
Abstract
Significance:
The global obesity epidemic is a significant risk factor for chronic metabolic diseases. Iron, an essential micronutrient involved in numerous biological processes, exhibits a dual role in health and disease. Understanding the mechanistic interplay between iron metabolism and adipocytes is crucial for elucidating the pathogenesis of obesity.
Recent Advances:
Emerging evidence indicates that iron plays a key role in adipose tissue physiology, and its dysregulation contributes substantially to obesity pathogenesis. Within adipocytes, iron exerts multifaceted effects, influencing lipid metabolism, differentiation, thermogenic capacity, secretory function, mitochondrial activity, and insulin sensitivity. Conversely, the metabolic status of adipose tissue reciprocally affects systemic iron homeostasis.
Goal of Review:
This review underscores the phenomenon of iron dysregulation in obesity. It examines the cellular mechanism describing iron demand and its dysregulation across adipose tissue subtypes. Following, it explores the functional pathology connecting iron imbalance to impaired energy metabolism, thermogenesis capacity, immune modulation, and secretion activity in adipose tissue, as well as iron-related oxidative stress, ferroptosis, heme toxicity, and cellular senescence within adipocytes in obesity. Then, it expands to the systemic level, highlighting the interorgan crosstalk in iron homeostasis, particularly among the liver, gut, and adipose tissue. Finally, it discusses the therapeutic strategies targeting iron homeostasis regulation to alleviate obesity and its related complications.
Future Direction:
Despite advancements, questions remain regarding depot-specific iron regulation and its relationship with metabolic dysfunction. Future studies should employ tissue-specific genetic models, well-designed human trials, and multiomics approaches to establish causality and translate the findings into targeted iron-modulating therapies for obesity. Antioxid. Redox Signal. 45, 44–62.

Introduction
Obesity, a major risk factor for a variety of chronic diseases, leads to a range of metabolic disorders, including metabolic dysfunction-associated steatotic liver disease, type 2 diabetes, hypertension, and atherosclerosis (Bray and Wilson, 2008; Piché et al., 2020; Polyzos et al., 2019). Its pathophysiology centers on adipose tissue dysfunction, which is characterized by chronic inflammation, adipocyte hypertrophy, metabolic inflexibility, and aberrant secretion of cytokines.
Iron is the most abundant transition metal in biology and indispensable for fundamental physiological processes. Mechanistically, complex crosstalk exists between iron homeostasis and energy metabolism, with iron serving as a critical regulator of glucose, lipid, and energy metabolism (Simcox and McClain, 2013; Hilton et al., 2023). Perturbations in iron metabolism are reliably correlated with metabolic disorders, as evidenced by altered expression of iron metabolism-related regulators in obesity. Transferrin receptor 1 (TfR1) level is negatively correlated with body mass index (BMI) and positively correlated with uncoupling protein 1 (UCP1) (Qiu et al., 2024). Ferritin and ferroportin (FPN) are upregulated, whereas transferrin (TF) is downregulated in adipose tissue from individuals with obesity (Moreno-Navarrete et al., 2014a). In addition, ceruloplasmin (Cp), hemojuvelin, and hepcidin (HAMP) are elevated (Luciani et al., 2011; Wu et al., 2014).
Adipose tissue has emerged as a key hub linking iron dysregulation to obesity and acts as an iron sensor and storage site (Aguree et al., 2023; Guglielmi et al., 2015; Qiu et al., 2024; Harrison et al., 2023). Iron deficiency may compromise adaptive thermogenesis capacity, thereby exacerbating obesity and its metabolic dysfunction (Yook et al., 2021a), while iron overload within adipocytes promotes oxidative stress and insulin resistance. Thus, there is a complex link between iron dysregulation and adipose dysfunction.
Given the complex role of iron in adipose tissue biology, understanding of its metabolism in adipose tissue is crucial for developing new therapeutic strategies for obesity. This review focuses on the paradox of iron dysregulation in obesity. It investigates the distinct iron demands across adipose tissue subtypes, linking the dysfunction of iron metabolism regulators to adipose pathology and detailing iron’s critical role in lipid metabolism, thermogenesis capacity, and secretion activity. This review also explores iron-mediated oxidative stress, ferroptosis, and senescence in dysfunctional adipocytes while revealing systemic connections among liver, gut, and adipose tissue crosstalk. Finally, emerging therapeutic strategies targeting iron homeostasis to mitigate obesity complications are discussed, offering mechanistic insights into iron metabolism and adipose tissue interaction for metabolic disease intervention.
Obesity: Focus on Iron Dysregulation
Obesity is characterized by an excessive accumulation of adipose tissues and is associated with altered iron metabolism. Studies show an inverse relationship between BMI and serum iron level (Laudisio et al., 2023), with iron deficiency commonly observed in individuals with obesity, particularly among pregnant women, adolescents, and children. This deficiency is not simply due to inadequate dietary intake, as insufficient iron absorption and increased metabolic demands also contribute (Mujica-Coopman et al., 2015; Zhao et al., 2015).
The relationship between obesity and iron is profoundly paradoxical (Fig. 1). Despite systemic iron deficiency, adipose tissue itself often exhibits iron overload (Pihan-Le Bars et al., 2016). This paradox is driven by obesity-related low-grade inflammation. Elevated HAMP and ferritin levels are closely linked to metabolic alteration and inflammation in adolescents with obesity (Rodriguez-Mortera et al., 2021). Inflammatory adipokines induce HAMP level (del Giudice et al., 2009), which blocks iron export from cells and accumulates iron in adipocytes with increasing expressions of iron uptake and storage-related genes (Oliveras-Cañellas et al., 2023). Reducing iron level specifically within adipocytes decreases overall body fat and leads to improvements in metabolic health (Zhang et al., 2021). Tissue iron overload promotes local oxidative stress and impairs insulin action. Therefore, understanding this paradox is crucial for the prevention and treatment of obesity and related diseases.
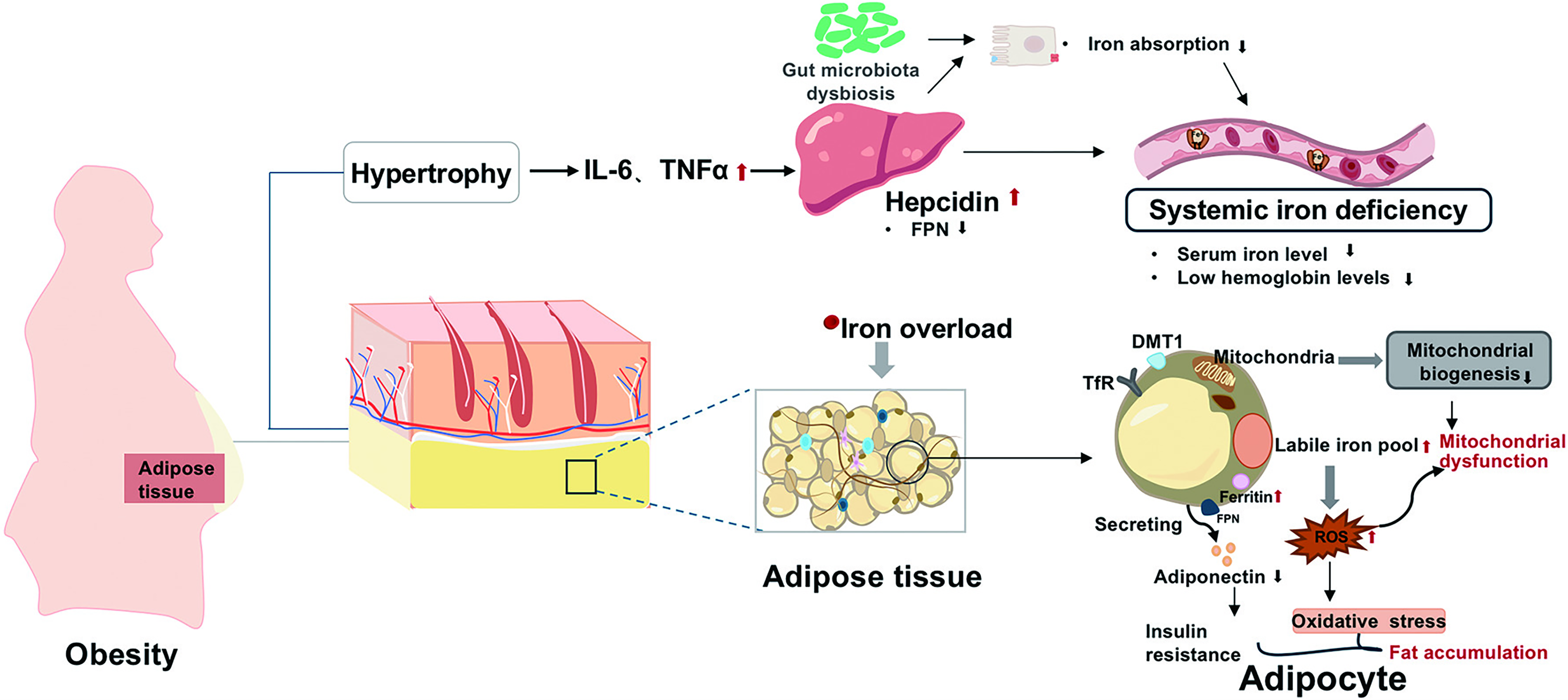
Iron Demand and Its Dysregulation Across Adipose Tissue Subtypes in Obesity
Adipose tissue exhibits remarkable heterogeneity and is classified into white (WAT), brown (BAT), and beige adipose tissue, each with distinct structural and functional characteristics, ranging from energy storage to dissipation (Min et al., 2019). Importantly, obesity perturbs iron metabolism in an adipocyte subtype-specific manner, differentially contributing to the pathogenesis of metabolic diseases. Iron handling properties across adipose tissues are summarized in Figure 2.
