
Abstract
Significance:
Cellular proteostasis is essential for cellular proteome integrity, which is exquisitely sensitive to the redox environment. Heat shock proteins (HSPs) are the central chaperones that sense and adapt to these redox fluctuations. Emerging evidence demonstrates dysregulation of cellular HSPs-modulated redox-proteostasis in protein aggregation diseases, including cancers, senescence, neurodegenerative diseases, limb-girdle muscular dystrophy type D1, and β-thalassemia, making HSPs promising therapeutic targets in disease treatment.
Recent Advances:
Redox post-translational modifications (PTMs) serve as master switchboards to dynamically modulate the structure and chaperone function of HSPs. Redox PTMs allow HSPs to participate in protein synthesis and folding, conformational maintenance, and degradation, thereby maintaining cellular proteostasis. Beyond their chaperone functions, HSPs also play critical roles in organelle-specific stress responses, such as mitochondrial unfolded protein response, endoplasmic reticulum (ER) stress, and unfolded protein response.
Critical Issues:
Despite the well-known contributions of HSPs to redox-proteostasis, the double-edged functions of HSPs in protein aggregation diseases remain unclear. The main issues covered in this review include the regulation of HSPs by redox PTMs, the important role of HSPs in proteostasis and organelle-specific stress responses, dual modulation of HSPs in protein aggregation diseases, and pharmacological agents targeting HSPs.
Further Directions:
The functional diversity of HSPs in redox-proteostasis makes them promising therapeutic targets in disease treatment. Further studies should focus on exploiting agents that precisely target cysteine residues modifications on HSPs with good blood–brain barrier (BBB) penetration and low toxicity. Antioxid. Redox Signal. 45, 78–112.
Introduction
Cellular proteome integrity is maintained by proteostasis, a dynamic network that balances protein synthesis, folding, and degradation (Balch et al., 2008; Hoppe and Cohen, 2020; Park and Hoang, 2017). This system is exquisitely sensitive to the cellular redox environment, where physiological redox fluctuations directly modulate disulfide bond formation, cysteine residues (Cys) modifications, and chaperone function, thereby controlling protein folding efficiency and aggregation propensity (Kadeřábková and Mavridou, 2025; Yang et al., 2016). When this integrated redox-proteostasis network becomes dysregulated, it triggers the accumulation of misfolded proteins and contributes to the pathogenesis of multiple human diseases, particularly cancer and neurodegenerative diseases (Xu et al., 2020; Yamashita et al., 2025). Therefore, identifying the key regulators that sense and adapt to these redox fluctuations is essential for the development of targeted therapies.
Heat shock proteins (HSPs) are central to stress-induced adaptive responses, and their chaperone function is dynamically regulated by cellular redox state. Following their initial discovery in Drosophila under thermal stress (Ritossa, 1962), studies in simpler model organisms have further confirmed the crucial function of HSPs in fundamental proteostasis: In bacteria, sequence homology between Drosophila HSP70 and E. coli DnaK provided direct molecular evidence of conservation across kingdoms (Bardwell and Craig, 1984). In yeast, HSP104 has been shown to play an important role in cell survival at extreme temperatures (Sanchez and Lindquist, 1990). Over 60 years of research, HSPs have been identified and categorized into six distinct families: Small HSP family (sHSP), HSP40, HSP60, HSP70, HSP90, and HSP100 (Wei et al., 2024). Critically, their activity is precisely regulated by redox post-translational modifications (PTMs) on specific Cys. These redox PTMs serve as molecular switches to rapidly modulate HSPs’ conformation, ATPase activity, and substrate-binding ability in response to redox fluctuations (Yang et al., 2020; Zhao et al., 2022b). This redox-encoded regulation of chaperone activity underpins the versatile functions of HSPs within the proteostasis network, where they function as holdase, foldase, sequestrase, aggregase, and disaggregase to maintain proteostasis (Hartl et al., 2011; Hipp et al., 2019). Beyond their chaperone functions, HSPs are also integral to organelle-specific stress responses. For instance, endoplasmic reticulum (ER)-resident glucose-regulated protein 78 (GRP78)/BiP is a master regulator of unfolded protein response (UPR), while mitochondria Tumour necrosis factor receptor-associated protein 1 (TRAP1) activates mitochondrial unfolded protein response (UPRMT) and participates in the maintenance of mitochondrial metabolic homeostasis through redox-sensitive tetramerization (Baqri et al., 2014; Braakman and Hebert, 2013; Faienza et al., 2025; Varone et al., 2021).
Notably, the function of HSPs in redox-proteostasis is context-dependent, exhibiting a double-edged sword (Wan et al., 2020). In cancers, HSPs are often overexpressed to combat the adverse microenvironment and support tumor survival, making their inhibition a rational strategy (Regimbeau et al., 2022). In neurodegenerative diseases, certain chaperones like HSP70 and sHSP can mitigate disease progression by facilitating the clearance of misfolded proteins, whereas others such as HSP90 may stabilize disease-associated proteins to aggravate pathology (Blair et al., 2013; Caramiello and Pirota, 2024; Hansson, 2021; Pirota et al., 2024; Subedi et al., 2022). Accordingly, selectively targeting specific HSPs has emerged as a promising therapeutic strategy for protein aggregation diseases (Fig. 1) (D’Annessa et al., 2020; Ma et al., 2023; Serwetnyk and Blagg, 2021; Song et al., 2021; Wang et al., 2022).
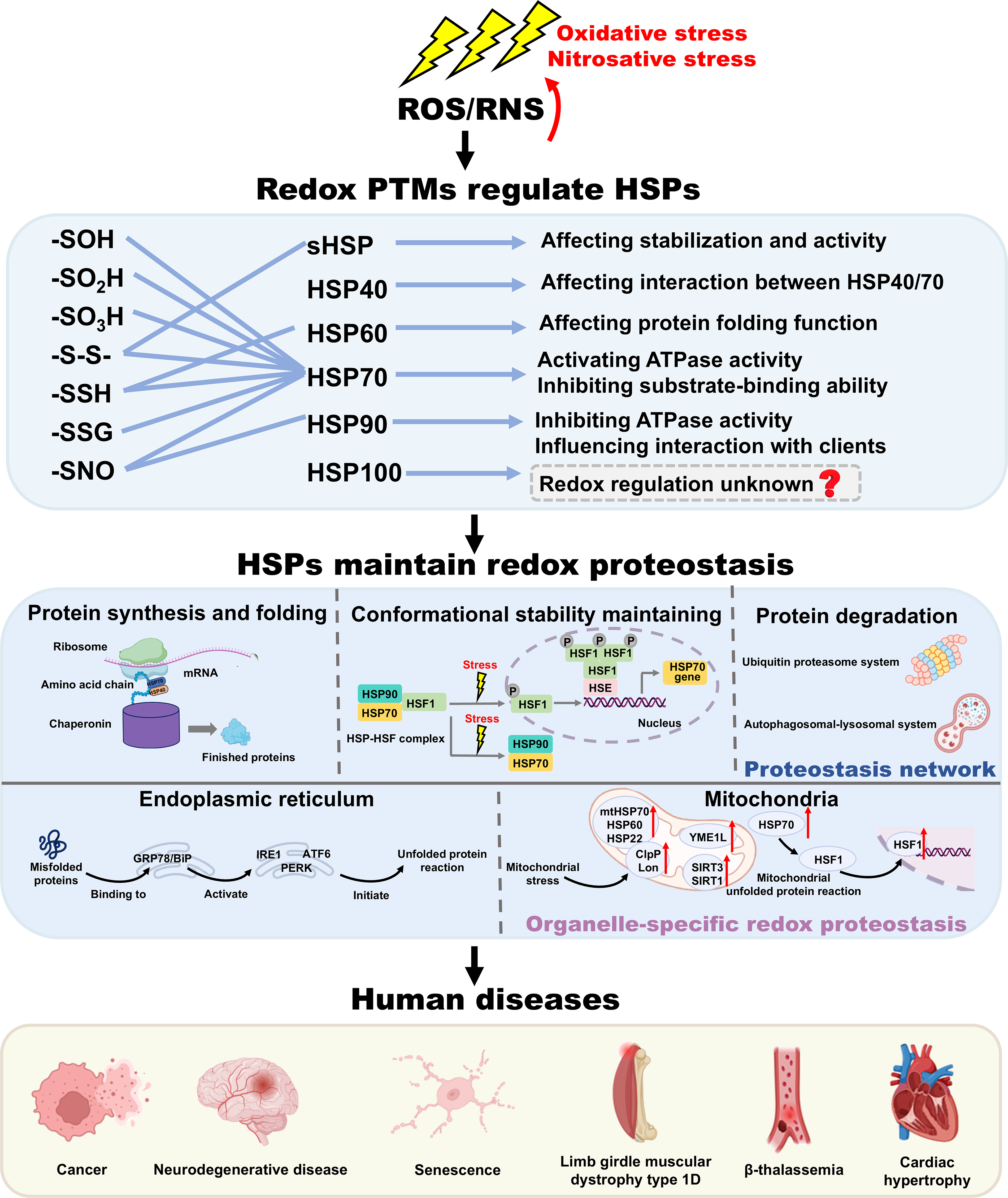
This review aims to summarize the current understanding of HSPs as central integrators of redox-proteostasis and critically evaluate their potential as therapeutic targets. We provide a comprehensive overview of how redox PTMs regulate HSPs structure and function, and elaborate on their critical role in proteostasis and organelle-specific stress responses. Moreover, a comprehensive summary of their molecular mechanisms and downstream signaling pathways is discussed. We also summarize preclinical study and clinical trials concerning compounds targeting HSPs, and highlight the dual role and therapeutic promise of targeting HSPs in protein aggregation diseases, which may contribute to the further development of small molecules targeting HSPs for disease treatment.
Redox PTMs as Master Switches of HSPs Function
Types of redox PTMs
Before discussing specific modifications, it is important to clarify key terms: “Redox fluctuations” refer to physiological redox events during which reactive oxygen species (ROS) act as signaling molecules. In contrast, “oxidative stress” and “redox imbalance” describe pathological states in which excessive ROS cause oxidative damage. Physiological redox fluctuations and pathological oxidative/nitrosative stress can both generate ROS and RNS (Morris et al., 2022). ROS/RNS selectively target specific Cys of HSPs and trigger redox PTMs, including S-sulfenation (-SOH), S-sulfination (-SO2H), S-sulfonation (-SO3H), disulfide bond formation (-S-S-), S-sulfhydration (-SSH), S-glutathionylation (-SSG), and S-nitrosylation (-SNO) (Yang et al., 2016). Each redox PTM serves as a molecular signal that rapidly adjusts chaperone function in response to cellular redox fluctuations, positioning HSPs as central integrators of redox proteostasis (Table 1) (Zhang et al., 2021c; Zhao et al., 2022c).
The Overview of Main Redox-PTMs of HSPs (Homo sapiens), Position, Effect on HSPs, Related Human Diseases, and Detection Approaches
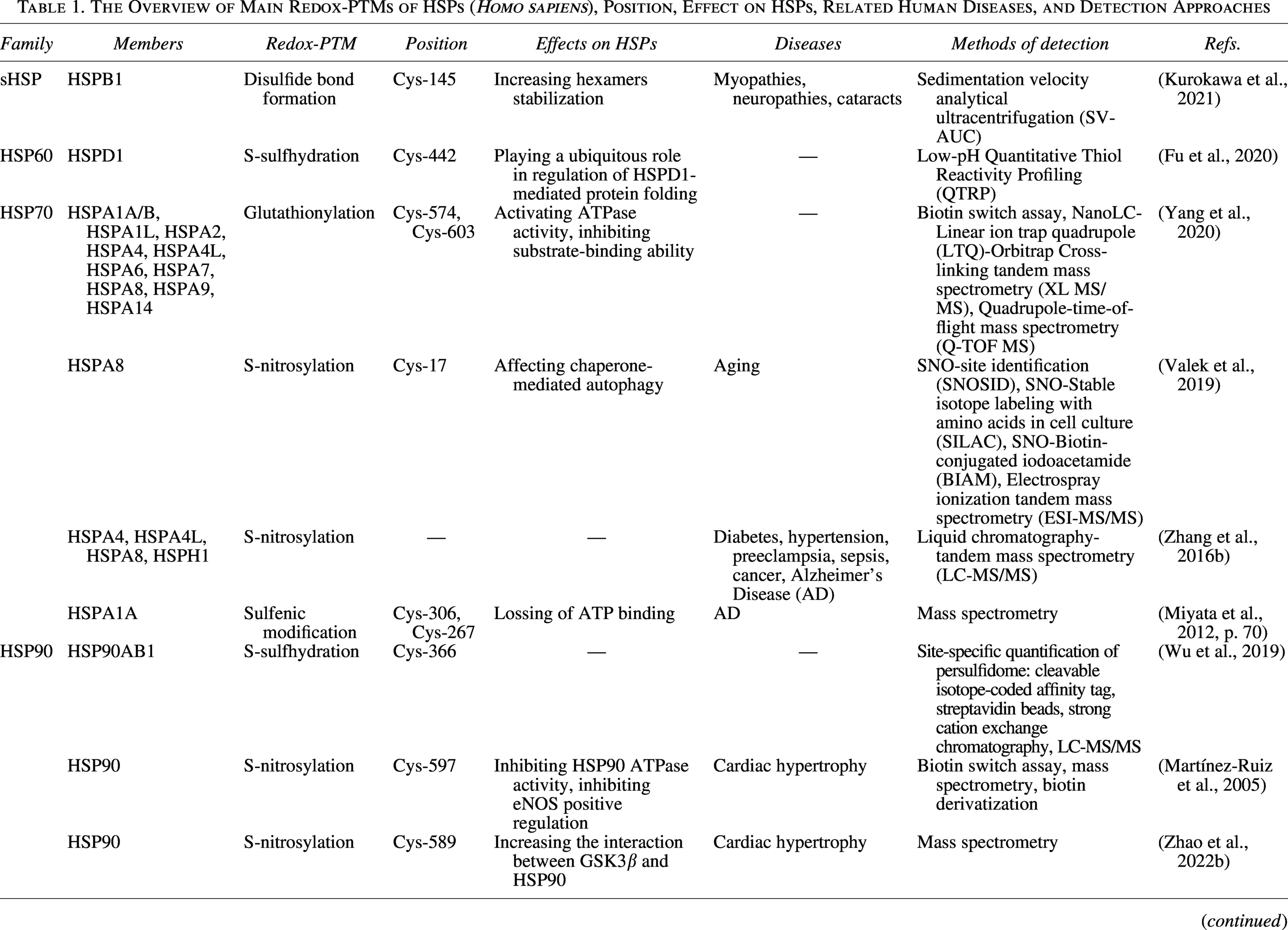

BIAM, Biotin-conjugated iodoacetamide; Cys, Cysteine residues; ESI-MS/MS, Electrospray ionization tandem mass spectrometry; HSP, Heat shock protein; LC-MS/MS, Liquid chromatography-tandem mass spectrometry; LTQ, Linear ion trap quadrupole; PTM, Post-translational modification; Q-TOF MS, Quadrupole-time-of-flight mass spectrometry; QTRP, Quantitative Thiol Reactivity Profiling; sHSP, Small HSP family; SILAC, Stable isotope labeling with amino acids in cell culture; SNOSID, SNO-site identification; SV-AUC, Sedimentation velocity analytical ultracentrifugation; XL MS/MS, Cross-linking tandem mass spectrometry.
