyang Hospital and were carried out in strict adherence to established laboratory animal care guidelines (2021-Animal-35,26 February 2021).
To generate myeloid-specific UBA1-deficient mice, Uba1fl/fl and Lyz2-CreERT2 transgenic mice (both from Beijing Viewsolid Biotechnology Co., Ltd.) were crossed to generate Uba1fl/fl:Lyz2-Cre (hereafter referred to as Uba1M-KO). Littermate Uba1fl/fl mice were used as wild-type (WT) controls (hereafter referred to as Uba1M-WT). Genotyping was performed by PCR using primers specific for the floxed Uba1 allele and Cre recombinase. For inducible Cre activation, tamoxifen (10540-29-1, Sigma-Aldrich, USA) was dissolved in corn oil (C7030, Solarbio, China) and administered intraperitoneally at 40 mg/kg body weight once daily for 5 days. Mice were allowed to rest for 7 days following the last injection to ensure complete recombination and clearance of residual tamoxifen before subsequent experimentation.
Only male mice were used in this study. This decision was based on the need to reduce variability in phenotype and avoid hormonal influences on tamoxifen-induced gene recombination. As estrogen levels fluctuate during the female estrous cycle, they may interfere with Cre activation efficiency. Whether similar findings would be observed in female mice remains to be determined.
CLP mouse model of sepsis
Polymicrobial sepsis was induced using the CLP model as previously described (Zhang et al., 2023) with minor modifications. Following isoflurane anesthesia, a 1–2 cm midline laparotomy was made to expose the cecum. The distal 20% of the cecum segment was ligated with a 4-0 silk suture and punctured once through-and-through with a 21-gauge needle. A minimal quantity of fecal content was gently extruded. The cecum was returned to the abdominal cavity, followed by a two-layer closure of the peritoneum and skin. Following the surgical procedure, 1 mL of pre-warmed sterile saline was administered subcutaneously for fluid resuscitation, and the mice were placed in a warm recovery chamber. Sham-operated mice underwent identical surgical procedures except for ligation and puncture. Mice were monitored for survival and euthanized at 24-, 48-, or 72-hours post-surgery for sample collection and subsequent analyses.
Administration of UBA1 inhibitor in mice
To assess the therapeutic efficacy of UBA1 inhibition in SA-AKI, wild-type C57BL/6J mice were intraperitoneally administered with 10 mg/kg PYR-41 (S7129, Selleck, China), a selective UBA1 inhibitor, 2 h before and 24 h after CLP. Control mice received an equal volume of vehicle. Mice were euthanized 48 h after CLP for sample collection and analysis.
Isolation of mouse bone marrow-derived macrophages (BMDMs)
Bone marrow cells were isolated from the femurs and tibias of 6-8-week-old C57BL/6J, Uba1M-WT, or Uba1M-KO mice under sterile conditions. The bones were flushed with pre-chilled Phosphate Buffered Saline (PBS) using a 21-gauge needle to extract the marrow. The cell suspension was passed through a 70 µm cell strainer and then resuspended in RPMI 1640 medium containing 10% Fetal Bovine Serum (FBS) (WB500T, Ausbian, Australia). Following a 3-hour incubation at 37°C in 5% CO2 to facilitate the adherence of fibroblasts and pre-existing macrophages, the suspension of non-adherent cells were then harvested and cultivated in RPMI 1640 basal medium supplemented with 10% FBS, 1% penicillin–streptomycin and 25 ng/mL recombinant murine macrophage colony-stimulating factor (M-CSF; 315-02, PeproTech) for 3 days to induce macrophage differentiation.
Co-culture of BMDMs and mRTECs
The mRTECs were purchased from BeNa Culture Collection (BNCC360478, BNCC, China) and maintained in RPMI 1640 culture medium supplemented with 10% FBS, 1% penicillin–streptomycin antibiotic solution at 37°C in a humidified atmosphere containing 5% CO2. BMDMs from Uba1M-WT or Uba1M-KO mice were differentiated as described above. For co-culture experiments, mRTECs were seeded in 6-well plates and grown to ∼80% confluency. BMDMs were added to the upper chambers of Transwell inserts (0.4 µm pore size, 3450, Corning) at a density of 1 × 106 cells per insert to allow paracrine interaction without direct contact. The co-culture system was stimulated with LPS (200 ng/mL; Sigma) for 24 h. mRTEC lysates were then collected for subsequent analyses, including qPCR, DHE staining, and TUNEL staining.