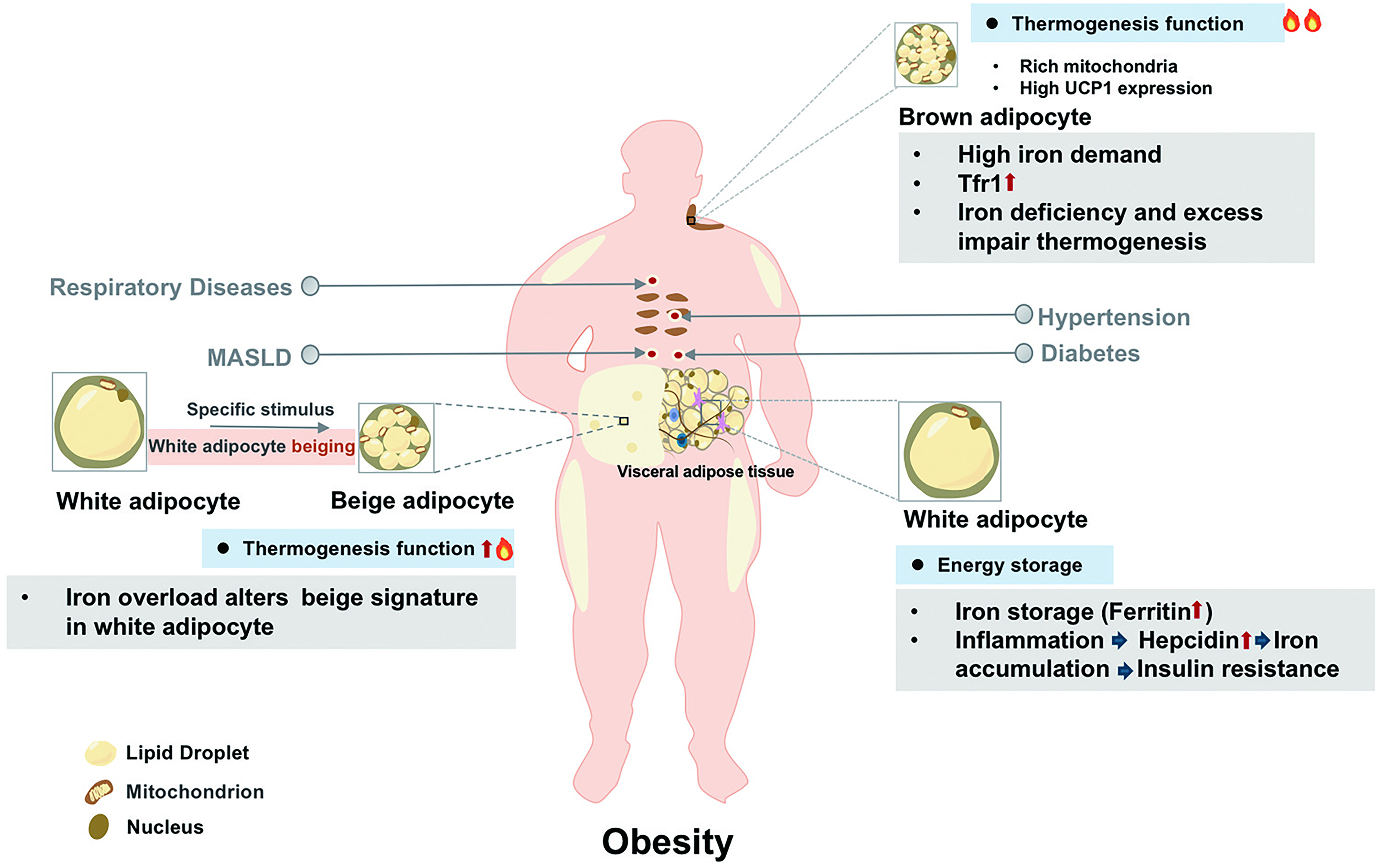
White adipose tissue
WAT serves as the primary energy storage site and the major secretory organ for adipokines. It uniquely expresses a higher level of iron storage protein ferritin. Under obese condition, WAT undergoes remodeling marked by hypoxia, inflammation, and fibrotic changes. Hypoxia upregulates ferritin heavy chain (FTH1), promoting iron retention that polarizes adipose tissue macrophages (ATMs) toward pro-inflammatory M1 phenotype (Ameka et al., 2022; Wang et al., 2025). Furthermore, obesity-associated inflammation upregulates HAMP, leading to iron retention in WAT through FPN degradation (Rodriguez-Mortera et al., 2021). Iron overload contributes to mitochondrial dysfunction, oxidative stress, and impaired insulin signaling.
Brown adipose tissue
BAT’s high energy expenditure for thermogenesis creates unique metabolic demands. It requires precise iron regulation to maintain mitochondrial function, relying on mitoferrin (Mfrn) for mitochondrial iron import, which is essential for iron–sulfur (Fe–S) cluster biogenesis and thermogenesis. This dependency creates a vulnerability to iron fluctuation. Iron deficiency disrupts UCP1 activity and impairs adaptive thermogenesis. TfR1 is expressed predominantly in thermogenic adipocytes (Qiu et al., 2020). Studies using adipocyte-specific Tfr1 knockout mice have confirmed the indispensable role of iron acquisition in sustaining thermogenesis and mitochondrial homeostasis (Li et al., 2020). Intriguingly, hamp deficiency also disrupts BAT function (Deschemin et al., 2023). High iron exacerbates metabolic risk by damaging BAT through impairing mitochondrial activity (Zhang et al., 2024). Enhanced iron regulation protein/iron-responsive element (IRP/IRE) interaction is increased in proportion to BAT, indicating the higher iron demand for thermogenic function (Yook et al., 2021c). Both iron deficiency and overload compromise thermogenesis, emphasizing the necessity for precise iron regulation in BAT.
Beige adipocytes
Beige adipocytes display remarkable plasticity, transitioning between white-like and brown-like phenotypes in response to stimuli. The browning process involves iron-dependent metabolic reprogramming, including increased mitochondrial biogenesis and heme oxygenase-1 (HO-1) activation (Choi et al., 2022). Activation of β3-adrenoreceptor (ADRβ3) drives beige adipogenesis through IRP/IRE signaling and multiorgan iron mobilization (Yook et al., 2021b). Iron overload by hamp knockout impairs thermogenesis by reducing UCP1 and mitochondrial respiration in beige adipocytes (Deschemin et al., 2023). Iron deficiency during differentiation disrupts mitochondrial respiratory complex assembly with milder effects on beige versus brown adipocytes (Yook et al., 2021c). Interestingly, iron chelation with deferasirox enhances beige fat quantity and activity under high-iron condition (Nazari et al., 2022). Collectively, it establishes iron as an active regulator for beige adipocyte plasticity. Its precise level appears critical for the transcriptional and metabolic remodeling required for thermogenesis, primarily through ensuring the integrity of mitochondrial electron transport chain and the function of UCP1.
Impact of Dysfunction in Molecular Regulators of Iron Metabolism on Adipocyte Function
Adipocytes express a complete set of iron metabolism regulators. In obesity, dysregulation of these regulators alters adipocyte biology through influencing lipid metabolism, insulin sensitivity, and thermogenic capacity (Fig. 3).