Family-specific redox PTMs
sHSP
sHSPs are Adenosine triphosphate (ATP)-independent chaperones and characterized by the conserved α-crystalline domain (Pfoh et al., 2015). Ten distinct forms are expressed in humans, including HSPB1 (HSP25, HSP27, HSP28), HSPB2 (MKBP), HSPB3 (HSPL27), HSPB4 (αA-crystallin), HSPB5 (αB-crystallin), HSPB6 (HSP20, p20), HSPB7 (cvHSP), HSPB8 (HSP22), HSPB9 (CT51), and HSPB10 (ODF1) (Janowska et al., 2019). As holdase chaperones, they sequester unfolded or misfolded proteins in the core of the complex to suppress aggregation and enable them to refold to the native state by HSP70/90 chaperon system (Haslbeck et al., 2019). Critically, their function as holdases is dynamically regulated by redox PTMs at specific Cys. For instance, oxidized HSPB1, containing a single disulfide bond, possesses higher rigidity and stability, and lower chaperone-like activity with certain model proteins (Chalova et al., 2014). Another research revealed that the disulfide bond between conserved Cys can strengthen HSPB1 hexamers stabilization (Kurokawa et al., 2021). However, it should be noted that these findings primarily rely on in vitro analyses of purified proteins, which may not fully reflect the dynamic regulation of HSPB1 within the complex physiological environment of living cells.
HSP40
HSP40 (J-domain proteins, JDPs/DnaJ) are divided into three groups, including class A (DnaJA), class B (DnaJB), and class C (DnaJC). They perform two important functions: they contribute to modulating HSP70 ATPase activity through their J domains, and they assist in the recognition and binding of unfolded proteins before transferring them to HSP70 for subsequent processing (Barriot et al., 2020; Biebl et al., 2022; Cho et al., 2021; Craig and Marszalek, 2017; Nillegoda et al., 2017; Piette et al., 2021). Numerous studies have found that redox PTMs modulate the interaction between HSP40 and HSP70. For instance, glutathionylation of DnaK (a well-studied model for HSP70) weakened the interaction of DnaK with DnaJ, GrpE, and σ(32) (Zhang et al., 2016a). Besides, a disulfide-bond-linked dimer of DnaK-A303C-H541C also exhibited severely impaired binding ability with HSP40, further illustrating how redox PTMs disrupt the interaction between chaperones (Liu et al., 2017).
HSP60
HSP60, also referred to as chaperonin or Cpn60, is categorized into two groups: Group I and Group II (Hartl and Hayer-Hartl, 2002). Group I is expressed in prokaryotes (known as GroEL), and eukaryotic mitochondria, nucleus, and chloroplasts. Group II is present in archaebacteria and eukaryotic cytosol (known as Tailless complex polypeptide 1-Ring Complex, TRiC or Chaperonin Containing Tailless complex polypeptide 1, CCT).
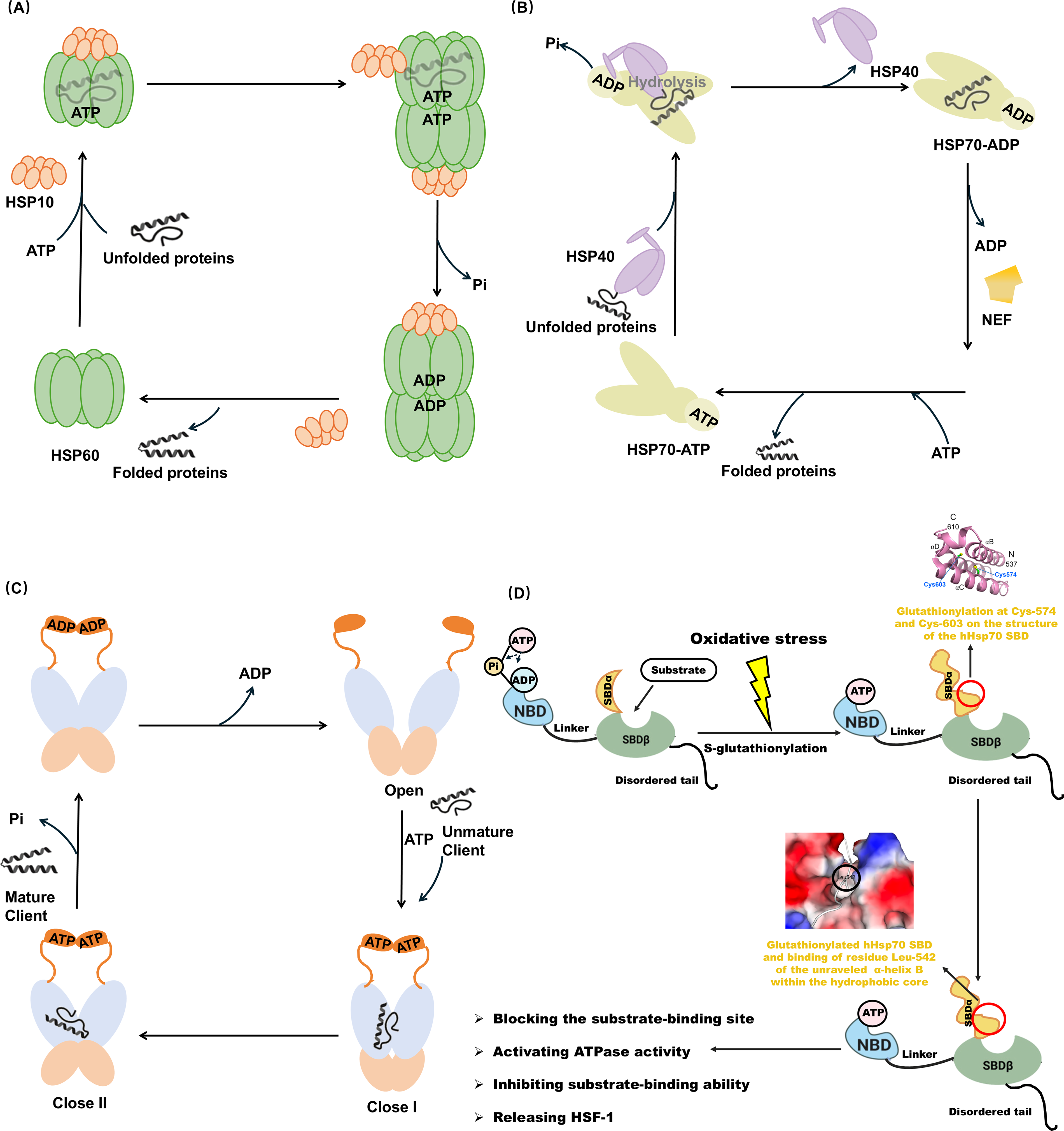
Group I contains three domains: an equatorial domain including ATP binding pocket, an intermediate hinge domain, and an apical domain complexed with HSP10 or substrate (Klebl et al., 2021). The oligomerization and double-ring formation of HSP60 depend on HSP10 and ATP (Gomez-Llorente et al., 2020; Ishida et al., 2018). HSP60 facilitates protein folding and assembly (Nie et al., 2024). They self-assemble into a stable heptamer single ring in the absence of ATP. Hydrophobic and exposed residues in its top domain drive the binding of unfolded proteins. Upon ATP binding, the HSP60 form a double ring structure and bind to HSP10 to form a football complex. ATP hydrolysis drives conformational alteration and supports the folding and release of proteins (Hu et al., 2022) (Fig. 2A). Group II possesses a unique structure, called co-chaperonin-like structure (Yamamoto et al., 2019). TRiC forms an octamer complex independent of cochaperone HSP10, whereas employ a protein folding mechanism resembling that of the HSP60-HSP10 complex (Cong et al., 2012).
Notably, the chaperone activity of HSP60 is finely modulated by redox PTMs at specific Cys. For instance, Ling Fu et al. developed a new low‐pH QTRP approach used to identify persulfidated sites on 994 proteins (Fu et al., 2020). They unveiled that HSPD1(HSP60) contained a persulfidation hotspot at Cys-442 (Fu et al., 2020). Given the essential role of this site in chaperone activity, this indicated that persulfidation likely played a widespread role in regulating the protein folding function of HSPD1 (Nagumo et al., 2005). Additional studies suggested that GroEL formed a stable complex with the CnoX, a protein combining a redox-protective function, which is important for protein folding (Dupuy et al., 2023). Despite these findings, direct evidence concerning how S-sulfhydration precisely regulates the HSP60 folding cycle remains unknown.
HSP70
As the highly abundant and ubiquitous chaperones, HSP70 exists in two forms mainly: one is constitutively expressed Heat Shock Cognate 70 (HSC70, known as HSPA8) essential for the maintenance of cellular proteostasis, the other is stress-induced HSP72 (known as HSPA1A) coping with adverse stimuli (Brocchieri et al., 2008; Daugaard et al., 2007; Rosenzweig et al., 2019). Structurally, HSP70 contains three domains, a 45-kDa nucleotide-binding domain (NBD) at N-terminal, followed by a 15-kDa substrate-binding domain (SBDβ), a 10-kDa helical lid domain (SBDα), and a disordered C-terminal. There is a flexible linker between NBD and SBDβ (Rosenzweig et al., 2019). In eukaryotic cytosolic and nuclear HSP70, the disordered C-terminal tail with an Glu-Glu-Val-Asp motif, which regulates interactions with specific cochaperones (Zuiderweg et al., 2017), such as tetratricopeptide repeat (TPR) co-chaperones including HSP40, C-terminal HSP70 interacting protein (CHIP), and HSPA8/HSP90-organizing protein (HOP) (Assimon et al., 2015).
HSP70 exerts chaperone function through ATP-dependent allosteric conformation. As a key kind of cochaperones, HSP40 recruits unfolded proteins and delivers to ATP-bound state of HSP70. HSP40-HSP70 interaction triggers ATP hydrolysis. Subsequently, SBDβ domain of HSP70 interacts with unfolded proteins, followed by nucleotide exchange factor-mediated ADP-by-ATP exchange (Solana et al., 2022). HSP70 in the ATP-bound state shows lower affinity for client proteins than ADP-bound state, thereby promoting the release of folded proteins (Höhfeld and Jentsch, 1997). Through interaction with HSP40, HSP70 returns to ADP-bound state and initiates another cycle (Fig. 2B).
Numerous redox PTMs have been identified in HSP70. In yeast, HSP70 Ssa1 serves as a sensor for the activation of heat shock factor 1 (HSF1)-mediated cytoprotection by thiol-reactive compounds, which appears to involve the modification of Cys-264 and Cys-303 (Wang et al., 2012). Importantly, a study first found that under oxidative stress, site-specific S-glutathionylation occurred at Cys-574 and Cys-603 within the C-terminal α-helical lid of human inducible HSPA1A. This caused the unfolding of SBDα and the exposure of the methyl groups of residue Leu-542. It is bound to the hydrophobic cleft of the SBDβ, thereby mimicking “substrate binding” and blocking the substrate-binding site. Further study revealed that this conformation activated ATPase activity and inhibited substrate-binding ability including HSF1 (Yang et al., 2020) (Fig. 2D). Moreover, S-nitrosylation at Cys-17 of HSPA8 has been shown to influence chaperone-mediated autophagy, leading to the imbalance of proteostasis (Valek et al., 2019).
HSP90
HSP90 is a highly preserved molecular chaperone, essential for the correct folding of client proteins (Lackie et al., 2017). In higher eukaryotes, HSP90 exists as four paralogs: HSP90α and HSP90β in the cytosol, GRP94 in the ER, and TRAP1 in mitochondria (Gupta et al., 2020). The HSP90 monomer is comprised of three functional domains, including N-terminal nucleotide-binding domain (NTD), the middle domain (MD), and the C-terminal domain (CTD) (Chiosis et al., 2023).