Ubiquitinome analysis by LC–MS/MS
Mouse bone marrow–derived macrophages (BMDMs) from wild-type and Uba1 knockout mice were lysed in urea buffer supplemented with protease inhibitors and the deubiquitinase inhibitor PR-619. Proteins were reduced, alkylated, and digested with trypsin. Ubiquitinated peptides were enriched using anti–K-ε-GG (diGly) antibody–based immunoaffinity purification and analyzed by LC–MS/MS on a timsTOF HT mass spectrometer (Bruker Daltonics). Raw MS data were processed using DIA-NN and searched against the UniProt Mus musculus database. Peptide and protein identifications were filtered at a false discovery rate (FDR) of 1%. Label-free quantification was performed, and differentially ubiquitinated sites were defined as those with an absolute log2 fold change ≥1 and p < 0.05. Functional enrichment analyses were subsequently conducted on the differentially ubiquitinated proteins. The MS proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD074268.
Bulk RNA sequencing and bioinformatics analysis
Total RNA was extracted from the kidneys of Uba1M-WT and Uba1M-KO mice using TRIzol reagent (15596026CN, Invitrogen, USA) in strict adherence to the manufacturer’s instructions. Following RNA quality assessment, cDNA libraries were prepared and subjected to single-strand circularization. Rolling circle amplification (RCA) utilizing phi29 DNA polymerase was performed on the prepared libraries to generate DNA nanoball (DNB) products. The DNBs were then loaded onto patterned nanoarrays and sequenced on the BGISEQ G400/T7/T10 platform (BGI-Shenzhen, China) to generate paired-end reads of 100 or 150 base pairs. Raw sequencing reads were aligned to the mouse reference genome (mm10) via HISAT2 (v2.2.1), and gene-level annotation was performed using Bowtie2 (v2.4.2) against the RefSeq gene database. Genes with a total read count below 20 across all samples were filtered to ensure robust quantification in downstream analyses.
Analysis of differential expression genes was performed using the DESeq2 R package (v1.34.0) (Love et al., 2014). Genes with a log2 fold change ≥0.2 or ≤ −0.2 and an adjusted q-value (Benjamini–Hochberg correction) <0.05 were classified as significantly differentially expressed. Visualization of differentially expressed genes (DEGs) included the generation of volcano plots using the ggplot2 package (v3.5.1; https://ggplot2.tidyverse.org), and heatmaps of the DEGs were constructed using the pheatmap package (v1.0.12; https://github.com/raivokolde/pheatmap). Normalized expression values were used for clustering. Gene Set Enrichment Analysis (GSEA) was carried out using the clusterProfiler package (v4.14.6) with the gseGO functions (Yu et al., 2012). Gene lists were ranked by log2 fold change of GSEA input. Enrichment data were considered significant with an adjusted p value <0.05 and a significant normalized enrichment score. Visualization of GSEA results was performed using the enrichplot package (v1.26.6). The raw RNA-seq data have been deposited in the NCBI BioProject database under accession number PRJNA1400645 and are scheduled for public release upon publication.
Histological analysis of kidney tissues
Kidney samples were fixed in 4% paraformaldehyde, paraffin-embedded and sectioned at 4 µm thickness. Sections were processed with H&E (G1120, Solarbio, China) for morphological assessment, or PAS (G1281, Solarbio, China) staining to visualize tubular basement membranes and glycogen deposition. A semi-quantitative scoring method was used to assess renal damage, with 10 nonoverlapping random fields per mouse analyzed under 200× magnification. Tubular damage was graded on a 0–5 scale according to the percentage of tubules exhibiting brush border loss in TECs, tubular dilation, and inflammatory cell infiltration: 0 = no lesion; 1 = <10%; 2 = 10%–25%; 3 = 25%–50%; 4 = 50%–75%; 5 = 75%–100% (Rong et al., 2011; Shi et al., 2023). The average score from the 5–10 fields was calculated for each animal and used for statistical comparison between groups. Quantitative analysis of PAS-positive areas and tubular injury was achieved through ImageJ software (National Institutes of Health, USA), and the percentage of positively stained areas and tubular injury index were measured and compared between groups.