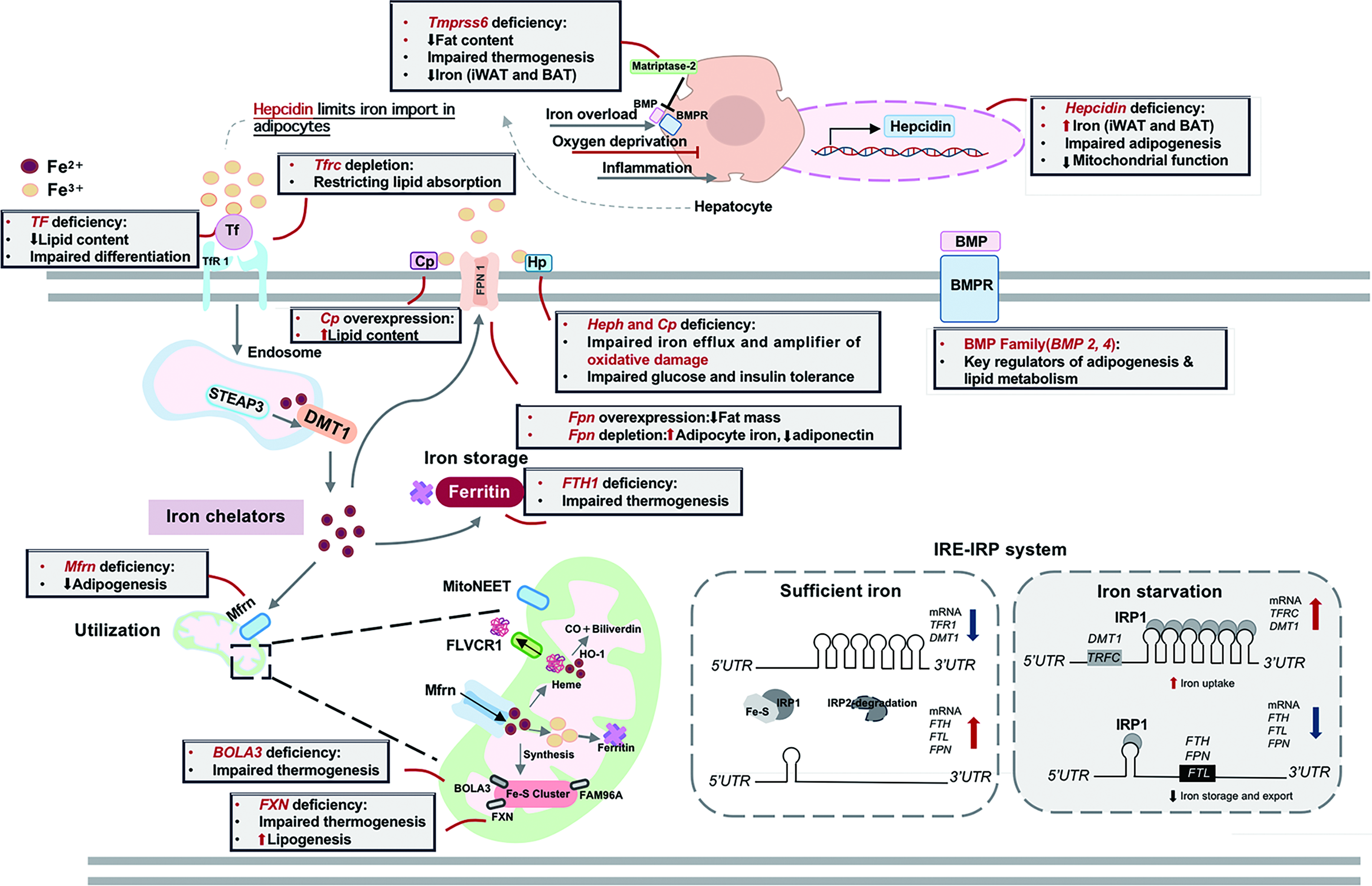
Systemic iron metabolism regulators
Hepcidin, encoded by HAMP gene, is a critical regulator of systemic iron homeostasis, which is primarily synthesized in liver and also expressed in adipose tissue. Serum hepcidin levels are elevated in obesity (Vuppalanchi et al., 2014). Studies using Hamp knockout mice reveal that iron accumulates in both inguinal WAT (iWAT) and BAT (Deschemin et al., 2023). Iron overload also alters the beige signature in iWAT through impairing de novo adipogenesis and decreasing mitochondrial respiration (Deschemin et al., 2023). Bone morphogenetic protein (BMP) signaling is the major upstream pathway inducing HAMP expression. BMP family members exhibit distinct effects on adipose tissue. BMP2 modulates energy storage and adipogenesis (Guiu-Jurado et al., 2016), while BMP4 promotes brown adipocyte whitening, reduces white adipocyte mass, and improves insulin sensitivity (Modica et al., 2016; Qian et al., 2013). Matriptase-2, encoded by Tmprss6, is a repressor of HAMP (Folgueras et al., 2008). Its deficiency not only reduces fat deposits and protects against obesity by increasing fat lipolysis (Folgueras et al., 2018; Wu et al., 2023) but also impairs cold-induced beige adipocyte formation and BAT thermogenesis (Li et al., 2020). Therefore, it acts as a modulator in adipose tissue phenotype and function (Wu et al., 2023).
Cellular iron metabolism regulators
Cellular iron uptake
TfR1 exhibits elevated expression in BAT compared with WAT, reflecting differential iron requirements for thermogenesis (Zhang et al., 2021). Adipocyte-specific Tfrc knockout protects against obesity by limiting lipid absorption in intestine, mechanistically through impaired vesicular transport within enterocytes, which reduces lipid flux into circulation and ameliorates fat accumulation (Zhang et al., 2021). Tf knockdown impairs adipocyte differentiation and decreases intracellular lipid accumulation through downregulation of adipogenic markers (Moreno-Navarrete et al., 2014b) while also altering the expression of genes involved in glucose transport, mitochondrial function, and obesity (McClain et al., 2018).
Cellular iron utilization
FTH1 plays a central role in iron sequestration. Its knockout in 3T3-L1 cells preserves adipogenic gene expression but reduces mitochondrial biogenesis markers (Moreno-Navarrete et al., 2014b) and disrupts thermal homeostasis by lowering BAT temperature and impairing adaptive thermogenesis (Blankenhaus et al., 2019).
Cellular iron efflux
FPN1 is the sole cellular iron exporter, and its manipulation in adipocytes yields complex outcomes. Adipocyte-specific Fpn overexpression alleviates metabolic dysregulation in obese mice through reducing body weight, decreasing fat mass, and improving glucose tolerance and insulin sensitivity (Zhang et al., 2021). Conversely, depletion of adipocyte Fpn increases adipocyte iron, decreases adiponectin expression, and increases insulin resistance (Gabrielsen et al., 2012), while another study finds no significant effects on iron level or insulin sensitivity (Britton et al., 2018). Multicopper ferroxidases (MCFs), including hephaestin (Heph) and Cp, oxidize ferrous iron to ferric iron to facilitate FPN1-mediated iron efflux. Cp-deficient mice exhibit increased macrophage infiltration, altered adipokine profile, and accumulation of subcutaneous adipose tissue without iron deposition (Raia et al., 2023). Heph−/−Cp−/− impairs iron efflux, causing adipocyte iron deposition, increased fasting glucose level, and impaired glucose and insulin tolerance (Zheng et al., 2018). These findings establish the cooperative role of MCFs in maintaining adipocyte iron balance and metabolic function.
Mitochondrial iron metabolism
MitoNEET is an outer mitochondrial membrane protein essential for Fe–S cluster maturation and regulation (Lipper et al., 2019). It protects against iron overload-induced insulin resistance by regulating mitochondrial iron level (Tam et al., 2023). Adipose-specific mitoNEET overexpression triggers chronic adipose tissue expansion and obesity development (Kusminski et al., 2012), yet paradoxically improves systemic lipid and carbohydrate homeostasis by altering mitochondrial matrix iron metabolism (Kusminski et al., 2014). When overexpression in perivascular adipose tissue, mitoNEET enhances cold resistance and upregulates thermogenic gene expression (Xiong et al., 2017), demonstrating its multifaceted role in mitochondrial iron handling and energy metabolism.
Mfrn transports iron into mitochondria for the biosynthesis of Fe–S clusters, heme, and other metalloenzyme cofactors. Mfrn depletion in preadipocytes suppresses adipogenic gene expression during differentiation process (Chen et al., 2015), while its overexpression reduces adipose lipase expression in WAT and BAT (Guan et al., 2024), linking mitochondrial iron delivery to adipogenesis and lipid metabolism. BolA-like protein 3 facilitates mitochondrial Fe–S cluster assembly and protects clusters from oxidative damage (Melber et al., 2016; Uzarska et al., 2016). Its deficiency suppresses UCP1 expression by impairing mitochondrial homeostasis but does not affect lipogenesis in differentiated beige adipocytes (Bai et al., 2020). Frataxin (FXN) is involved in Fe–S cluster synthesis. BAT-specific deficiency increases oxygen consumption, disrupts mitochondrial structure, promotes lipid accumulation, increases lipogenesis, and impairs thermogenesis (Turchi et al., 2020). Similarly, WAT-specific deficiency leads to metabolic dysfunction characterized by adipocyte hypertrophy, lipid accumulation, impaired lipolysis, and dysregulated lipid metabolism (Turchi et al., 2023).
Iron’s Role in Adipocyte Function and Metabolic Risk
Obesity impairs systemic metabolism homeostasis by disrupting the integrated metabolic, immune, and endocrine functions of adipose tissue. Iron, an essential cofactor for metabolic enzymes and mitochondrial energy production, is critically involved in regulating adipose tissue biology. Both iron deficiency and overload trigger pathological cascades that compromise adipocyte metabolic homeostasis, increasing susceptibility to obesity-associated metabolic disorders (Fig. 4).