HSP90 adopts an open V-shaped conformation in the absence of ATP, and the unfolded client proteins bind to the MD of HSP90. ATP binding leads to the closure of the lids, which induces NTD dimerization (closed 1 state) and twisting of the HSP90 monomers (closed 2 state) (Schopf et al., 2017). ADP release promotes HSP90 returning the conformation back to open V-shaped conformation, and the correctly folding proteins are released in native conformations (Zierer et al., 2016) (Fig. 2C). Various co-chaperones associate with conformational cycle of HSP90, including HOP, cell division cycle 37 homologue (CDC37), HSP90 ATPase homologue 1 (Aha1), and p23 (Hessling et al., 2009; Schopf et al., 2017, 2017; Verkhivker, 2022).
Many studies have revealed that redox PTMs altered the chaperone function of HSP90 and consequently impacted cellular processes. For example, S-nitrosylation of Cys-597 (located in the CTD of HSP90) in human HSP90A has been demonstrated to inhibit its ATPase and endothelial nitric oxide synthase (eNOS) regulatory activities, which may have negative effects on the cardiac hypertrophy model (Martínez-Ruiz et al., 2005). Similarly, another study found that S-nitrosylation of Cys-589 elevated the interaction between glycogen synthase kinase 3β (GSK3β) and HSP90, increased GSK3β phosphorylation, and decreased eukaryotic translation initiation factor 2Bε (eIF2Bε) phosphorylation, thereby exacerbating cardiac hypertrophy (Zhao et al., 2022b). Nevertheless, researchers also noted that the specific function of HSP90/GSK3β axis in cardiac hypertrophy is waiting for further exploration (Zhao et al., 2022b). Additional research revealed that the inhibition of HSP90 S-nitrosylation at Cys-589 alleviated cardiac fibrosis through Transforming growth factor-beta (TGFβ)/SMAD family member 3 (SMAD3) signaling pathway (Zhang et al., 2021c). In addition, S-nitrosylation at Cys-521 of HSP90 acts as a conformational switch, destroying HSP90-Aha1 interaction while facilitating the recruitment of CDC37, thereby aggravating atherosclerosis (Zhao et al., 2022c). In TRAP1, S-nitrosylated Cys-501 can form a disulfide bridge with Cys-527, referred to as population shift mechanism (Papaleo et al., 2023).
HSP100
HSP100 is a large super-family of energy-driven conformational “machines” known as AAA + ATPases. ClpB (in the bacteria) and HSP104 (in the yeast) are widely-studied members of HSP100 (Zolkiewski et al., 2012). Both ClpB and HSP104 share similar domain architecture: an NTD, two nucleotide-binding AAA + domains (NBD1 and NBD2), and a coiled-coil MD. Differently, HSP104 contains a unique CTD, while it is not found in ClpB (DeSantis and Shorter, 2012; Schirmer et al., 1996). As a kind of co-chaperon of HSP70, HSP100 plays a significant role in protecting proteins from aggregation (Easton et al., 2000; Zolkiewski et al., 2012). Despite the critical role of HSP100 in proteostasis, research on its redox regulation remains scarce, representing a promising avenue for future exploration.
Collectively, redox PTMs precisely modulate the conformation stability and chaperone activity of HSPs in response to redox fluctuations, playing an important role in the maintenance of redox proteostasis. However, determining which redox PTMs will occur at a given cysteine residue of HSPs, and their distinct physiological importance remain undetermined (Yang et al., 2020). Under different conditions, the underlying regulatory mechanisms for disparate redox PTM sites in HSPs are also worthy to be studied further (Zhao et al., 2022c).
Targeting HSPs in Proteostasis Network
HSPs are the central players of proteostasis network, which ensure protein synthesis and correct folding, maintain conformational stability, and participate in protein degradation, including ubiquitin proteasome system (UPS) and autophagosomal-lysosomal system (Table 2). Moreover, small molecules selectively targeting HSPs have demonstrated their regulatory impact on protein proteostasis and have been applied to the treatment of protein aggregation diseases (Tables 3 and 5).
The Role of HSPs in Proteostasis

Structures and Sources of Small-Molecule Modulators Targeting HSPs

HSPs in protein synthesis and folding
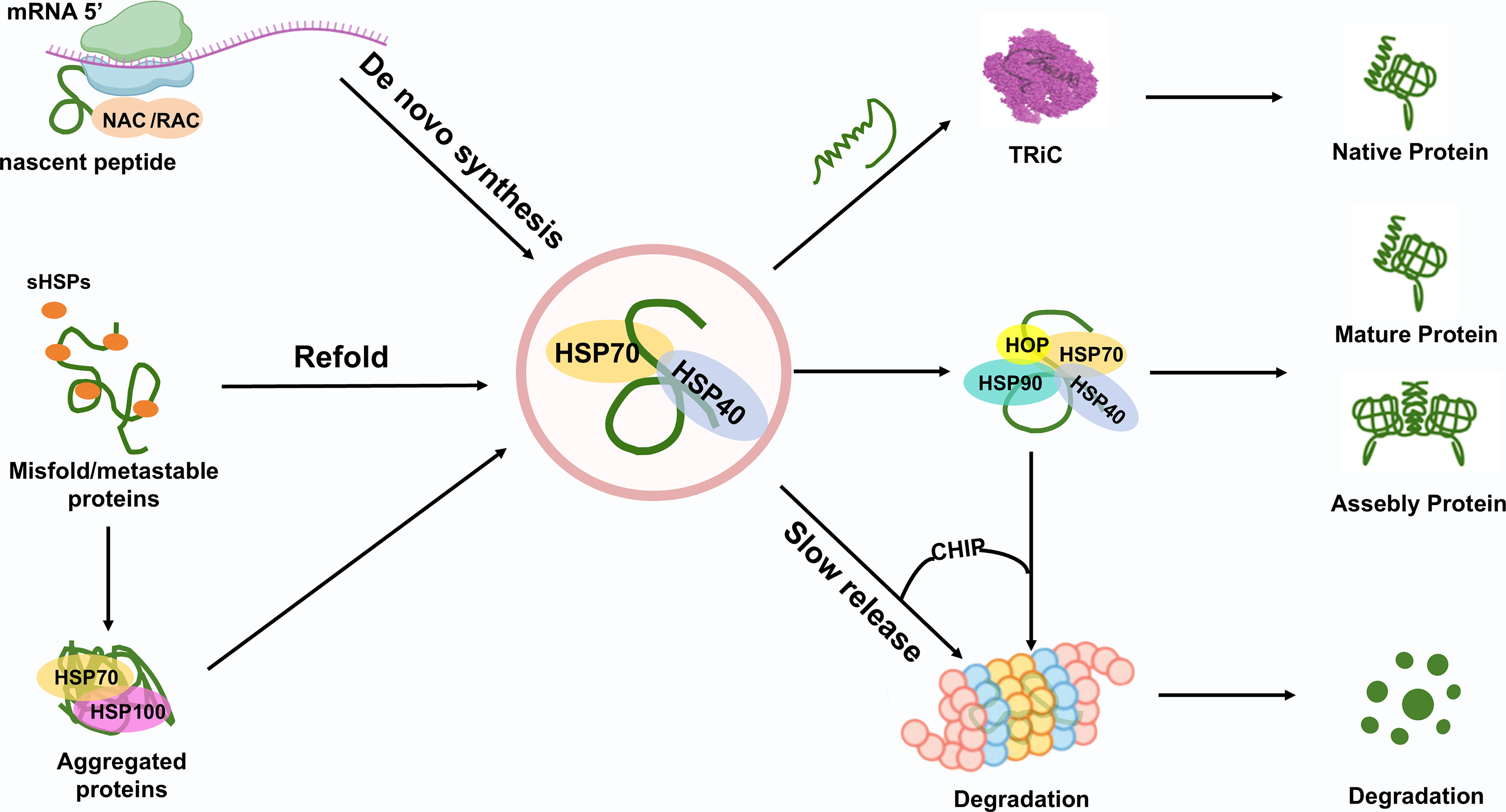
Nascent chain-associated complex (NAC) and ribosome-associated complex (RAC) bind to nascent polypeptides as they are synthesized on ribosomes (Kirstein-Miles and Morimoto, 2013; Preissler and Deuerling, 2012). The correct folding of nascent proteins, particularly those with complicated structures or multidomain, necessitates the assistance of HSPs network. Unfolded proteins are subsequently transferred to HSP70-HSP40 complex, which functions as a central hub (Hu et al., 2022). Depending on structural complexity and folding status, client proteins are delivered to disparate pathways: (1) HSP70 directly facilitates the folding of unfolded proteins to the native state. (2) Some proteins with domains specific folding intermediates folded by HSP70 are transferred to the TRiC and are further folded to the native protein (Hu et al., 2022). (3) HSP70 transfers partially folded proteins to HSP90 via HOP for further folding (Baindur-Hudson et al., 2015), such as protein kinases vital for cellular signaling transduction (Taipale et al., 2012). (4) sHSP and HSP100 play a pivotal function in preventing proteins from aggregation (Haslbeck et al., 2019; Zolkiewski et al., 2012). (5) Except for nascent proteins and the denatured proteins induced by stress, the irreversibly misfolded proteins are degraded through UPS and autophagosomal-lysosomal system with the assistance of cochaperones, including B cell lymphoma 2-associated athanogene-1 (BAG-1) and CHIP (Hantouche et al., 2017; Zhang et al., 2015) (Fig. 3).
HSPs in the maintenance of conformational stability
Owing to the exposure of hydrophobic amino acid residues and unpaired β-strands, polypeptides in non-native conformations tend to aggregate, such as nascent chains, folding intermediates, and misfolded proteins (Hipp et al., 2019). To inhibit these protein aggregations, the proteostasis network requires the assistance of HSPs. Therefore, the heat shock response serves as the primary response to stress within the cytosol. As a typical stress sensor, HSF1 dynamically controls the expression of HSPs (Murshid et al., 2018). Under normal conditions, HSF1 is maintained inactive in the cytoplasm via forming a complex with HSP90 and HSP70 (Fig. 4A) (Zheng et al., 2016; Zou et al., 1998). In the presence of destabilizing factors (including mutations, oxidative stress, and toxic compounds), the disruption of protein conformation leads to cytotoxic protein aggregation. During these processes, the HSF1-HSP complex is dissociated, which enables HSF1 to release, trimerize, and translocate to the nucleus. Subsequently, they bind to HSE elements located in the promoters of HSP genes and potentiate their transcription (Fig. 4B) (Gomez-Pastor et al., 2018; Mahat et al., 2016).
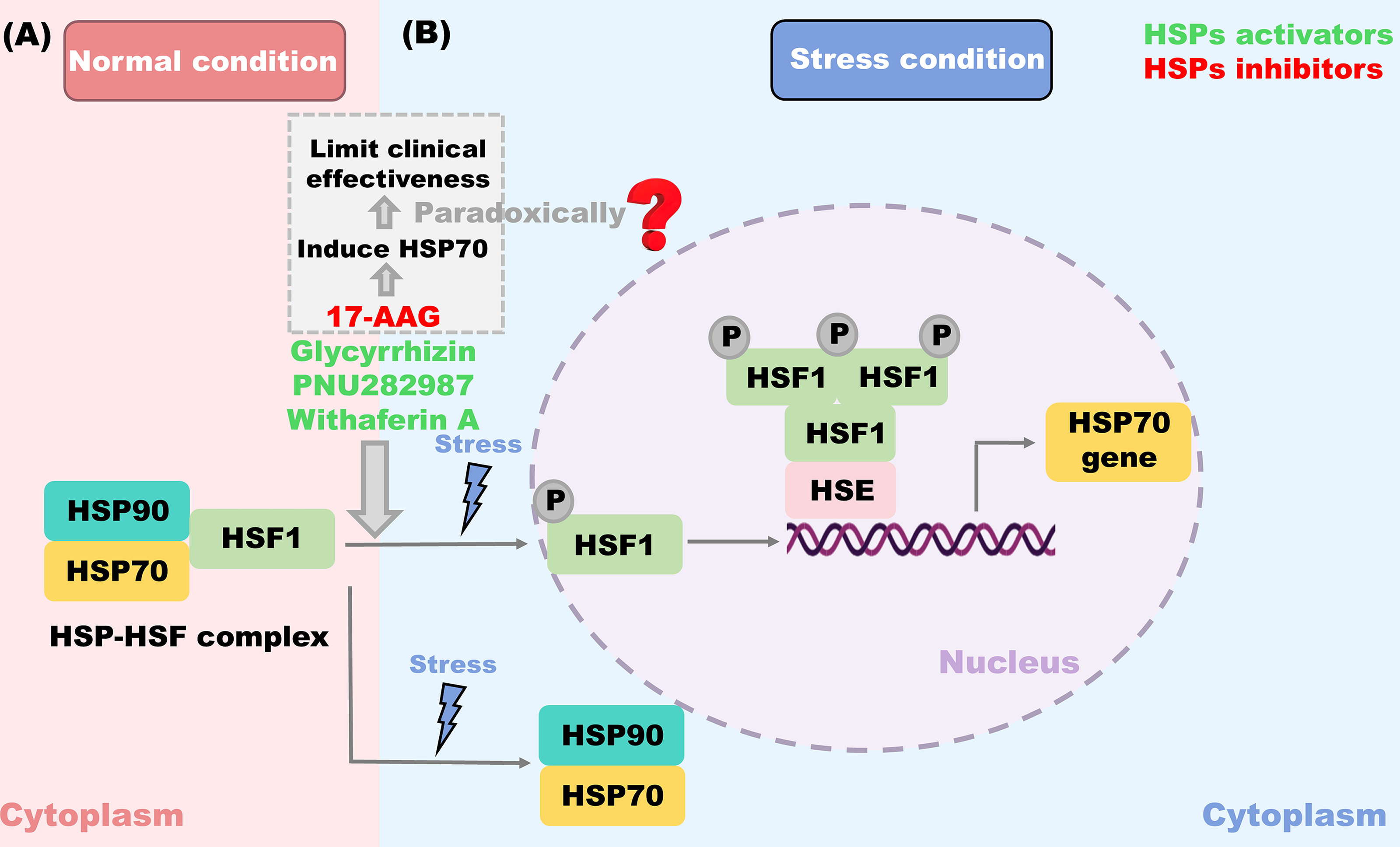
The induction of proteostasis network members suppresses general protein synthesis, thereby limiting the production of new clients of the proteostasis network. While the cessation of proteotoxic stress and the re-establishment of enough free chaperone capacity promote the formation of the HSF1-HSP complex, the system restores homeostasis (Gomez-Pastor et al., 2018). Consequently, HSF1 serves as a pivotal target for compounds targeting HSPs. There are some HSP70 activators that have been reported to induce HSF1 and alleviate protein aggregation, such as Glycyrrhizin (Yan et al., 2004), PNU282987 (Ren et al., 2020), and Withaferin A (Joshi et al., 2021). Interestingly, some HSP90 inhibitors, like 17-allylamino-geldanamycin (17-AAG), also trimerize HSF1 and drive HSP70 transcription (Zhang et al., 2021a). Unfortunately, this HSP70 induction effect paradoxically limits their clinical effectiveness (Bagatell et al., 2000; Pacey et al., 2012; Zou et al., 1998).