Immunofluorescence staining
Kidney samples were fixed in 1% paraformaldehyde at room temperature for 1 h, followed by overnight incubation in 20% sucrose and subsequent dehydration in 30% sucrose. After optimal cutting temperature compound embedding, tissues were cryosectioned at 4 µm thickness. For immunofluorescence analysis, sections were permeabilized using 0.5% Triton X-100 in PBS, followed by blocking for 1-hour with goat serum (ZSGB-BIO, Cat# ZLI-9056, China) at ambient temperature. The sections were then subjected to overnight incubation at 4°C with primary antibodies against F4/80 (1:200, ab6640, Abcam, USA), UBA1 (1:200, 67198-1-lg, Proteintech, USA), Collagen IV (1:200, A25131, ABclonal, China), Nup35 (1:200, NB100-93322, Novus Biologicals, USA), p65 (1:200, 8242, Cell Signaling Technology, USA), or IκBα (1:200, ab32518, Abcam, USA). For the detection of ROS, kidney sections were incubated with 5 µM DHE at 37°C for 30 min. Cell apoptosis was detected with the TUNEL Apoptosis Assay Kit (12156792910, Roche, Switzerland) following the manufacturer’s protocol. Finally, all sections were counterstained with 4′, 6-diamidino-2′-phenylindole (C1006, Beyotime, China) for nuclear staining and imaged using a fluorescence microscope (DM2500, Leica, Germany). For quantitative analysis, the number of TUNEL-positive cells was systematically counted in five randomly selected high-power fields (200×) per section. The mean fluorescence intensities of F4/80, UBA1, Nup35, p65, IκBα, and DHE signals were measured with the ImageJ software (National Institutes of Health, USA) using identical exposure settings across all samples.
Renal function assessment
Renal function was evaluated by measuring serum BUN, serum creatinine (SCR), and urinary protein levels. Fresh mouse blood samples were collected and centrifuged at 3,000 rpm for 10 min to obtain serum. Serum BUN concentrations were determined using a Urea Nitrogen Assay Kit (Urease Method) (C013-2-1, Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer’s instructions. For the assay, serum samples were diluted 1:80 with distilled water prior to measurement. SCR levels were measured using a Creatinine Assay Kit (C011-2-1, Nanjing Jiancheng Bioengineering Institute, China) following the manufacturer’s protocol. Urinary protein concentrations were quantified using a Urinary Protein Assay Kit (UP) (E2086, Beijing Applygen Gene Technology Co., Ltd., China) according to the manufacturer’s instructions.
siRNA transfection
The siRNAs targeting mouse NUP35 and a non-targeting control siRNA were chemically synthesized by Sicagene Biotechnology Co., Ltd. (Beijing, China) and transfected into BMDMs using Lipofectamine™ 3000 (L3000015, Thermo Fisher Scientific, USA). Briefly, BMDMs were seeded in 6-well plates and at 60%–70% confluency, cells were transfected with siRNAs at a final concentration of 150 nM. After 48 h of incubation, cells were harvested for downstream analyses, including RT-qPCR and western blotting to assess knockdown efficiency or for further functional assays.
The siRNA sequences targeting NUP35 were as follows:
sense: 5′-GUCCUCUUGUUGGAGCUACAA(dTdT)−3′
antisense: 5′-UUGUAGCUCCAACAAGAGGAC(dTdT)−3′.
CHX chase assay
To assess the stability of the NUP35 protein, BMDMs were treated with CHX (10µM, HY-12320, MedChemExpress, China) to inhibit de novo protein synthesis. Cells were collected at 0, 6, 12, and 24 h post-treatment, lysed in RIPA buffer supplemented with protease and phosphatase inhibitor cocktails, followed by immunoblotting analysis with anti-NUP35 (1:1000, NB100-93322, Novus Biologicals, USA).