Lipid metabolism
The capacity of adipocytes for triglyceride (TG) synthesis and storage as well as for lipolysis-mediated free fatty acid release is influenced by iron status. Iron exerts potentially pro-adipogenic effects through stimulating body weight gain, increasing fat mass, and enlarging adipocyte size in high-fat diet (HFD)-fed rats (Tinkov et al., 2013). Iron-dextran administration disturbs lipid metabolism by increasing serum TG and low-density lipoprotein cholesterol while decreasing high-density lipoprotein cholesterol (Tang et al., 2021). Mechanistically, iron impacts lipid metabolism through three key processes, including lipid accumulation during adipogenesis, lipid release during lipolysis, and lipid peroxidation (Hubler et al., 2015). Consequently, iron overload leads to dyslipidemia manifesting as suppressed adiponectin synthesis, inhibited lipoprotein lipase and hepatic lipase activities, and elevated serum TG level in adipocytes (Tang et al., 2021). Overall, iron overload appears to increase lipogenesis, reduce lipolysis, and promote oxidative stress in adipocytes. However, contradictory findings highlight that iron’s role depends on various conditions. High-iron diet-fed db/db mice exhibit widespread and smaller lipid vacuole deposition in parenchymal cells (Ma et al., 2019). In other models, a high-iron diet decreases fat accumulation and lipid deposition by downregulating adipogenesis and upregulating lipolysis in adipose tissues of mice (Xiong et al., 2022). Addition of TF or TF-bound iron promotes lipolysis in adipocytes (Rumberger et al., 2004). These discrepancies suggest that the metabolic impact of iron depends on experimental models, iron dosage, and metabolic context. Therefore, iron regulates adipocyte lipid metabolism through complex and multifaceted mechanisms in obesity.
Adipocyte differentiation
Iron also plays an indispensable role in adipocyte differentiation (Ma et al., 2021), the process by which undifferentiated precursor cells become mature adipocytes essential for energy storage and metabolic homeostasis. This process is tightly regulated by iron availability, with requirements that vary across adipocyte subtypes and differentiation stages. Brown adipocytes have a higher iron demand than white or beige adipocytes during differentiation, as evidenced by the fact that chelation of labile iron pool dampens H1B1B brown adipocyte differentiation but has milder effect on C3H/10T1/2 beige adipocyte differentiation (Yook et al., 2021c). Such subtype-specific iron requirements reveal a complex relationship between iron availability and adipogenesis.
Iron overload has detrimental effects on 3T3-L1 adipocyte differentiation, whereas iron deprivation by knocking down Tf or using deferoxamine (DFO) inhibits differentiation, adipogenesis, and mitochondrial biosynthesis in adipocytes (Moreno-Navarrete et al., 2014b). It emphasizes that precise iron homeostasis is essential for optimal adipocyte differentiation. The molecular mechanisms linking iron to adipogenesis involve regulation of key iron-handling proteins. During differentiation, TF expression and secretion are increased to facilitate iron uptake to meet rising metabolic demands (Moreno-Navarrete et al., 2014b). Ferritin level is increased, and IRP2/IRE complex is more abundant in 3T3-L1 adipocytes maturing into functional adipocytes (Festa et al., 2000). This coordinated upregulation suggests an adaptation in iron metabolism to support the growing energy demands for adipocyte maturation.
Beyond its metabolic roles, iron also exerts epigenetic regulation on adipogenesis. Iron-dependent epigenetic enzymes such as Jumonji domain-containing 1A and 10–11 translocation 2 require nuclear iron transport via poly(rC)-binding proteins to activate pro-adipogenic gene networks during early stages of adipocyte differentiation (Suzuki et al., 2023). Consequently, subcellular iron depletion, whether due to inadequate iron supply or impaired subcellular iron transport, reduces iron-dependent histone and DNA demethylation and suppresses the terminal adipocyte differentiation (Suzuki et al., 2023). Thus, iron also functions as an epigenetic modulator of adipogenic transcriptional programming.
In summary, adipocyte differentiation depends on iron that acts as a metabolic cofactor supporting energy and biosynthesis and as an essential regulator of epigenetic remodeling. Disruptions in iron homeostasis compromise adipogenic efficiency, illustrating the critical balance required for healthy adipose tissue development.
Thermogenic capacity
Thermogenesis is critical for maintaining body temperature and energy homeostasis. Adaptive thermogenesis, a mitochondria-dependent and iron-intensive process, requires rapid mitochondrial biogenesis (Yook et al., 2021c) and is mechanistically governed by UCP1 in brown and beige adipocytes (Chang et al., 2019). Adipocyte browning, the induction of thermogenic phenotype, is an iron-dependent process (Shi et al., 2023). Thermogenic stimuli trigger cell-autonomous iron uptake and mitochondrial compartmentalization as well as enhanced mitochondrial respiration. ADRβ3 agonist suppresses hepcidin expression, mobilizing splenic macrophage iron store to enhance systemic iron availability for thermogenesis (Yook et al., 2021b). Moreover, iron supplementation promotes thermogenesis through peroxisome proliferator-activated receptor γ coactivator 1 α (PGC-1 α) and adipose TG lipase (ATGL) –mediated lipolysis (Mai et al., 2024). Conversely, iron deficiency impairs mitochondrial biogenesis, attenuates adaptive thermogenesis, and further exacerbates obesity (Yook et al., 2021a). Enhanced IRP/IRE interaction and mitochondrial iron influx are essential for thermogenic adipocyte differentiation. Iron homeostasis balances adipocyte thermogenic capacity and lipid storage. Tight regulation of iron level ensures optimal mitochondrial function, UCP1 activity, and systemic energy expenditure, highlighting iron as a key modulator of metabolic heat production.
Mitochondrial function
Obesity disrupts mitochondrial homeostasis in adipocytes, leading to reduced mitochondrial mass and impaired oxidative capacity. Mitochondria are central to adipocyte functions, including differentiation, thermogenesis, and the regulation of systemic insulin sensitivity (De Pauw et al., 2009; Kusminski and Scherer, 2012). Mitochondrial biogenesis is not only essential for metabolic process in adipocyte differentiation but also a driving force for iron influx during brown adipocyte formation (Yook et al., 2021c). Manipulation of mitochondrial iron levels has an impact on energy metabolism. DFO activates mitochondrial biogenesis in epididymal adipose tissue of ob/ob mice, partially ameliorating metabolic dysfunction (Yan et al., 2018). The functional integrity of mitochondria critically depends on iron-associated pathways. Fe–S clusters are essential cofactors for metabolic enzymes in tricarboxylic acid cycle and electron transport chain complexes I–IV. Mfrns maintain mitochondrial iron equilibrium, and its deficiency attenuates mitochondrial respiratory complexes’ expression, reduces mitochondrial respiration rate, and decreases adenosine triphosphate content in the differentiated 3T3-L1 adipocytes (Chen et al., 2015). Thus, mitochondrial function in adipocytes is tightly linked to iron homeostasis, as iron fluctuation impairs metabolic outcome and adipogenic capacity.
Secretion activity
Adipocytes function as endocrine cells secreting adipokines that regulate systemic metabolism (Scheja and Heeren, 2019). Iron imbalance disrupts this endocrine function, particularly affecting the production of leptin and adiponectin. Acute iron overload decreases the secretion of adipocytokines, including adiponectin, leptin, and resistin, in adipocytes (Tang et al., 2021). Leptin suppresses appetite and improves insulin sensitivity. Ferric ammonium citrate downregulates leptin mRNA in 3T3-L1 and primary mouse adipocytes, an effect reversed by DFO (Gao et al., 2015). Mechanistically, iron alters leptin secretion by regulating cyclic adenosine monophosphate (cAMP)-response element-binding protein signaling (Gao et al., 2019). Adiponectin, which protects against hepatic steatosis and metabolic dysfunction, is inversely correlated with iron content in obese adipose tissue (Pihan-Le Bars et al., 2016). FeSO4 treatment suppresses adiponectin in 3T3-L1 and primary adipocytes (Gabrielsen et al., 2012). Thus, iron excess acts as a potent negative regulator of both leptin and adiponectin. In summary, iron plays a role in regulating the secretion activity of adipocytes, thereby affecting food intake, insulin sensitivity, and systemic metabolic health.
Macrophage polarization
Immune-metabolic crosstalk within adipose tissue reveals macrophage polarization as a critical determinant of iron homeostasis. Two macrophage populations exist, differing in iron content and expression of iron-metabolism-related proteins (Ikeda et al., 2022). M1 macrophages that are characterized by low iron status drive inflammatory responses, while iron-rich M2 macrophages resolve inflammation by sequestering excess iron and releasing it in response to localized iron deficiency, protecting adipose tissue from iron overload-induced lipotoxicity and insulin resistance (Hubler et al., 2015). Concurrently, iron handling by ATMs may regulate not only local but also systemic homeostasis. Through M2 macrophage-mediated iron storage or release, macrophages maintain tissue iron balance while modulating inflammatory cytokines that influence systemic insulin sensitivity. A phenotypic shift in ATMs from anti-inflammatory M2 to pro-inflammatory M1 phenotype disrupts iron metabolism, further exacerbating chronic inflammation and insulin resistance, which are hallmarks of obesity-related metabolic complications (Xu et al., 2023). Taking together, macrophage polarization links adipocyte hypertrophy and insulin resistance via its regulation of iron handling in macrophages.