HSPs in the protein degradation
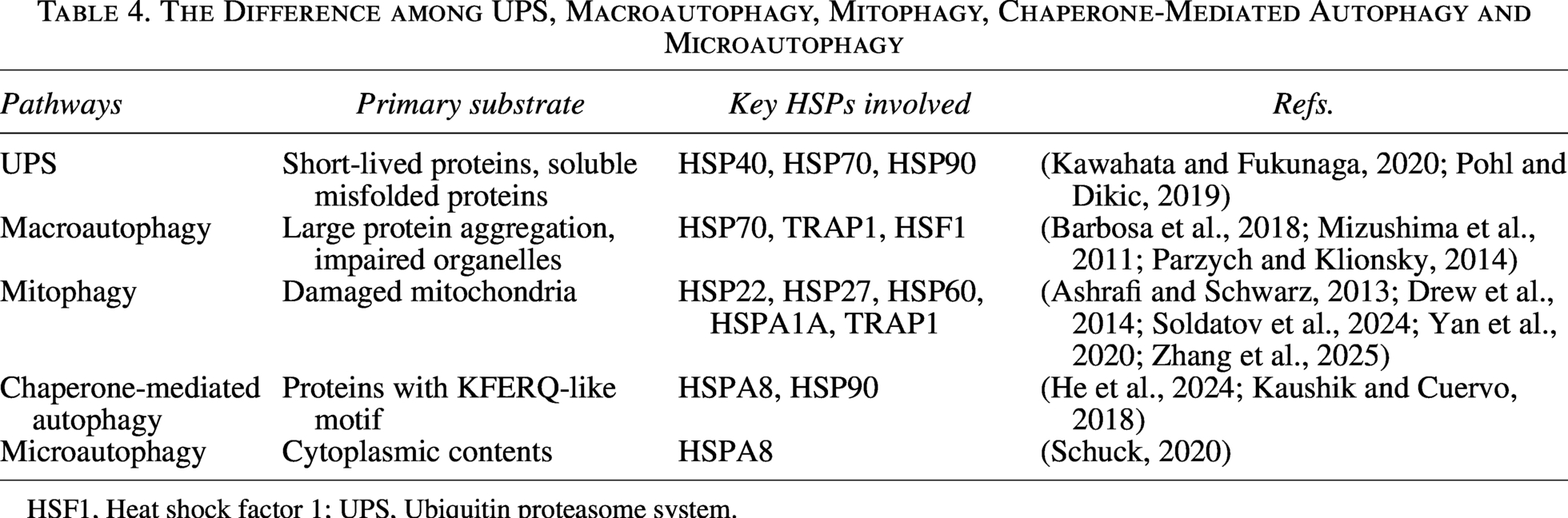
Despite the precision of protein folding machinery, cellular stress leads to the accumulation of misfolded and denatured proteins. Specific cooperation between HSPs and cochaperones fosters the degradation of protein aggregation and alleviates cytotoxicity. Herein, we concentrate on two main HSP-mediated protein degradation pathways, including the UPS and autophagosomal-lysosomal system (Table 4). The specific regulation mechanisms are shown in Table 5 and Figures 5, 6.
The Difference among UPS, Macroautophagy, Mitophagy, Chaperone-Mediated Autophagy and Microautophagy
HSF1, Heat shock factor 1; UPS, Ubiquitin proteasome system.
The Effects of Regulating HSPs on Protein Aggregation and Their Underlying Mechanisms
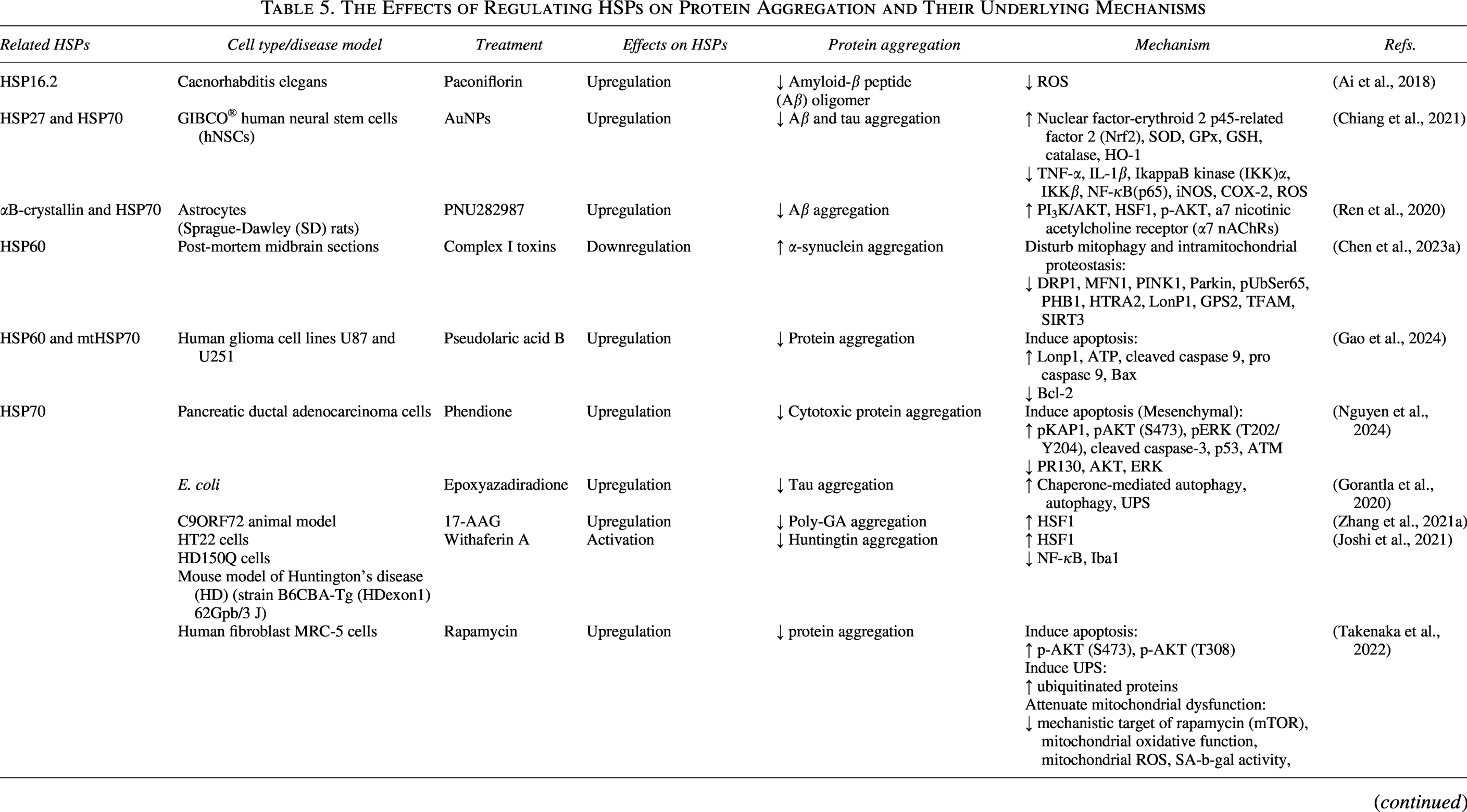
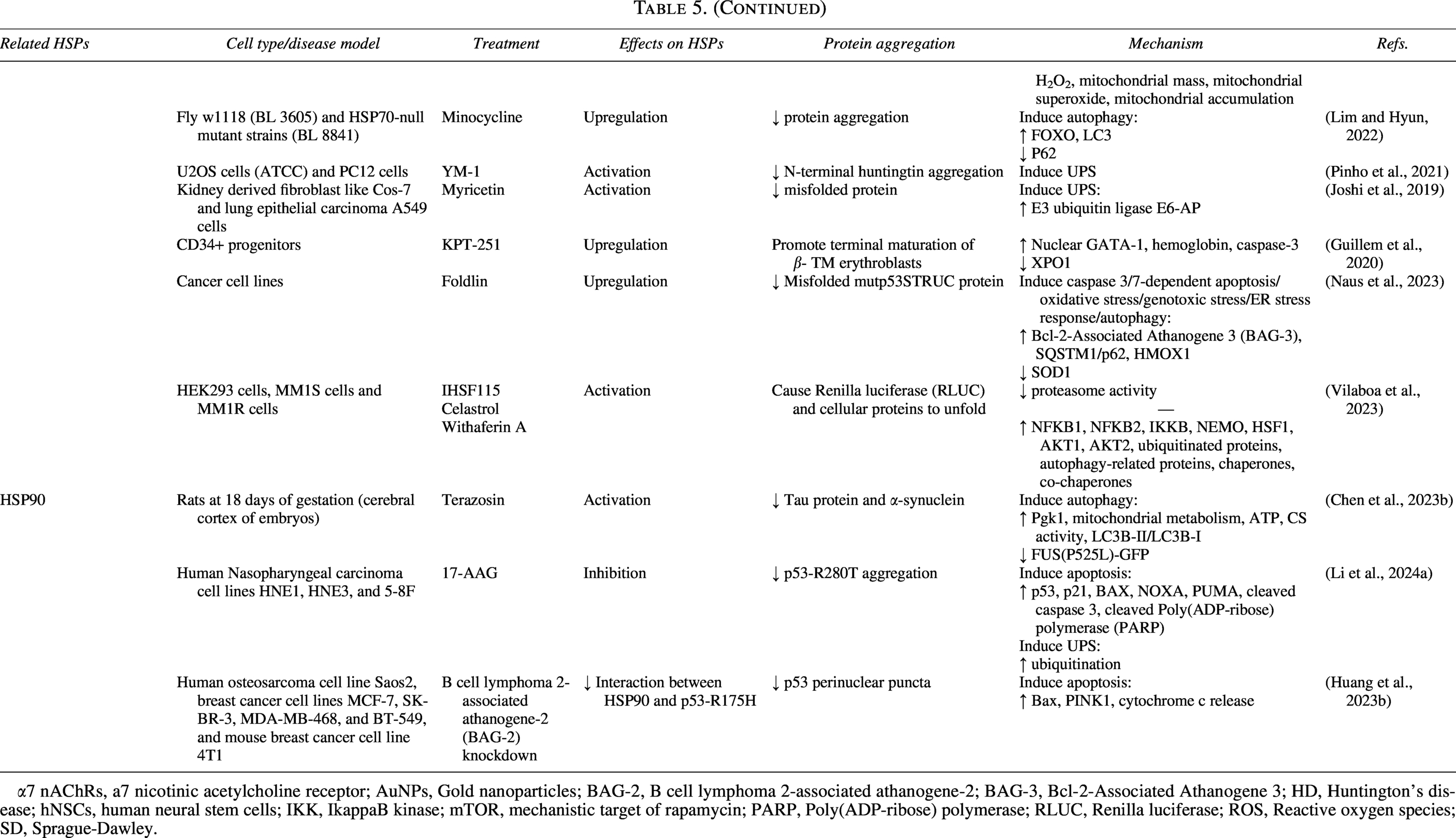
α7 nAChRs, a7 nicotinic acetylcholine receptor; AuNPs, Gold nanoparticles; BAG-2, B cell lymphoma 2-associated athanogene-2; BAG-3, Bcl-2-Associated Athanogene 3; HD, Huntington’s disease; hNSCs, human neural stem cells; IKK, IkappaB kinase; mTOR, mechanistic target of rapamycin; PARP, Poly(ADP-ribose) polymerase; RLUC, Renilla luciferase; ROS, Reactive oxygen species; SD, Sprague-Dawley.