Quantitative real-time PCR
Total RNA was extracted from kidney tissues or BMDMs using RNAiso Plus reagent (9109, Takara, Japan) according to the manufacturer’s instructions. cDNA was synthesized from 1 µg RNA using the Hifair® III 1st Strand cDNA Synthesis SuperMix reverse transcription kit (11141ES60, Yeasen, China). RT-qPCR was performed using SYBR Green Master Mix (11202ES08, Yeasen, China) on the QuantStudioTM3 Real-time PCR System (Applied Biosystems). Gene expression was normalized to mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and calculated by the 2–ΔΔCt method. Primer sequences are listed in Supplementary Table S1.
Western blotting and IP
BMDMs or kidney tissues were lysed in RIPA buffer containing protease and phosphatase inhibitors. Protein quantification was determined via the bicinchoninic acid assay. Equal amounts of protein were separated by 7.5% or 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Following blocking in Tris-buffered saline containing 0.1% Tween-20 containing 5% (w/v) skim milk for 1 h, membranes were incubated overnight at 4°C with the following primary antibodies: anti-UBA1 (1:1000, MA5-32402, Thermo Fisher Scientific, USA), anti-NUP35 (1:1000, NB100-93322, Novus Biologicals, USA), antiphospho-p65 (1:1000, 310013, ZEN-BIOSCIENCE, China), anti-p65 (1:1000, 8242, Cell Signaling Technology, USA), anti-phospho-IκBα (1:5000, ab92700, Abcam, USA), anti-IκBα (1:5000, ab32518, Abcam, USA), and anti-GAPDH (1:3000, 60004-1-lg, Proteintech, USA). After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:10000, Jackson ImmunoResearch, USA) for 1 hour at room temperature. Immunoreactive bands were visualized using enhanced chemiluminescence detection reagents.
For IP, BMDMs or kidney specimens were homogenized in IP lysis buffer (P0013, Beyotime, China) supplemented with protease inhibitors. The lysates were incubated overnight at 4°C with anti-UBA1 (2 µg/IP, ab264179, Abcam, USA), anti-NUP35 (2 µg/IP, NB100-93322, Novus Biologicals, USA), or control IgG (0.25 µg/mg, 30000-0-AP, Proteintech, USA) on a rotating platform. Immunocomplexes were captured by incubation with 10 µL protein A/G magnetic beads (88803, Thermo Fisher Scientific, USA), followed by three washes with PBS. Bound proteins were eluted by boiling in 1 × SDS loading buffer at 100°C for 15 min and analyzed by western blotting as described above.
Statistics
All data are expressed as mean ± SEM. Statistical analyses were performed using GraphPad Prism version 9.5.1 (GraphPad Software, La Jolla, CA, USA). Intergroup differences were assessed by unpaired two-tailed Student’s t-test for comparisons between two groups. For analyses involving multiple groups, either one-way or two-way ANOVA was employed, followed by Bonferroni’s multiple comparisons post hoc test. Survival data were analyzed using the Kaplan–Meier method with log-rank (Mantel–Cox) testing. A p value <0.05 was considered statistically significant.
Authors’ Contributions
S.-Y.L.: Conceptualization, investigation, data curation, formal analysis, and writing—original draft. C.C.: Conceptualization, methodology, formal analysis, writing—review and editing, and funding acquisition. L.-Y.J.: Data curation. W.-X.J.: Formal analysis. H.-H.L.: Conceptualization, funding acquisition, writing—review and editing, and supervision. All authors reviewed and approved the final version of the article.
Footnotes
Acknowledgments
The authors gratefully acknowledge the core facilities of their institution for technical support.
Data Availability
The data supporting the findings of this study are available within the article and its Supplementary Data. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE repository under accession number PXD074268. The RNA sequencing data have been deposited in the NCBI BioProject database under accession number PRJNA1400645 and will be publicly available upon publication. Additional data are available from the corresponding author upon reasonable request.
Author Disclosure Statement
All authors declare no conflict of interest.
Funding Information
This work was supported by
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.