Insulin sensitivity
Insulin resistance is a key early feature of metabolic dysfunction, accelerating adipose tissue pathology and type 2 diabetes pathogenesis (James et al., 2021). A complex link exists between iron overload and insulin resistance (Sobieska et al., 2024). Clinical observations show that serum ferritin and TF levels are inversely associated with adipocyte insulin resistance (Wlazlo et al., 2013). In human adipose tissue biopsies, Tf expression positively correlates with insulin sensitivity, while FTH1 shows a negative correlation (McClain et al., 2018). Obesity induces iron overload in adipose tissues, which subsequently results in adipocyte insulin resistance. DFO improves insulin sensitivity in epididymal adipose tissues of ob/ob mice by increasing phosphorylation of insulin receptor, protein kinase B, and glycogen synthase kinase 3β (Yan et al., 2018). Collectively, excessive iron might diminish insulin sensitivity, highlighting iron status as a modulator of adipose tissue function in metabolic health.
Pathological Mechanisms Linking Iron Dysregulation to Obesity
Iron dysregulation and obesity are interconnected through a complex network of pathological mechanisms centered on adipose tissue dysfunction. Central to this nexus are iron-mediated oxidative stress and ferroptosis driven by lipid peroxidation. Concurrently, hemoglobin-derived heme toxicity exacerbates inflammation and mitochondrial impairment in adipocytes. Furthermore, iron overload accelerates cellular senescence, promoting a pro-fibrotic and insulin-resistant microenvironment in adipocytes. These processes form a vicious cycle wherein iron accumulation promotes metabolic deterioration, and metabolic dysfunction further disrupts iron homeostasis.
Mechanisms of obesity-driven oxidative stress
Redox equilibrium in adipose tissue is critical for metabolic health. Under physiological condition, reactive oxygen species (ROS) homeostasis is maintained by balancing pro-oxidant systems, including mitochondrial electron transport chain and nicotinamide adenine dinucleotide phosphate oxidases, with antioxidant defenses, such as glutathione system, superoxide dismutase (SOD), and peroxiredoxin (PRX) enzymes. However, obesity disrupts this equilibrium, leading to excessive ROS accumulation and decreased antioxidant defenses (Sun et al., 2020; Yu et al., 2023)
Mitochondria are the primary source of ROS. Their dysfunction in adipocytes triggers oxidative stress, which in turn exacerbates metabolic dysregulation and systemic inflammation. Moreover, obesity-induced adipocyte hypertrophy creates a hypoxic and metabolically stressed microenvironment, impairing oxidative phosphorylation (OxPhos) and amplifying oxidative stress (Baldini et al., 2021).
The regulation of oxidative stress in adipocytes is increasingly recognized as a critical determinant of metabolic health. Adipocyte-specific inhibition of OxPhos via CR6-interacting factor 1 knockout activates mitochondrial stress response that upregulates fibroblast growth factor 21 and growth differentiation factor 15, improving systemic energy expenditure and glucose homeostasis while protecting against obesity and insulin resistance (Choi et al., 2020). Conversely, adipocyte-specific deletion of SOD triggers an adaptive mitochondrial stress response that enhances fatty acid oxidation, mitochondrial biogenesis, and energy expenditure, thereby counteracting obesity (Han et al., 2016). Nuclear factor erythroid 2-related factor 2 (NRF2) ablation alleviates oxidative stress-induced lipid accumulation in obese adipose tissue (Sun et al., 2020). Prx3 deficiency exacerbates obesity-linked mitochondrial oxidative stress, adipocyte hypertrophy, and metabolic dysfunction, while Prx5 suppresses adipogenesis by modulating cytosolic/mitochondrial ROS and adipogenic gene expression, with its loss worsening diet-induced obesity and hepatic steatosis (Huh et al., 2012; Kim et al., 2018). Solute carrier family 7 member 10 (SLC7A10) maintains glutathione level and mitochondrial respiration, and its deficiency exacerbates obesity, adipocyte hypertrophy, and insulin resistance (Jersin et al., 2021). These molecular mechanisms govern adipose resilience to oxidative damage and offer promising avenues for therapeutic intervention, including natural products with antioxidant capacity for ROS scavenging in adipocytes to combat obesity (Perez-Torres et al., 2021). Moving forward, it will be crucial to understand how iron dysregulation, often coexisting with obesity, interacts with oxidative stress in adipocytes.
Iron-mediated oxidative stress and ferroptosis in adipose dysfunction
Oxidative stress disrupts lipid metabolism and activates inflammatory signaling pathways, promoting lipogenesis while inhibiting lipolysis and β-oxidation in adipocytes. This pro-oxidative state is further exacerbated by iron-driven Fenton reaction, which promotes lipid peroxidation and cellular membrane damage (Kakhlon and Cabantchik, 2002). Chronic inflammation in adipose tissue further exacerbates this dysregulation. Adipocyte inflammation hinders adipogenesis and leads to iron accumulation (Oliveras-Cañellas et al., 2023). Downregulation of antioxidant system induces accumulation of lipid aldehydes, including trans-4-hydroxy-2-nonenal and trans-4-oxo-2-nonenal, enhancing protein carbonylation and promoting metabolic dysfunction (Long et al., 2013).
Ferroptosis, an iron-dependent form of regulated cell death driven by lipid peroxidation, plays a critical role in adipose tissue dysfunction in obesity (Fig. 5). Obesity-driven inflammation upregulates HAMP, creating systemic iron deficiency concurrent with cellular iron accumulation, an imbalance that ultimately promotes ferroptosis (Catapano et al., 2025). Bioinformatics analyses have identified key ferroptosis-related genes in obesity, including interleukin-6 (IL-6), vascular endothelial growth factor A, signal transducer and activator of transcription 3, and prostaglandin endoperoxide synthase 2, revealing their association with immune cell infiltration (Li et al., 2023). In addition, aldo-keto reductase family 1-member C1, nuclear receptor coactivator 4, and glutamate-cysteine ligase catalytic subunit have emerged as predictive biomarkers for weight control outcomes, linking ferroptosis-related inflammation to long-term metabolic regulation (Li et al., 2023).
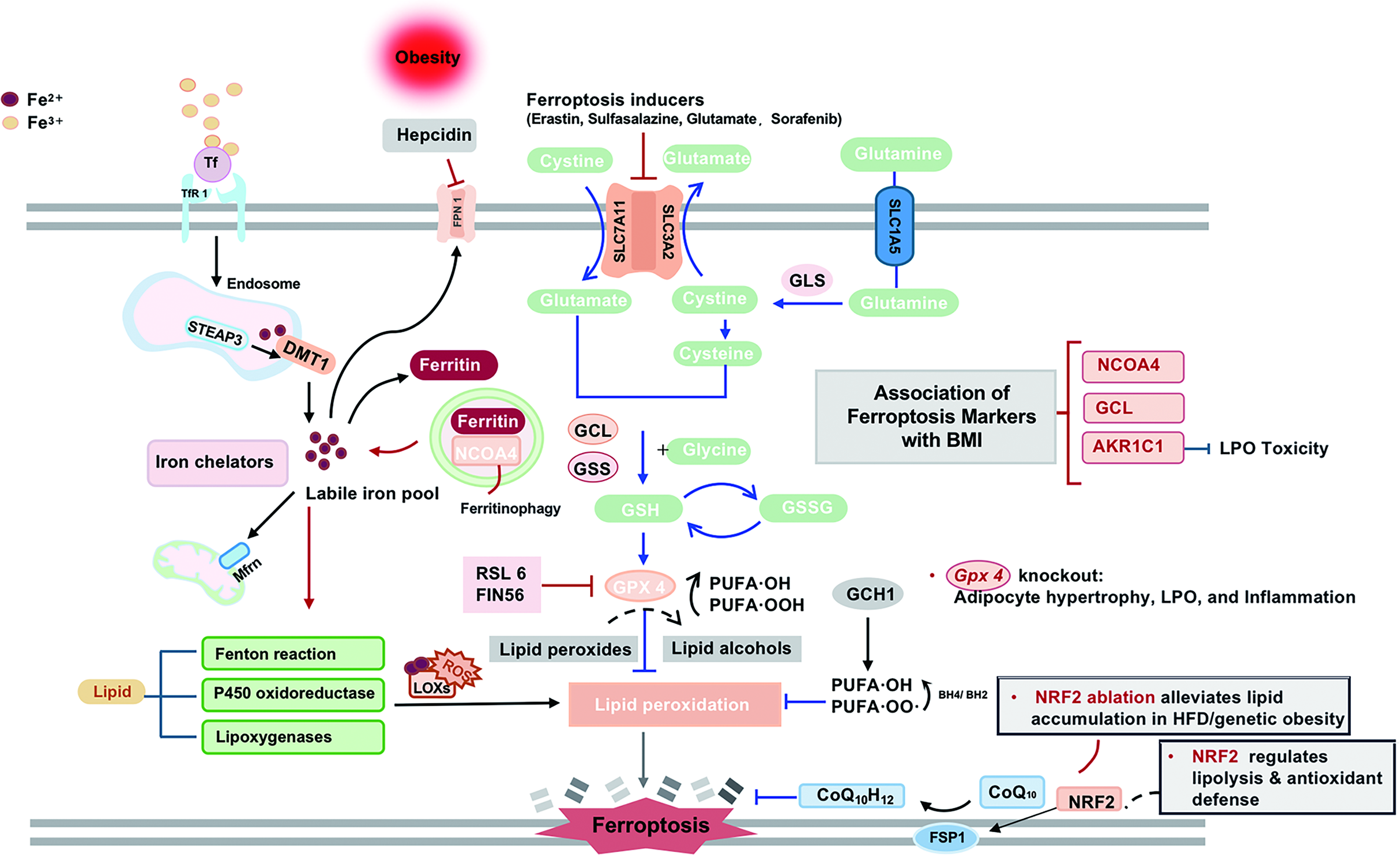
Key molecular regulators of ferroptosis include glutathione peroxidase 4 (GPX4), which protects cells from oxidative damage by neutralizing lipid peroxidation. Adipocyte-specific Gpx4 knockout causes hypertrophy, lipid peroxidation accumulation, and macrophage infiltration (Schwarzler et al., 2022). NRF2 inhibition impedes lipolysis and antioxidant in 3T3-L1 adipocytes with downregulation of adipose triacylglyceride lipase, ferroptosis suppressor protein 1 and SOD2 (Hu et al., 2024). While the precise mechanism of ferroptosis in adipose tissue is largely unknown, existing evidence supports its involvement in obesity pathogenesis.