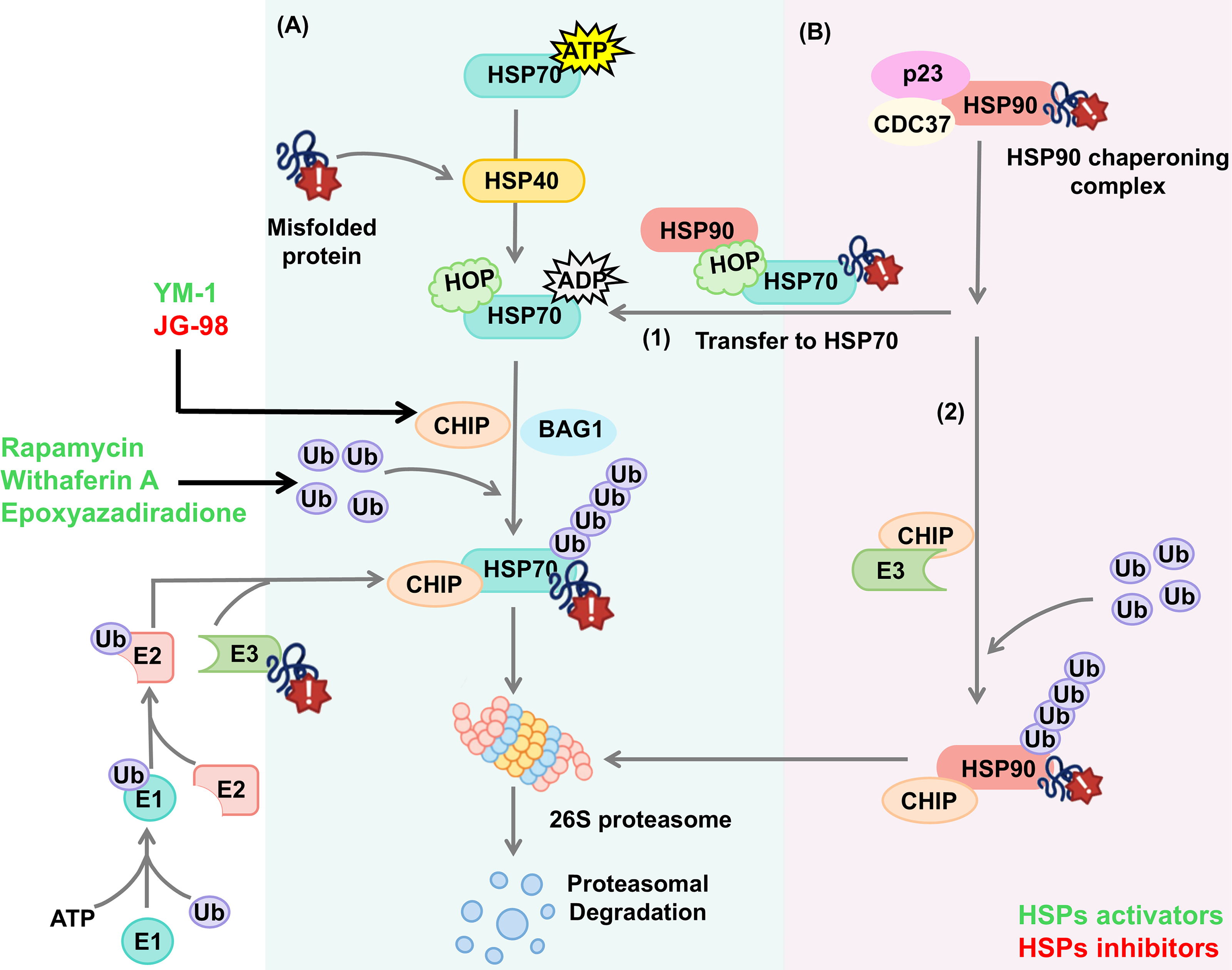

Targeting HSPs in UPS
The UPS is responsible for the clearance of short-lived proteins and soluble misfolded proteins (Kawahata and Fukunaga, 2020; Pohl and Dikic, 2019). HSP70 and HSP90 bind with E3 ligase-CHIP to trigger the ubiquitylation of misfolded proteins and subsequent proteolysis (Zhang et al., 2015).
In this pathway, the ubiquitin protein is connected to protein aggregation through sequential reaction: The E1 ubiquitin-activating enzyme (E1) activates and transfers ubiquitin protein to the E2 ubiquitin-conjugating enzyme (E2), which then cooperates with the E3 ubiquitin ligase (E3)-binding protein aggregation and transfers ubiquitin protein to the protein aggregation (Eldridge and O’Brien, 2010; Yuan et al., 2018). Through iterative cycles of this process, the protein aggregation is polyubiquitinated, which is then recognized and degraded by the 26S proteasome (Zhao et al., 2022a).
Numerous studies have revealed the pivotal role of HSP70-HSP40 in UPS, involving the following key steps (Fig. 5A): (1) With the assistance of HSP40, HSP70 selectively recognizes and binds protein aggregation, forming HSP40-HSP70-protein aggregation complex. (2) Either CHIP competes with HOP and binds to the C-terminal of HSP70, or BAG-1 competes with HIP and binds to the ATPase domain of HSP70 (Kim et al., 2020), promoting the transition of the complex from the folding state to degrading state. (3) The protein aggregations are polyubiquitinated, fostering their disaggregation by 26S proteasome (Zhao et al., 2022a).
Owing to the similarity of HSP70 and HSP90 cochaperones, the role of HSP90 in UPS is closely relevant to HSP70. HSP90 client proteins are stabilized through the formation of HSP90-cochaperones (p23 and CDC37) complex. When HSP90 client proteins fail to fold correctly or HSP90 is pharmacologically inhibited, it will be degraded through the following two pathways: (1) the CHIP binds to the TPR domain of HSP90 (Ko et al., 2009; Xu et al., 2002), and the client proteins are ubiquitinated by E3, fostering their disaggregation by the proteasome (Didelot et al., 2007). (2) HOP binds to Met-Glu-Glu-Val-Asp motif of HSP90 and transfers client proteins to HSP70 for subsequent ubiquitination degradation (Yamamoto et al., 2014) (Fig. 5B).
Small molecules targeting HSPs have been utilized to regulate UPS. For instance, both YM-1 and JG-98 have been confirmed to recruit CHIP and enhance the affinity with protein aggregation, thereby promoting their ubiquitination and facilitating their degradation by proteosome (Assimon et al., 2013; Pinho et al., 2021; Wang et al., 2013). Epoxyazadiradione and Rapamycin activated HSP70, upregulated ubiquitinated proteins, and accelerated ubiquitination progression (Gorantla et al., 2020; Takenaka et al., 2022). After Myricetin treatment, the level of HSP70 and E3 ubiquitin ligase E6‐AP was induced, fostering the UPS (Joshi et al., 2019). Besides, upregulation of HSPA1A using Withaferin A activated UPS and caused the cellular proteins to unfold (Vilaboa et al., 2023). In conclusion, exploiting drugs targeting HSPs as well as the ubiquitination steps such as CHIP, ubiquitin enzymes, and proteasome will be potential treatment strategies. However, the major limitation of UPR-targeted strategies, such as Proteolysis-targeting chimera (PROTACs), is their toxicity due to their mechanism of total protein degradation versus simple activity inhibition (Zhao et al., 2022a).
Targeting HSPs in autophagosomal-lysosomal system
Autophagy is responsible for the clearance of misfolded proteins, large protein aggregation, and whole damaged organelles (Klionsky and Codogno, 2013; Mputhia et al., 2019). HSPs regulate distinct autophagy, including macroautophagy, mitophagy, chaperone-mediated autophagy, and microautophagy (Fig. 6).
Macroautophagy involves five distinct stages: initiation, nucleation, maturation, fusion, and degradation (Yu et al., 2018). In this process, autophagosomes are formed through engulfing protein aggregation or impaired organelles, and are transported to lysosomes along microtubules (Mizushima et al., 2011; Parzych and Klionsky, 2014). They fuse to form autolysosome, and then the captured contents are degraded (Menzies et al., 2012). Studies reveal that the formation of autophagosomes necessarily needs the participation of HSP70 and HSF1. Besides, p62 forms a complex with Bcl-2-Associated Athanogene 3 (BAG-3), a HSPA8–HSP70 co-chaperone, promoting the macroautophagy degradation of unfolded proteins (Gamerdinger et al., 2009). As a special macroautophagy, mitophagy means selectively transporting mitochondria to lysosomes for subsequent degradation, which is mediated by the serine–threonine kinase PTEN-induced kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin (Ashrafi and Schwarz, 2013). The main feature of Chaperone-mediated autophagy is the cargo selectivity, which means that only the proteins possessing KFERQ-like motif can be recognized by HSPA8 (Kaushik and Cuervo, 2018). After motif recognition, the complex is transferred to the lysosome for its degradation through subsequent binding to Lysosome associated membrane protein type 2 A (Lamp2A) (Tekirdag and Cuervo, 2018). Microautophagy, refers to the direct engulfment of cytoplasmic contents by lysosomal membrane invagination, and then the vacuole is degraded (Schuck, 2020). In mammals, the invagination is associated with the recognition between KFERQ-like motif in the proteins and HSPA8 (Rios et al., 2020). Notably, recent studies have highlighted the role of p97/VCP, an AAA + ATPase chaperone, in organelle-specific autophagy such as autophagic peroxisomal degradation (pexophagy). They reported that Fas-associated factor family member 2 (FAF2)/UBXD8 (a p97/VCP cofactor) cooperates with p97/VCP to extract PMP70 (a ubiquitylated protein) from peroxisomes, thereby inhibiting nonessential pexophagy (Koyano et al., 2024). In addition, emerging evidence discovered that Cdc48 (in yeast) and p97/VCP facilitate DNA damage response through targeting SUMOylated chromatin substrates and ubiquitination (Noireterre and Stutz, 2024).
Notably, the modulation of autophagy through HSPs has emerged as a promising strategy for degrading protein aggregation. For example, upregulation of HSP70 via Epoxyazadiradione activated macroautophagy and chaperone-mediated autophagy, preventing tau aggregation (Gorantla et al., 2020). Minocycline can also induce HSP70 and macroautophagy, with the upregulation of LC3 and the downregulation of p62, thereby inhibiting protein aggregation (Lim and Hyun, 2022). It was also reported that Foldlin-induced HSP70 and autophagy activation along with the upregulation of Sequestosome 1 (SQSTM1)/p62, massively degrade the misfolded mutp53STRUC proteins (Naus et al., 2023). Terazosin, an activator of HSP90, reduced Tau protein and α-synuclein levels by enhancing the interaction between HSP90 and Unc-51-like autophagy-activating kinase 1 (ULK1), a master regulator of autophagy, along with the upregulation of microtubule-associated protein 1 light chain 3beta (LC3B)-II/LC3B-I (Chen et al., 2023b). Moreover, histone deacetylase 6 (HDAC6)-mediated HSP90 deacetylation activated chaperone-mediated autophagy and mitigated the aggregation and toxicity of α-synuclein protein (Du et al., 2021). TRAP1 was reported to inhibit KAT8 regulatory NSL complex subunit 3 (KANSL3) acetylation and active mitophagy, thereby mitigating mitochondrial dysfunction (Zhang et al., 2025). Conversely, inhibition of HSP60 through Complex I toxins disturbed mitophagy along with the downregulation of PINK1, Parkin, and pUbSer65, finally causing α-synuclein aggregation (Chen et al., 2023a). Moderate autophagy clears protein aggregation and promotes cell survival, while excessive autophagy triggers cell death (Chaabane et al., 2013; Vidal et al., 2014). Therefore, targeting autophagy alone may limit clinical effects due to its dual role on cell fate (Ghavami et al., 2014). Given the interconnected association between HSPs and autophagy, conjoined therapies may be a promising direction for protein aggregation diseases.
HSPs in stress granule (SGs)
SGs are ribonucleoprotein (RNP) condensates induced by various stress conditions, including heat shock, arsenite, and osmosi (Aulas et al., 2017; Panas et al., 2016; Protter and Parker, 2016). Emerging study noted that HSPs and their co-chaperones serve as significant determinants of SG composition, dynamics, and disassembly.
Misfolded proteins can also co-localize with the SGs, which include nuclear SGs composed of HSF1/2 and pre-mRNA processing factors, and cytoplasmic SGs composed of proteins and mRNAs (Alastalo et al., 2003; Buchan and Parker, 2009; Sandqvist and Sistonen, 2004). Recent evidence has elucidated that HSPs play a role in SGs assembly and disassembly. During assembly, HSP90 is shown to mediate the localization of eukaryotic translation initiation factor 4E (eIF4E) and its binding partner eIF4E-T into SGs (Suzuki et al., 2009). HSP22 cooperates with HSP27 to inhibit the aggregation of misfolded proteins in SGs, thereby preventing aggresome formation (Verma et al., 2021). HSP22, HSP40, HSP70 and HSP104 promote the disassembly of SGs (Cherkasov et al., 2013; Verma et al., 2021; Walters and Parker, 2015). Moreover, HSP40-HSP70 complexes are reported to induce SG entering autophagy (Walters et al., 2015) (Fig. 7). In summary, these preliminary studies reflect the importance of HSPs in SGs, positioning HSPs-SGs axis as a potential therapeutic target in neurodegeneration and cancer. Further studies in this direction are warranted to reveal the specific mechanisms of HSPs-SGs axis and their relationship with diseases (Verma et al., 2021).

Targeting HSPs in Organelle-Specific Redox Proteostasis
In addition to chaperone functions, HSPs also operate as redox-responsive switches to directly regulate stress responses within the ER and mitochondria, thereby maintaining organelle-specific redox proteostasis. Small molecules targeting HSPs have been exploited to modulate stress response, offering therapeutic potential for protein aggregation diseases (Table 5 and Fig. 8).