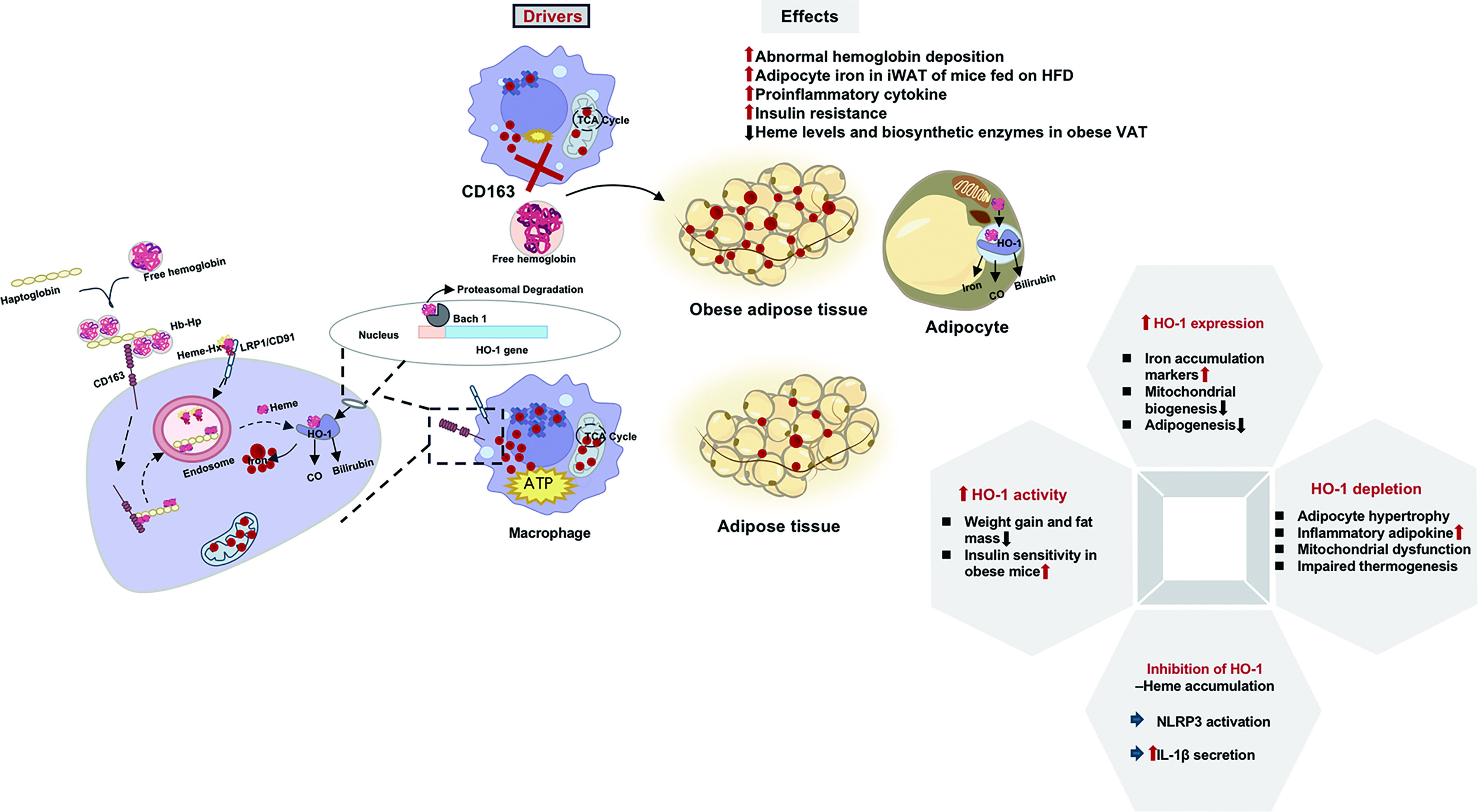
Hemoglobin-derived heme toxicity
Adipose tissue iron homeostasis is critically regulated through the hemoglobin degradation pathway by specialized macrophage populations. Under physiological condition, tissue-resident macrophages phagocytose senescent erythrocytes and process hemoglobin-derived heme via HO-1 pathway. However, obesity disrupts this delicate equilibrium through multiple mechanisms. Impaired microcirculation in obese adipose tissue leads to abnormal hemoglobin deposition. CD163+ macrophages normally clear hemoglobin–haptoglobin complexes through receptor-mediated endocytosis, but CD163 deficiency elevates adipocyte iron in iWAT of HFD-fed mice (Schleh et al., 2024) and triggers proinflammatory cytokine release that exacerbates insulin resistance (Etzerodt and Moestrup, 2013; Schleh et al., 2024).
HO-1, the rate-limiting enzyme in heme catabolism, plays significant roles in adipocyte function. Its expression positively correlates with adipocyte iron accumulation markers and negatively correlates with mitochondrial biogenesis and adipogenesis markers (Moreno-Navarrete et al., 2017). Increasing HO-1 activity reduces weight gain, decreases fat mass, remodels adipose tissue, improves insulin sensitivity, and increases serum adiponectin in mice with obesity (Burgess et al., 2010; Li et al., 2008; Nicolai et al., 2009). Adipocyte-specific HO-1 depletion causes larger fat pads, adipocyte hypertrophy, elevated inflammatory adipokines, mitochondrial dysfunction, and impaired thermogenesis (Singh et al., 2017). Adipocytes themselves express HO-1 at levels comparable to macrophages, enabling direct processing of extracellular heme.
Heme overload in adipocytes drives metabolic impairment. HO-1 inhibition causes heme accumulation, which induces interleukin-1 beta secretion through NOD-like receptor thermal protein domain-associated protein 3 inflammasome activation (Chen et al., 2019; Li et al., 2014). Free heme activates Toll-like receptor 4/nuclear factor κB signaling, creating a pro-inflammatory microenvironment that exacerbates metabolic dysfunction (Drummond et al., 2019; Kang et al., 2021). Progesterone receptor membrane component 2 deficiency in brown adipocyte impairs mitochondrial respiratory capacity, compromises body temperature maintenance, and affects systemic glucose homeostasis (Galmozzi et al., 2019).
Hemoglobin-derived heme serves as both a crucial iron source and potential metabolic disruptor. Macrophage-adipocyte crosstalk through CD163/HO-1 signaling regulates iron homeostasis and inflammation (Fig. 6). Therapeutic modulation of heme metabolism may improve obesity-related metabolic dysfunction.
Adipocyte senescence
Iron-induced senescence has profound pathological consequences (Zeidan et al., 2021). Excess iron drives ROS generation, causing DNA damage and activating p53 pathway to trigger cellular senescence (Xu et al., 2023; Zhou et al., 2018). The senescent phenotype manifests as increased lipid accumulation and is characterized by secretion of pro-inflammatory mediators including tumor necrosis factor-α and IL-6 (Ahmed et al., 2024; Maus et al., 2023). Obesity drives adipocytes to senescence by activating a cell cycle program (Li et al., 2021). Evidence reveals an interplay between iron dysregulation and adipocyte senescence in obesity pathogenesis. Aged adipose tissue and macrophage express high FTH level, indicating increased intracellular iron (Zhu et al., 2025). A significant correlation exists between cellular Fe2+ level and senescence-associated β-galactosidase activity in senescent adipose tissue (Shu et al., 2022), suggesting direct evidence of iron’s role in metabolic deterioration. These findings may establish iron as one of the key drivers of adipocyte senescence.