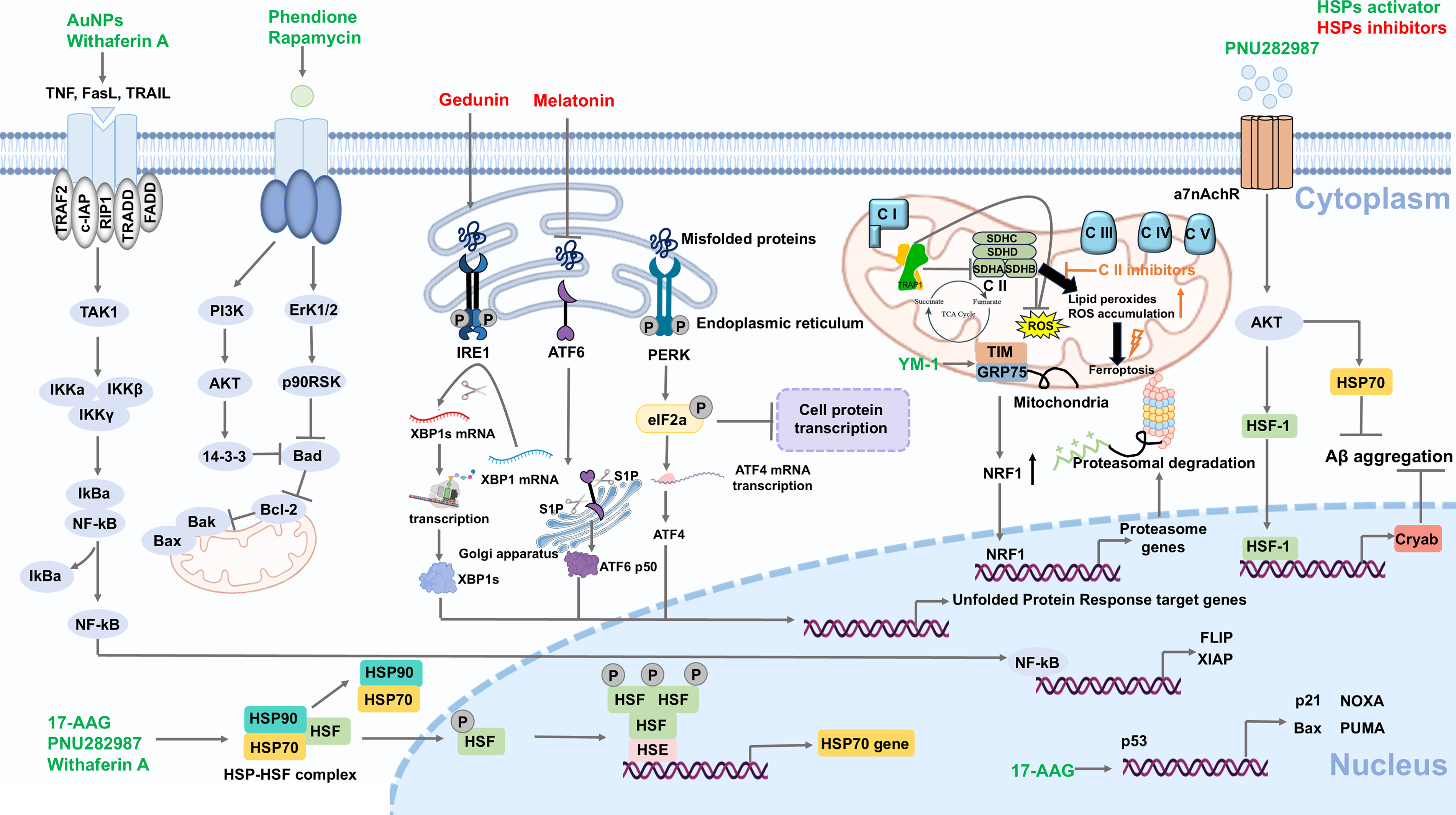
Targeting HSPs regulating ER-specific redox proteostasis
The accumulation of misfolded proteins within ER will cause ER stress and then unfolded protein reaction (Oakes and Papa, 2015). This adaptive signaling network is coordinated by three principal ER transmembrane sensors: Protein kinase R (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). Under normal conditions, these sensors are maintained in an inactive state through binding to GRP78/BiP, a member of the HSP70 (Braakman and Hebert, 2013; Varone et al., 2021). Upon ER stress, the GRP78/BiP is released to bind to protein aggregation, resulting in the activation of PERK, ATF6, and IRE1, as well as subsequent initiation of UPR (Malhotra and Kaufman, 2007).
Among these signaling branches, IRE1α–X-box-binding protein 1 (XBP1) pathway has been shown to be particularly important by numerous studies. IRE1 activation triggers RNase activity, which cleaves XBP1 mRNA at specific sites (Hetz et al., 2020; Shamu and Walter, 1996; Walter and Ron, 2011; Welihinda and Kaufman, 1996). The latest research findings uncovered a phase transition mechanism by which IRE1α dynamically coalesces with SGs through liquid–liquid phase separation (LLPS) at ER membrane (Liu et al., 2024). This establishes a workstation to enrich XBP1 mRNA splicing machinery component in response to ER stress, revealing a novel biophysical dimension about UPR regulation (Liu et al., 2024).
Intriguingly, a host of studies have highlighted the correlation between HSPs and ER stress. For example, the downregulation of GRP94 by hesperidin inhibits ERS-induced inflammation through decreasing key UPR components, including p-PERK, ATF6, ATF4, and p-IRE1α (Xie et al., 2022). In neuronal cells, silencing of Pirh2 reduces HSP90 and attenuates ER stress, along with the downregulation of GRP78, CHOP, and ATF4 (Singh et al., 2023). Similarly, Melatonin also inhibits the expression of HSF1, HSP70, GRP78, and ATF6, alleviating ER stress and the subsequent unfolded protein reaction (Bona et al., 2022). In contrast, the overexpression of HSPA6 can activate ER stress, with the significant upregulation of ATF6 (Zhu et al., 2024). Moreover, further studies reveal the molecular interactions between HSPs and the main UPR components. For instance, HSPA1A forms a stable complex with the cytosolic domain of IRE1α, thereby facilitating its activation (Gupta et al., 2010). HSP47 binds to IRE1α and displaces its negative regulator GRP78/BiP to promote its oligomerization (Sepulveda et al., 2018).
Growing evidence shows the influence of redox PTMs on ER stress sensors. For example, nitric oxide (NO) has been shown to modify Cys-931 and Cys-951 on IRE1α, which selectively abrogates its ribonuclease activity without affecting its oligomerization, thereby disrupting UPR (Ohkubo et al., 2016). In contrast, S-nitrosylation also triggers the IRE1α–XBP1-mediated UPR, which is crucial for maintaining hematopoietic progenitors through inducing cell-cycle arrest in Drosophila (Cho et al., 2024). Similarly, ATF6α activity is regulated by a redox-sensitive disulfide-bond switch. The specific intermolecular disulfide bonds (C467-C467 and C618-C618) lock ATF6α in inactive dimeric forms, and ER stress stimulates their single reduction for rapid activation (Koba et al., 2020). Collectively, these findings highlight redox PTMs as switches that directly modulate the activity of UPR sensors, contributing to the maintenance of redox proteostasis. Among the pathways discussed, IRE1α–XBP1 is highly druggable due to its disease-specific roles.
Targeting HSPs regulating mitochondria-specific redox proteostasis
Mitochondria are particularly crucial for maintaining redox proteostasis. As a redox-sensitive HSP90 paralog, TRAP1 was unveiled forming tetramers via disulfide bonds (Cys-261 and Cys-573), thereby maintaining mitochondrial metabolic homeostasis and promoting pro-neoplastic activity (Faienza et al., 2025). Besides, TRAP1 can decrease stress-induced mitochondrial ROS production by binding to the subunit A of succinate dehydrogenase and inhibiting its activity (Dekker and Rüdiger, 2021; Guzzo et al., 2014). However, TRAP1 triggers metabolic rewiring and protects cancer cells from oxidative damage, thereby contributing to tumor progression (Somatilaka et al., 2022). Notably, UPRMT is a protective transcriptional response against mitochondrial proteostasis, which regulates HSPs to mitigate protein aggregation. According to reports, HSP22 interact with HSP60, mitochondrial HSP70, and TRAP1 to participate in the UPRMT, which may contribute to regulating the accumulation of mitochondrial ROS (Morrow et al., 2000; Morrow et al., 2016). In Acetaminophen-induced acute liver injury, TAR DNA-binding protein-43 (TDP-43) accumulation activated UPRMT along with the upregulation of the mitochondrial protease mtHSP70, HSP60, and caseinolytic protease P (ClpP, a mitochondrial protease) (Liu et al., 2024). In another study, Resveratrol alleviated the serious glucose-induced mitochondrial damage via inducing the key member of mitochondrial proteases Lon peptidase 1 (Lonp1), along with the upregulation of HSP27, HSP60, and HSP90 (Kalvala et al., 2020). Consequently, mitochondria critically modulate protein proteostasis through mitophagy and HSPs-mediated UPRMT, making it a significant therapeutic target for drug development.
Targeting HSPs regulating apoptosis
During oxidative stress, HSPs modulate apoptosis along with the clearance of protein aggregation. For instance, Fas apoptosis inhibitory molecule-HSP27 interaction prevented the formation of protein aggregation effectively (Kaku et al., 2022). Besides, enhancing HSP70 through Rapamycin promoted the progress of apoptosis and mitigated protein aggregation, with the upregulation of p-AKT (Takenaka et al., 2022). In pancreatic ductal adenocarcinoma cells, Phendione-induced HSP70 upregulation was shown to activate apoptosis with the upregulation of pAKT and pERK, thereby preventing cytotoxic protein aggregation (Nguyen et al., 2024). Furthermore, HSP60 and mtHSP70 were increased by Pseudolaric acid B, followed by apoptosis activation and protein aggregation degradation, with the upregulation of cleaved caspase 9 and Bax (Gao et al., 2024). A research unscored that 17-AAG activated HSP70 and dissociated p53-R280T aggregation, followed by the transcriptional restoration of p53 downstream key target genes such as p21, BAX, NOXA, and PUMA (Li et al., 2024a). Nevertheless, current small molecules targeting HSPs may non-selectively modulate HSPs-mediated pro-apoptosis and HSPs-mediated pro-survival pathway, making it uncontrollable for actual effect in protein aggregation diseases, which may be a research direction for further works.
Signaling Molecules and Pathways Regulating HSPs
HSPs modulate redox-proteostasis through four interconnected signaling pathways, including AKT, mechanistic target of rapamycin (mTOR), AMP-activated protein kinase alpha (AMPKα) and nuclear factor-erythroid 2 p45-related factor 2 (NRF2), which collectively maintain cellular redox-proteostasis (Fig. 8 and Fig. 9).
AKT
As a central mediator of intracellular signal transduction, ATK is frequently activated by phosphorylation in response to diverse stimuli (Haddadi et al., 2018; Pritchard and Hayward, 2013). HSPs prevent protein aggregation through activating the AKT signaling pathway to support cell cycle progression, which is beneficial for cells to recover proteostasis (Takenaka et al., 2022). For instance, HSP70, DNAJB3, and HSP90 have been reported to upregulate AKT (Abu-Farha et al., 2015; Lee et al., 2015; Zhang et al., 2019), whereas HSP20 overexpression blocked the AKT and MAPK signaling pathways, thereby hampering breast carcinogenesis (Yang et al., 2022). Besides, a study revealed that PNU282987 suppressed Aβ aggregation through upregulation of astrocytic endogenous αB-crystallin and HSP70 via the α7AChR, PI3K/AKT/HSF1 signaling axis (Ren et al., 2020). Similarly, Rapamycin accelerated the degradation of misfold protein by activating AKT and upregulating HSP70 (Dibble et al., 2009; Harston et al., 2011; Takenaka et al., 2022). Phendione resulted in an increase of pKAP1, pAKT, pERK and HSP70, thus preventing cytotoxic protein aggregate formation (Nguyen et al., 2024). Moreover, Withaferin A has reported to upregulate AKT1 and AKT2, thereby caused Renilla luciferase (RLUC) and cellular proteins to unfold with the activation of HSPA1A (Vilaboa et al., 2023). However, Deguelin, a HSP90 inhibitor, was suggested to suppress AKT signaling, as well as decrease survivin and cyclin-dependent kinase 4 (Cdk4) through hampering their interact with HSP90 (Yang et al., 2013). Accordingly, it may be a feasible method to promote protein aggregation degradation through specifically activating AKT or enhancing interacting with HSPs.
mTOR
As the downstream of AKT, mTOR is a key regulator of cell cycle control and protein synthesis, with the major downstream targets of the translational machinery (Chou et al., 2012). As HSP90 client proteins, Epidermal growth factor receptor (EGFR)-PI3K-AKT-mTOR signaling mediates cell apoptosis pathways (Freudlsperger et al., 2011). Gedunin, a HSP90 inhibitor, was shown to decrease the p-PI3K, p-AKT, and p-mTOR (Subramani et al., 2017). Besides, studies demonstrate that mTOR inhibition interrupts HSPs synthesis, thus enhancing HSP90 inhibitor activity (Acquaviva et al., 2014). Another study found that AKT-mTOR signaling potentiated protein degradation via the UPS and autophagosomal-lysosomal system (Bodine and Baehr, 2014). Interestingly, Rapamycin suppressed mTOR signaling, while positively regulating HSP70 expression and increasing the level of ubiquitinated proteins, restoring proteostasis and accelerating cell proliferation (Gorantla et al., 2020; Santagata et al., 2012; Schopf et al., 2017). Consequently, as a HSP90 client protein, mTOR regulates both HSPs synthesis and protein aggregation degradation such as UPS and autophagosomal-lysosomal system, playing a crucial role in protein quality control.
AMPK
AMPK serves as a proverbial energy sensor of oxidative stress response and cell metabolism (Hardie et al., 2012; Herzig and Shaw, 2018; Ido et al., 2015; Nakano et al., 2010). Yan-li Yang et al. found that Deguelin inhibited HSP90 and induced ceramide production, which activated AMPK and then phosphorylated Ulk1, ultimately resulting in autophagy (Yang et al., 2013). Conversely, another study demonstrated that HSP90 inactivation suppressed AMPK signaling while facilitating the degradation of LKB1 protein (Ye et al., 2023). Therefore, HSPs inhibition has a dual effect on AMPK, which may be attributed to the influence of AMPK targeting other potential pathways or the various mechanisms of different drugs necessitating further systemic investigation.
NRF2
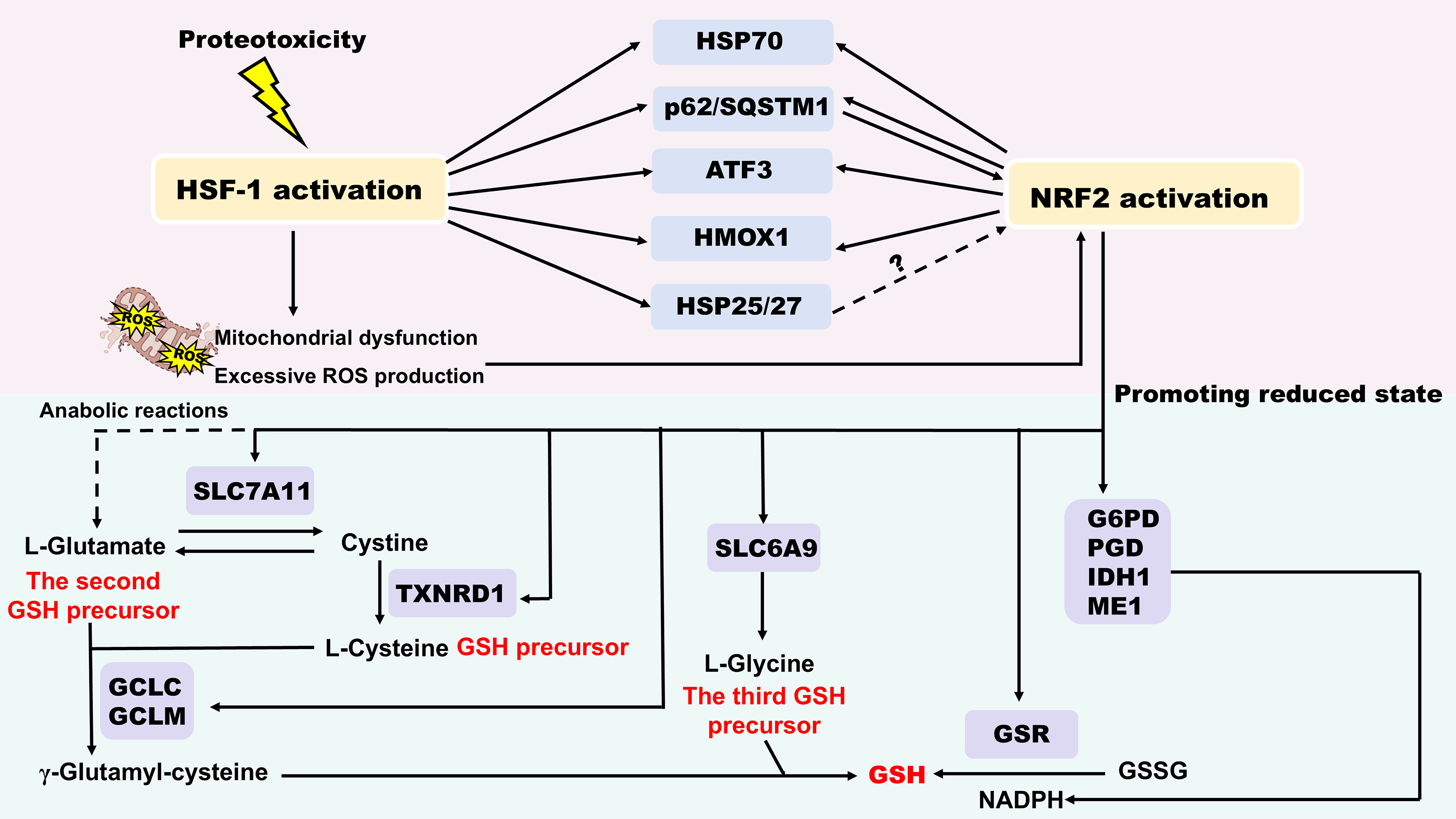
Similar to HSF1, NRF2 remains at low basal levels under normal conditions, but is strongly activated by diverse stimuli (Dayalan Naidu et al., 2015). Notably, HSF1 and NRF2 function as a double-edged sword in redox-proteostasis network. On the one hand, HSF1 and NRF2 constitute an idiotypic defense network against proteotoxic and oxidative stress. They regulate overlapping target genes, such as HSP70 (Almeida et al., 2010), the autophagy cargo protein sequestosome 1 (p62/SQSTM1) (Komatsu et al., 2010), activating transcription factor 3 (ATF3) (Dziunycz et al., 2014; Hoetzenecker et al., 2011; Kim et al., 2010; Takii et al., 2010, p. 3), heme oxygenase 1 (HMOX1, also known as HSP32) (Prestera et al., 1995) and HSP25/27 (Dayalan Naidu et al., 2015; Rajasekaran et al., 2007), and in certain cases, may compensate for each other. HSF1 and NRF2 influence the redox balance through promoting a more reduced state. HSF1 activates NRF2 through the following mechanisms: (1) Various kinds of cellular stressors such as proteotoxicity cause HSF1 activation and mitochondrial dysfunction, resulting in excessive ROS production, which in turn activates NRF2 (Dayalan Naidu et al., 2015). (2) HSF1 enhances the expression of HSP70, p62/SQSTM1 and ATF3, HMOX1. p62 competes with NRF2 for binding to Kelch-like ECH associated protein 1 (Keap1), thereby displacing NRF2 from Keap1 and leading to NRF2 activation (Ichimura et al., 2013, p. 62). (3) Sequestration of Keap1 within protein aggregation possibly induces NRF2 activation (Dayalan Naidu et al., 2015). Activation of NRF2 significantly regulates the intracellular levels of reduced glutathione (GSH), a key small-molecule antioxidant. It upregulates the cystine/glutamate antiporter (SLC7A11, system xc-), enhancing cystine uptake (Sasaki et al., 2002). Intracellular cystine is then reduced to cysteine (a GSH precursor) by the NRF2-dependent enzyme thioredoxin reductase 1 (TXNRD1). Furthermore, NRF2 supports the flux of glutamine into anabolic pathways to provide glutamate (the second GSH precursor) (Mitsuishi et al., 2012), and increases glycine (the third GSH precursor) import via the transporter SLC6A9, another NRF2-regulated gene (Hirotsu et al., 2012). NRF2 activation strengthens the expression of GCLC and GCLM, the two subunits of the γ-glutamylcysteine ligase, which catalyzes the rate-limiting step in the GSH biosynthesis (Wild and Mulcahy, 2000). The maintenance of GSH in its reduced state relies on glutathione reductase (GSR), an enzyme regulated by NRF2 and using NADPH as a cofactor. The supply of NADPH is also under the control of NRF2, which regulates the gene expression of the four key NADPH-generating enzymes, including glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (PGD), isocitrate dehydrogenase 1 (IDH1), and malic enzyme 1 (ME1) (Mitsuishi et al., 2012; Singh et al., 2013) (Fig. 9). On the other hand, persistent activation may destroy homeostasis balance, resulting in cardiomyopathy, facilitating carcinogenesis and even reducing longevity (Dayalan Naidu et al., 2015; Wang et al., 2012). Consequently, precise modulation of their interactive network emerges as novel therapeutic strategies.
Targeting HSPs to Regulate Protein Aggregation in Human Diseases
Proteostasis is disturbed in multiple pathological conditions, mainly including protein aggregation diseases, such as cancer and neurodegenerative diseases. HSPs are closely associated with the progression of these diseases owing to their critical role in maintaining cellular proteostasis. Emerging studies have identified small molecules targeting HSPs as promising therapeutic strategies (Table 6 and Fig. 10).

Clinical Trials of Small Molecules Targeting HSPs

Data from https://www.clinicaltrials.gov/.
Cancers
Cancer pathogenesis is fundamentally characterized by redox imbalance, which necessitates a constitutively elevated level of HSPs to maintain survival (Alimardan et al., 2023; Gorrini et al., 2013). Within the tumor microenvironment, dysregulated redox homeostasis generates numerous ROS that specifically modify Cys on HSPs (Faienza et al., 2025; Glorieux et al., 2024). These redox PTMs allow HSPs to exhibit oncogene-like functions and modulate “non-oncogene addiction” of stressed cancer cells adapting to an adverse microenvironment, making it a promising sensitization strategy target in cancer therapy (Jego et al., 2019). For instance, disulfide bonds-mediated tetramerization of TRAP1 has been shown to promote its antioxidant and pro-neoplastic activity (Faienza et al., 2025).
Growing evidence revealed that the occurrence and poor prognosis of malignancies is usually accompanied by the overexpression of HSPs (Lianos et al., 2015). A clinical trial has identified serum HSP70 as an additional biomarker for early detection of pancreatic cancer (Dutta et al., 2012). Therefore, the development of HSF1 inhibitors and HSPs inhibitors is a tendency in cancer therapy field. For instance, HSF1 inhibitors, including KRIBB11, DTHIB and Rohinitib-Cantharidin hybrid ligands have been exploited for cancer (Agarwal et al., 2015; Xu et al., 2025; Yoo et al., 2021). Some HSPs inhibitors caused protein aggregation and killed cancer cells, like okadaic acid and JG-98 (Backe et al., 2020; Nguyen et al., 2024). Clinical trials have demonstrated the positive role of HSP90 inhibitor AUY922 in advanced non-small cell lung cancer (Felip et al., 2018). 17-AAG, another inhibitor of HSP90, dissociated p53-R280T aggregation and restored p53 tumor suppressor function, thus blocking tumor growth (Li et al., 2024a). Nevertheless, the toxicity profile and poor penetration of the blood–brain barrier (BBB) of 17-AAG result in the termination of lots of clinical trials (NCT00104897) (Pacey et al., 2012; Saif et al., 2013).
Subsequent development has yielded improved agents. For example, TAS-116 (pimitespib), a selective HSP90 inhibitor, has received approval in Japan as a fourth-line therapy for gastrointestinal stromal tumors (Naoki et al., 2024). Feifei Qin et al. designed a HSP90/PI3Kα dual inhibitor for cutaneous melanoma (Qin et al., 2019). Besides, some HSP90-CDC37 protein–protein interaction disruptors have been developed for cancer treatment, including Celastrol-triazole derivative, 18 b-Glycyrrhetinic acid derivative, DDO-5994, and K-Ras-selective HSP90–Cdc37 interface inhibitors (Huang et al., 2023a; Li et al., 2021; Siddiqui et al., 2021; Zhang et al., 2021b). Fei-Ya Yang designed a Prostate-specific membrane antigen (PSMA)-targeted nano-PROTACs, which precisely degraded HSP90 and androgen receptor (AR) receptor, exhibiting a high tumor growth inhibition value of up to 78% (Yang et al., 2025).
Of particular significance, combining HSP inhibitors with other therapies has emerged as a significant trend in clinical trials. For instance, a Phase I trial showed that combining Trastuzumab with 17-AAG exhibited manageable safety profiles, tolerances, and antitumor activity for Trastuzumab-refractory HER-2–overexpressing breast cancer (Modi et al., 2007). Moreover, a Phase II Trial showed that combining 17-AAG and Trastuzumab was effective in patients with HER2-positive metastatic breast cancer (Modi et al., 2011). In a Phase Ib trial, the combination of TAS-116 and nivolumab was observed to be safe and active for solid tumors, particularly microsatellite stable colorectal cancers (Kawazoe et al., 2021). Maggie J Phillips et al. conducted a Phase Ib/II clinical trial, which demonstrated that the combination of pembrolizumab (anti-PD-1) and XL888 (HSP90 inhibitor) can activate local and systemic immune activation in patients with advanced colorectal cancer (Phillips et al., 2025).
Collectively, HSPs are positively related to tumor metastasis, poor prognosis, and resistance to chemotherapy (Yang et al., 2021). Therefore, further investigation should focus on exploiting HSP inhibitors with low side effects and exploring combination treatment strategies, which will make a valuable contribution to enhancing the accuracy of cancer diagnosis and fostering the development of efficient anti-cancer avenues.
Senescence
Senescence is associated with dysfunction of proteostasis and highly cross-linked protein aggregation (Höhn and Grune, 2013; Reeg and Grune, 2015), promoting the development of neurodegenerative diseases. While the HSF1-HSPs system in senescence is still unclear, necessitating a further elucidation of the specific mechanisms (Murshid et al., 2013). It is noteworthy that upregulation of HSP70 facilitated by Rapamycin or Minocycline attenuated protein aggregation (Lim and Hyun, 2022; Takenaka et al., 2022), which underscores that targeting HSP70 will provide potential strategies for aging-related phenotypic alteration.
Neurodegenerative diseases
The pathogenesis of neurodegenerative diseases, such as Huntington’s disease (HD), Parkinson’s disease (PD), AD, Amyotrophic lateral sclerosis (ALS), and Frontotemporal dementia (FTD), shares common pathogenic hallmarks characterized by the pathological protein aggregation and aberrant proteostasis within the central nervous system (Wilson et al., 2023). This proteostasis collapse is frequently accompanied by impaired heat shock response and redox imbalance (Jacob-Tomas et al., 2022). Consequently, the major therapeutic is to restore proteostasis through modulating key HSPs. The potential therapeutic strategy for neurodegenerative diseases includes activating HSP70 and inhibiting HSP90 (Gupta et al., 2020).
Huntingtin (HTT)-polyglutamine (polyQ) aggregation
HD is characterized by an expanded Cytosine-Adenine-Guanine (CAG) trinucleotide repeat in exon 1 of the HTT gene, leading to the expanded polyQ repeat of mutant huntingtin (mHTT). The mHTT exerts toxic gain-of-function (i.e., excitotoxicity, dopaminergic toxicity, abnormal protein aggregation, dysregulated autophagy, mitochondrial dysfunction, and oxidative stress), ultimately resulting in neuronal dysfunction and cell death (Kim et al., 2021; Rodríguez-González et al., 2020). Expansion of the CAG sequence beyond the normal range of 6–26 repeats leads to instability, with further expansion possible in subsequent generations, a process particularly associated with paternal transmission (Dayalu and Albin, 2015; Semaka et al., 2013).