Therapeutic potential is emerging through natural compounds like saponins, which alleviate age-related adipose inflammation by selective downregulation of FTH1 in senescent adipocytes (Zhu et al., 2025). Targeting iron metabolism in senescent cells could be a promising strategy for obesity-related metabolic disorder. Nevertheless, the link between iron accumulation and adipocyte senescence in obesity still remains unanswered.
Cross-Tissue Iron Communication Through Liver–Adipose Tissue–Gut Axis in Obesity
Obesity pathophysiology involves complex interorgan crosstalk with gut microbiota dysbiosis, compromised intestinal barrier, adipose tissue inflammation, and hepatic metabolic disturbances acting together. These interconnected alterations promote secretion of proinflammatory cytokines and metabolites into circulation, exacerbating metabolic dysfunction (Konrad and Wueest, 2014). Iron metabolism regulation in obesity involves interactions among multiple organs, particularly through the liver–adipose tissue–gut axis, an integrated network with feedback loops that become dysfunctional in obesity (Fig. 7). Evidence shows that mitoNEET deficiency triggers the secretion of adiponectin from adipose tissue and fibroblast growth factor (Fgf15) from the gut, which then acts on the liver to reduce inflammation and injury, proving the role of iron metabolism in this axis (Hu et al., 2016).
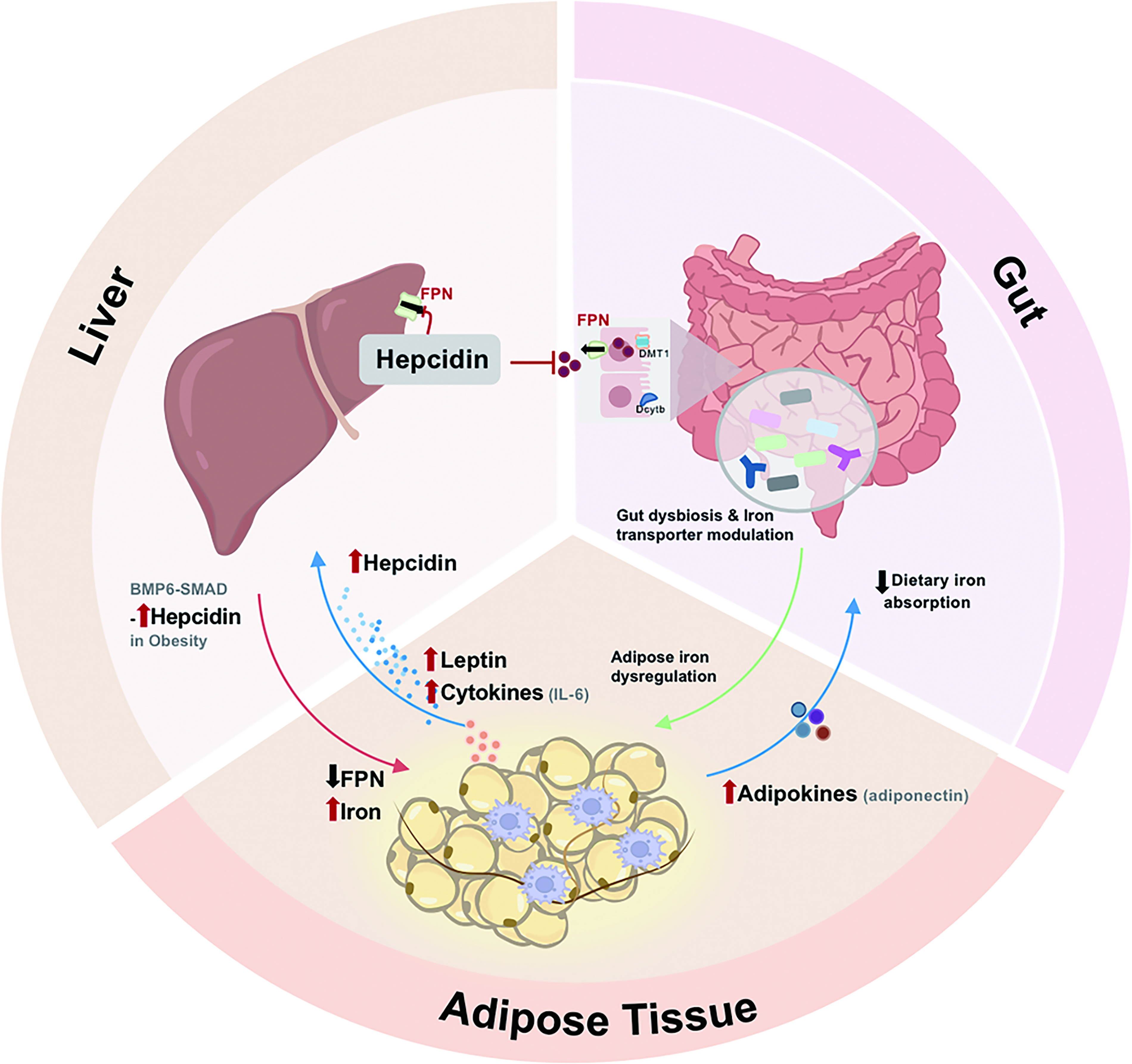
The liver serves as the central regulator of systemic iron metabolism through hepcidin production. Liver sinusoidal endothelial cells sense systemic iron status and produce BMP6. This, in turn, activates the BMP6-mothers against decapentapleigic hmolog (SMAD) pathway in hepatocytes to modulate hepcidin secretion. Obesity-associated inflammation, in which cytokines such as IL-6 further stimulates excessive hepcidin production, leads to pathological iron retention in adipose tissue. Importantly, adipocyte-derived leptin potently enhances hepatic hepcidin expression (Chung et al., 2007; Coimbra et al., 2013), thereby driving the vicious cycle of the liver-adipose tissue axis in obesity.
The gut–adipose tissue axis represents another site of interaction for iron homeostasis regulation. HFD-induced alterations in gut microbiota composition significantly impact iron metabolism. Dysbiosis may worsen iron deficiency by competing for dietary iron or impairing its absorption (Sun et al., 2024; Yan et al., 2024). Microbial metabolites such as butyrate modulate intestinal iron absorption and macrophage FPN expression (Xiao et al., 2024), providing a direct molecular link between the gut microbiome and systemic iron distribution. The microbial changes coordinate with altered expression of intestinal iron transporters to influence whole-body iron distribution. Experimental evidence demonstrates that lowering iron by adipocyte-specific Tfrc depletion protects against metabolic disturbances through restricting intestinal lipid absorption (Zhang et al., 2021), while adipose tissue iron deficiency triggers a signal to the gut that attenuates lipid absorption (De Siqueira and Villanueva, 2021; Robertson et al., 2021). It reveals an endocrine function of adipocytes in regulating gut function based on their iron status.
The gut–adipose tissue axis coordinates systemic iron metabolism in obesity. Integrative multiomics reveal how specific microbial metabolites correlate with host iron metabolism networks and metabolic outcomes in obesity (Xiao et al., 2024). Study supports the involvement of gut–adipose tissue–liver axis in systemic regulation of glucose metabolism (Castells-Nobau et al., 2025), and similar integrative mechanisms may exist in iron regulation. Gut microbiota regulates systemic iron metabolism through mechanisms beyond absorption, with microbial dysbiosis potentially exacerbating obesity-related metabolic impairments (Mayneris-Perxachs et al., 2022).
The effectiveness of utilizing microbial therapies, particularly probiotics involved in iron metabolism, has been proved for the treatment of iron-related disorders (Sun et al., 2024; Yan et al., 2024), highlighting the therapeutic potential of targeting the gut within this axis. Thus, iron may act as a key player that transmits signals and exerts metabolic effects between organs. A deeper understanding of how iron communication across the gut–adipose tissue–liver axis affects obesity-associated metabolic dysfunction will be important.
Therapeutic Strategies Targeting Iron Homeostasis in Obesity
A mount of evidence positions iron metabolism as a critical therapeutic target for obesity. Precision interventions including iron chelation, lifestyle intervention, ferroptosis regulation, and molecular targeting of iron metabolism regulators provide new perspectives for the prevention and treatment of obesity and related metabolic diseases (Fig. 8).

Iron chelation
Iron chelation therapy represents a promising avenue for obesity treatment, and iron chelators are becoming the focus of researches. Elevated iron levels in adipose tissue of both genetic and diet-induced obese models implicate iron dysregulation in unhealthy adipocyte hypertrophy. The efficacy of iron chelators in inhibiting adipogenesis and ameliorating adipose dysfunction demonstrates therapeutic potential in obesity management (Rodrigues de Morais and Gambero, 2019). The translational potential of this approach is underscored by consistent positive outcomes across animal studies. DFO decreases adipocyte size in epididymal fat of KKAy mice and shifts the distribution of adipocyte size toward an increased proportion of small-sized adipocytes (Tajima et al., 2012). Similarly, DFO ameliorates fat accumulation and improves adipocyte function in the epididymal adipose tissues of ob/ob mice (Yan et al., 2018). Lower iron level has a positive impact on shaping the function of adipose tissue (Zhang et al., 2021). Beyond reducing fat mass, iron chelation also decreases adipose macrophage infiltration and oxidative stress (Tajima et al., 2012) and enhances beige fat differentiation and metabolic activity to counteract HFD-induced weight gain (Nazari et al., 2022). Iron chelators already have an established clinical safety profile from decades of use in other diseases, which could potentially facilitate their repurposing for metabolic conditions (Farr and May, 2020). However, a significant limitation remains their systemic administration, which may cause anemia when used long-term for chronic conditions like obesity. To address this, future efforts should focus on adipose tissue-specific delivery systems. Strategies in nanomedicine and targeted therapeutics, such as those already being designed for cancer (Lang et al., 2019), could enable precise iron regulation within adipocytes, minimizing systemic side effects on iron metabolism and offering a safer and more effective treatment for obesity and related metabolic disorders.