To investigate the complicated pathophysiology of HD, a variety of experimental models have been established, including transgenic truncated HD mouse models, transgenic full-length HD mouse models, knock-in HD mouse models, etc. (Kim et al., 2021). Since the pathogenesis of HD prominently involves the disruption of proteostasis and mitochondria integrity, which are also regulated by HSPs. Thus, targeting HSPs has emerged as a rational therapeutic strategy for HD. Accordingly, numerous studies have demonstrated the HSP activators’ regulatory impact on HD. For example, NG-094 up-regulates HSP16.11, HSP16.2, and HSP70, reducing polyQ aggregation and inhibiting polyQ-mediated animal paralysis (Haldimann et al., 2011). With the ability to activate HSP70, YM-1, and Withaferin A are shown to attenuate Huntingtin aggregation and alleviate the pathological processes of HD (Joshi et al., 2021; Pinho et al., 2021). Furthermore, a recent study reported that downregulating HSP70-binding protein 1 (HSPBP1), an HSP70/CHIP inhibitor, prevented mutant HTT accumulation in neuronal cells, offering a new target for HD treatment (Jing et al., 2021). Strikingly, clinical trials have been conducted to research the role of Minocycline (a HSP70 activator) in HD (NCT00029874).
α-synuclein
The pathological accumulations of α-synuclein (Lewy bodies) and the detection of substantia nigra parscompacta (SNpc) neuronal degeneration are the definitive diagnosis of PD (Obeso et al., 2017; Tripathi, 2020). Given their significant role in inhibiting protein aggregation, HSPs have been considered crucial therapeutic targets for PD. For instance, SNX-0723 induces brain HSP70 and inhibits α-synuclein oligomer formation, representing an outstanding therapeutic strategy for PD (Putcha et al., 2010). Similarly, 17-AAG inhibits HSP90 and activates HSP70, thereby preventing the oligomerization of α-synuclein and rescuing neurotoxicity (Putcha et al., 2010). In contrast, the therapeutic mechanism of some drugs also involves the activation of HSP90. African walnut ethanolic extract, which activates both HSP70 and HSP90, has been shown to block α-synuclein misfolding, oligomerization, and aggregation (Tokunbo et al., 2023). Terazosin (an agonist of HSP90) has entered clinical trials for the treatment of PD (NCT04386317), which marks a critical step in clinical translation.
Aβ and tau protein
Aβ and Tau protein accumulation are the main pathological hallmarks of AD (Gorantla et al., 2020). Multiple studies have reported the beneficial effects of activating sHSP and HSP70 in AD. For instance, Paeoniflorin upregulates HSP16.2 and attenuates soluble Aβ oligomer levels, thereby inhibiting Aβ aggregation and alleviating AD (Ai et al., 2018). Gold nanoparticles activate HSP27 and HSP70, reduce inflammation and oxidative stress, and inhibit insoluble Aβ and tau aggregation (Chiang et al., 2021; El-Naggar et al., 2015; Sandhir et al., 2015; Shaheen et al., 2016). Furthermore, Epoxyazadiradione has demonstrated significant efficacy in upregulating HSP70 and abrogating the Tau aggregation in the form of neurofibrillary tangles (Gorantla et al., 2020). Notably, some HSP70 activators have advanced clinical trials for the treatment of AD, such as Rapamycin (NCT04629495, NCT04200911) and Minocycline (NCT01463384).
In contrast to the protective roles of sHSP and HSP70, HSP90 plays a more complex role in AD pathogenesis. HSP90 promotes the misfolding and accumulation of tau, and may even strengthen their toxicity (Blair et al., 2013). Therefore, inhibiting HSP90 has emerged as a major therapeutic strategy for AD. For example, HSP90 inhibitor OS47720 alleviates synaptic dysfunction and memory loss in symptomatic Tg2576 mice (a model of AD) (Wang et al., 2017). A clinical trial demonstrated that with the downregulation of HSP90, Lactoferrin improved AD pathology and cognitive decline through targeting the p-Akt/PTEN pathway, which modulated oxidative stress and inflammatory processes in the disease (Mohamed et al., 2019). Interestingly, seemingly paradoxically, evidence also suggests that HSP90 activation may have benefits under certain conditions. In mouse models of APP/PS1AD, treatment with the HSP90 activator terazosin decreased tau and α-synuclein levels, thereby ameliorating memory deficit (Chen et al., 2023b). Collectively, all these findings underscore the importance of HSP regulators in AD, highlighting the duality and complexity of targeting the HSPs for therapy.
Poly-GA and TDP-43
The most common genetic cause of ALS and FTD is the excessive GGGGCC repeat expansion in the intron of the chromosome 9 open reading frame 72 (C9ORF72) gene, which leads to the mass aggregation of neurotoxic dipeptide repeat proteins, like Poly-GA (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The cytoplasmic aggregation of TDP-43 is another key pathological hallmark, a process recently suggested to be exacerbated through methionine oxidation (Ozguney et al., 2025). Together, these pathologies cause a profound burden on cellular proteostasis.
Given this central role of proteostasis failure, enhancing HSPs function, especially HSP70, has been a rational therapeutic strategy. Preclinically, upregulation of HSP70 induced by 17-AAG can downregulate Poly-GA aggregation and alleviate behavioral defects in adult Poly-GA-expressing mice (Zhang et al., 2021a). Initial two-phase 2 clinical trials showed that arimoclomol, a HSP70 inducer with the function of clearing protein aggregation, was a safe and effective treatment strategy for ALS (Benatar et al., 2018; Cudkowicz et al., 2008). However, a subsequent phase 3 trial concluded that arimoclomol did not significantly ameliorate ALS progression (Benatar et al., 2024). Moreover, other HSP70 activators including Rapamycin (NCT03359538) and Minocycline (NCT00047723) have also entered clinical trials for the treatment of ALS. In conclusion, despite this clinical setback, the underlying scientific rationale for targeting HSP70 remains compelling, underscoring the continued importance of developing more effective HSP70 activators for ALS.
N-acetylaspartate (NAA)
Canavan disease is a severe progressive neurodegenerative disorder caused by loss of enzymatic activity in aspartoacylase (ASPA) gene, resulting in the toxic accumulation of NAA (Gersing et al., 2021; Wei et al., 2022). Unlike other neurodegenerative diseases driven by toxic protein aggregation, Canavan disease pathogenesis is primarily characterized by enzymatic deficiency. Importantly, studies concerning the disease-associated ASPA C152W mutations have shown that its functional loss arises from enhanced proteasomal degradation, which is highly dependent on HSP70 and its cochaperone HSP110 (Gersing et al., 2021). Consequently, inhibiting the HSP70 system, particularly HSP110, represents a rational therapeutic strategy to reduce the degradation of functional ASPA (Gersing et al., 2021). Accordingly, recently developed HSP110 inhibitors, including Foldamers 33 and 52, have demonstrated therapeutic potential in experimental models, offering a promising targeted strategy for Canavan disease (Abildgaard et al., 2019; Clausen et al., 2020; Gozzi et al., 2020).
In summary, HSPs function as a complex double-edged sword in the pathogenesis and progression of neurodegenerative diseases. On the one hand, HSPs can alleviate disease progression through promoting the clearance of misfolded proteins, such as HSP70 and sHSP. On the other hand, HSPs may aggravate disease progression through stabilizing disease-related proteins, such as HSP90. Therefore, selectively targeting specific HSPs has emerged as a promising therapeutic strategy. Nevertheless, several critical challenges impede its clinical translation: (1) Most studies are limited to animal experiments, reminding researchers to accelerate the pace of clinical trials. (2) Achieving a precise therapeutic balance between the closely interconnected HSP70 and HSP90 chaperone systems is inherently difficult (Gupta et al., 2020). (3) Some small molecules targeting HSPs such as 17-AAG fail to cross BBB, influencing their practical efficacy (Egorin et al., 2001). 4) The clinical translation of small molecules has been significantly constrained due to their hepatotoxicity, which may be addressed through strategic structural modifications (Banerji et al., 2005).
Limb-girdle muscular dystrophy type D1
DNAJB6 mutation leads to protein aggregation, which is a prominent marker of limb-girdle muscular dystrophy type D1 (LGMDD1) (Harms et al., 2012). Recently, Ankan K. Bhadra et al. first elucidated the impact of LGMDD1 mutants on the ATPase cycle of HSP70/40 (Bhadra et al., 2022). Similarly, Meital Abayev-Avraham et al. also proved that in C. elegans, high-affinity and non-productive coactions with mutant DNAJB6 decreased HSP70 level and destroyed protein homeostasis (Abayev-Avraham et al., 2023). Therefore, exploiting specific inhibitors targeting HSP70/DNAJ cycle or binding blockers targeting DNAJB6-HSP70 will contribute to reversing the myopathy-associated disease phenotype.
β-thalassemia
β-thalassemia presents the feature of a quantitative defect in hemoglobin β-globin chains synthesis, leading to free a-globin chains accumulation and ineffective erythropoiesis. As a chemical inhibition of XPO1, KPT-251 is suggested to upregulate HSP70 and improve terminal maturation of β-thalassemia erythroblasts, which will be a potential therapeutic schedule for β-thalassemia (Guillem et al., 2020).
Others
Yuchen Li et al. found that TRAP1 suppressed ferroptosis via interacting with ferroptosis-related proteins, such as acyl-CoA synthetase long-chain family member 1, acyl-CoA synthetase long-chain family member 4, and cytochrome b5 reductase 1, thereby mitigating hyperglycemia-induced mitochondrial damage. This study confirmed that TRAP1 played a significant role in regulating mitochondrial injury during early diabetic retinopathy (Li et al., 2025). Besides, TRAP1-driven metabolic reprogramming elevated lactate-dependent H4 lysine 12 lactylation by down-regulating HDAC3, thereby promoting SASP transcription and aggravating vascular smooth muscle cell (VSMC) senescence. Therefore, targeting TRAP1 presents a novel therapeutic target for diabetic retinopathy and VSMC senescence (Li et al., 2024b).
Conclusions and Perspectives
Based on the current research progress, HSPs are central integrators of redox-proteostasis, and their dysregulation is a hallmark of numerous diseases. As this review underscores, their function in human diseases is context-dependent. Typically, HSP inhibitors represent a therapeutic strategy for cancer, causing numerous protein aggregation and killing cancer cells. However, HSPs serve as a complex double-edged sword in neurodegenerative diseases, making their precise regulation an important direction for future research. Considering their essential role in maintaining proteostasis, therapeutic interventions must avoid global inhibition or activation. Therefore, further studies should focus on site-specific redox pharmacology, aiming to exploit agents that precisely target Cys modifications on HSPs.
Although attention to the important role of HSPs in redox-proteostasis has increased significantly in recent years, there are still several questions that remain unresolved: (1) The molecular mechanisms of protein folding by HSPs remain unclear, owing to the complexity of dynamic and transient binding structures of HSPs complexed with clients (Hu et al., 2022). (2) Most research has concentrated on HSP70 and HSP90, whereas other HSP families, particularly the sHSP, remain largely understudied. (3) Which redox PTMs occur at specific cysteine sites of HSPs and how they are regulated under different conditions remain unclear (Yang et al., 2020; Zhao et al., 2022c). (4) The redox regulation of HSP100 within proteostasis remains scarce, necessitating further investigation. (5) The close interconnection between the HSP70 and HSP90 chaperone systems makes achieving a precise therapeutic balance inherently difficult (Gupta et al., 2020). (6) The therapeutic potential of targeting redox PTMs of HSPs remains unexplored in drug development. (7) HSF1 and IRE1α–XBP1 should be prioritized as promising drug targets due to their disease-specific roles. (8) No HSPs-targeted agents have been approved by the FDA due to their poor penetration of the BBB and potential toxicity (Fig. 11). However, this challenge is not insurmountable. As a novel modality, PROTACs have obtained considerable attention, which successfully delivers into the brain across the BBB (Jiang et al., 2025; Tashima, 2023). HSP90-targeting PROTACs, such as NX-5948 and NX-2127, have entered clinical trials, offering promising avenues for overcoming the BBB challenge in drug development (Table 6).

In conclusion, the present review summarized how redox PTMs regulate HSPs, how they maintain cellular proteostasis, and deal with organelle-specific stress responses. We also highlight the advantages and challenges of HSP-targeted agents in protein aggregation disease. As important integrators of redox-proteostasis, targeting HSPs offers a promising avenue to improve the efficacy of disease treatment.
Authors’ Contributions
K.G.: Writing—review and editing, writing—original draft, formal analysis, data curation. X.L.: Writing—review and editing, writing—original draft. Q.L.: Writing—review and editing, writing—original draft. C.C.: Writing—original draft, investigation, formal analysis, conceptualization. J.L.: Writing—original draft, investigation, formal analysis, conceptualization. T.Z.: Writing—review and editing, investigation, formal analysis, funding acquisition, conceptualization. C.S.: Writing—review and editing, investigation, formal analysis, funding acquisition, conceptualization. N.K.: Conceptualization, formal analysis, funding acquisition, project administration, resources, software. F.Q.: Conceptualization, formal analysis, funding acquisition, project administration, resources, software. Q.Z.: Writing—review and editing, conceptualization, funding acquisition, software, supervision, validation, visualization.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (No. 82030116 to Feng Qiu), Young Faculty’s Research and Innovation Capability Development in Tianjin Higher Education Institutions Initiated by the Ministry of Education (Chengpeng Sun); National Natural Science Foundation of China (No. 82274221 to Ning Kang; No. 82104461 to Qiang Zhang), Natural Science Foundation of Tianjin (No. 24JCZDJC00570 to Ning Kang), National College Students’ Innovation and Entrepreneurship Training Program (No. 202410063021 to Tiancheng Zhan).