Lifestyle interventions
Exercise and dietary iron management synergistically regulate adipose iron homeostasis. Exercise reduces iron stores in adults with obesity, potentially contributing to the improvement of iron homeostasis disorders (Ryan et al., 2021). An in-depth exploration of how dietary iron intake patterns and exercise dynamically regulate iron homeostasis in adipose tissue will provide a basis for the development of scientifically sound obesity prevention and management programs (Sun et al., 2024). The formulation of personalized dietary iron recommendations based on metabolic or iron status could prevent obesity-associated iron overload while meeting physiological iron needs. It advocates the integrated lifestyle, combining exercise and nutrition management to fight against obesity from the daily routine.
Molecular targeting of iron metabolism regulators
The key regulators of iron metabolism play a pivotal role in impaired adipogenesis and contribute significantly to the pathogenesis of obesity. Adipocyte-specific iron restriction, such as FTH1 knockout, enhances insulin sensitivity, glucose tolerance, and adaptive thermogenesis (Lu et al., 2024; Wang et al., 2025), while reducing adiposity and improving systemic metabolism through controlling intestinal fat absorption (Zhang et al., 2021). Tissue-targeted interventions address the iron deficiency and overload paradox in obesity. In the future, researchers may focus on the development of small molecule modulators that specifically regulate adipocyte iron uptake storage and utilization-related proteins to correct obesity-driven metabolic abnormalities. Hepcidin plays a central role in the regulation of iron homeostasis and has potential clinical applications in iron overload or restrictive iron utilization disorders. The development of therapeutic approaches to regulate HAMP expression holds promise for improving iron overload conditions associated with obesity. TfR1-mediated iron acquisition varies across adipocyte subtypes (Qiu et al., 2020). Targeting adipocyte TfR1 may mitigate hyperlipidemia-driven metabolic disorders while requiring careful balance to preserve adaptive thermogenesis.
Targeting ferroptosis
Current ferroptosis inhibitors primarily target lipid peroxidation and iron metabolism. Liprostatin-1 or ferrostatin-1 ameliorates hepatic steatosis in preclinical studies, providing proof for ferroptosis inhibition as a metabolic therapy (Qi et al., 2020; Li et al., 2020). Vitamin E protects adipose tissue against inflammation and oxidative stress and shows promise in antiferroptosis therapy (Alcala et al., 2015; Bhatti et al., 2018; Hu et al., 2021). While large-scale clinical trials specifically targeting ferroptosis in obesity are still lacking, clinical evidence demonstrates that vitamin E supplementation improves liver function in nonalcoholic fatty liver disease, a condition closely linked to adipose tissue dysfunction (Podszun et al., 2020). Iron chelators like DFO effectively reduce labile iron in adipocytes (Yan et al., 2018), but their systemic administration often leads to side effects, necessitating targeted delivery approaches.
Emerging strategies focus on addressing adipocyte-specific vulnerabilities in ferroptosis regulation. Adipocyte-specific GPX4 expression prevents spontaneous metabolic dysregulation and reduces chronic low-grade inflammation through ferroptosis-independent mechanisms (Schwarzler et al., 2022). GPX4 activators like RSL3 derivatives may counteract obesity-associated antioxidant enzyme downregulation. Activation of ferroptosis signaling by RSL3 suppresses TG accumulation in adipose tissues (Wang et al., 2025). Inhibitors of lipoxygenase or cyclooxygenase-2, enzymes preferentially expressed in adipose tissue during obesity development, show promise by blocking lipid peroxidation (Banhos Danneskiold-Samsoe et al., 2019; Sun et al., 2021; Yang et al., 2016).
The translation of these agents hinges on overcoming the challenge of tissue specificity. Advanced nanodelivery systems, such as ferroptosis regulator nanoparticles encapsulated in liposomes, may improve adipose tissue targeting. Applying such strategy to deliver liproxstatin-1 or potent iron chelator could achieve depot-specific iron modulation.
Glucagon-like peptide-1 receptor agonists and iron metabolism
Glucagon-like peptide-1 receptor agonists (GLP-1RAs), originally developed for the treatment of type 2 diabetes, have demonstrated promise in promoting weight loss in obesity. Beyond their established metabolic benefits, evidence also underscores their significant impact on systemic iron homeostasis. Recent findings indicate that semaglutide reduces dietary iron absorption in patients with diabetes, an effect independent of its effects on gastric emptying (Melis et al., 2025). The observation provides a direct human-relevant link between GLP-1RA therapy and iron regulation. While GLP-1RAs offer therapeutic benefits, they may also elevate the potential risk of anemia in individuals with obesity and diabetes. This underscores the necessity for a precision medicine approach, where monitoring iron status becomes an integral part of GLP-1RA therapy, particularly in patients with pre-existing risk factors for anemia.
Experimental studies reveal that liraglutide has the potential to reduce cellular iron content by decreasing TfR1 expression and increasing FPN expression, thereby inhibiting hepatic ferroptosis in diabetic models (An et al., 2023; Song et al., 2022). Liraglutide improves glucose tolerance and reduces body weight while simultaneously decreasing circulating and stored iron levels in a mouse model of hereditary hemochromatosis, suggesting its dual therapeutic potential for managing both metabolic dysfunction and iron overload (Bozadjieva-Kramer et al., 2024). Furthermore, GLP-1RA also exhibits protective effects on type 2 Wolfram syndrome by reducing mitochondrial labile iron and associated oxidative stress while enhancing β-cell function (Danielpur et al., 2016). These observations from clinical and mechanistic evidence highlight the impact of GLP-1RAs on systemic iron homeostasis in diabetes, which is mediated through multiple tissue-specific pathways. Despite these advances, the mechanisms underlying GLP-1RAs-mediated iron regulation remain poorly understood, particularly with respect to their effects on adipocyte function. Further research is warranted to elucidate the tissue-specific iron-regulating effects of GLP-1RAs and their implications in metabolic disorders.
Conclusion and Future Perspectives
Iron homeostasis is a core regulator of adipose tissue biology and metabolic health, intricately controlling lipid metabolism, mitochondrial respiration, thermogenesis, immune response, and adipokine secretion in different adipocyte subtypes. Dysregulation of iron triggers pathogenic pathways, including oxidative stress, ferroptosis, and cellular senescence, which contribute to adipose tissue dysfunction and systemic metabolic complications. The emerging concept of interorgan crosstalk via the liver–adipose tissue–gut axis further reveals that systemic iron metabolism and adipose function are fundamentally linked, making iron pathways a promising and multifaceted therapeutic target for obesity.
Despite significant progress, several critical knowledge gaps and challenges remain:
The drivers and metabolic consequences of iron heterogeneity among adipocyte subtypes are poorly understood as most studies analyze adipose tissue as a whole rather than conducting comparative analyses across different depots. Strong correlations exist, but causal evidence is still lacking. Whether iron dysregulation drives adipose tissue dysfunction or merely reflects the obese state? The precise mechanism by which adipose tissue hypoxia and inflammation influence iron metabolism need to be unraveled. The role of ferroptosis in obesity remains unclear; reliable biomarkers for adipocyte ferroptosis require validation for treatment monitoring.
To address these gaps, developing transgenic mouse models with adipocyte-specific manipulation of iron metabolism regulators will be essential for defining cell-specific mechanism and causal relationship. Well-designed human intervention trials are crucial to evaluate new pharmacological agents and explore the potential iron-modulating effects of existing drugs, such as GLP-1RAs. Finally, integrating multiomics analyses of iron-modulated adipose tissue will provide a system-level understanding of how iron remodels adipose function and identify novel regulatory nodes for therapeutic intervention.
Authors’ Contributions
B.W. drafted the initial article, formed and drew the figures, and integrated the figures with the text. X.D., M.S., and Y.X. were specifically responsible for the literature search process and format improvement in revising and refining the article. O.U.A. reviewed and edited the revised article. C.L. and J.Q. collaborated with other authors in discussing and validating the structural details. H.L. conceptualized the review framework, wrote the draft and initial article, supervised the entire review process, contributed to the overall interpretation and synthesis of the literature, and oversaw the final preparation of the article.
Footnotes
Acknowledgments
The authors would like to express sincere gratitude to the anonymous reviewers for their insightful suggestions and rigorous evaluation.
Author Disclosure Statement
The authors declare that there is no conflict of interest.
Funding Information
This work was carried out with the support of the