
Abstract
Background:
Sepsis-associated acute kidney injury (SA-AKI) is a frequent and severe complication in critically ill patients, yet effective targeted therapies are lacking. Ferroptosis has been implicated in various forms of organ injury, but its role in SA-AKI and underlying regulatory mechanisms remain unclear.
Methods:
A SA-AKI mouse model was established using cecal ligation and puncture (CLP). Renal histopathology, kidney function assays, and spatial proteomics were employed to assess ferroptosis activation. In vivo and in vitro models were subjected to lipopolysaccharide (LPS) stimulation to evaluate ferroptosis-related markers, including reactive oxygen species (ROS), lipid peroxidation, ferrous iron levels, and mitochondrial membrane potential. DNA pull-down coupled with mass spectrometry identified potential upstream regulators of HO-1. Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) and dual-luciferase reporter assays were used to validate transcriptional regulation by SMAD4. Functional studies assessed the impact of SMAD4 on HO-1 expression, ferroptosis, and renal function.
Results:
Ferroptosis was markedly activated during SA-AKI progression. LPS stimulation induced significant ROS accumulation, lipid peroxidation, elevated ferrous iron levels, mitochondrial membrane potential disruption, and robust upregulation of heme oxygenase-1 (HO-1). SMAD4 was identified as a transcriptional repressor of HO-1. ChIP-qPCR and dual-luciferase assays confirmed SMAD4 binding to the HO-1 promoter and suppression of its transcription. SMAD4 overexpression reduced HO-1 expression, alleviated ferroptosis, and improved renal function in both in vivo and in vitro models.
Conclusions:
SMAD4 mitigates ferroptosis by transcriptionally repressing HO-1, exerting a protective effect in SA-AKI. This study identifies a novel SMAD4–HO-1 regulatory axis and suggests a potential therapeutic target for sepsis-induced kidney injury. Antioxid. Redox Signal. 45, 28–43.
Keywords
Introduction
Sepsis is associated with a cascade of physiological, pathological, and biochemical abnormalities (Singer et al., 2016). Sepsis-associated acute kidney injury (SA-AKI) is a severe clinical complication frequently observed in critically ill patients and is closely linked to high morbidity and mortality (Chang et al., 2022). Moreover, SA-AKI significantly increases the long-term risk of chronic kidney disease and dialysis dependence (Mayeux and MacMillan-Crow, 2012). Despite its clinical importance, the pathophysiological mechanisms underlying SA-AKI remain incompletely understood, and effective targeted therapies are currently lacking. Therefore, elucidating the molecular mechanisms of SA-AKI is urgently needed to guide the development of novel therapeutic strategies.
Innovation
SA-AKI is a severe complication of sepsis characterized by excessive oxidative stress and ferroptotic tubular injury, posing a significant threat to patient survival. The main aim of this study was to elucidate the transcriptional regulatory role of SMAD4 in ferroptosis during SA-AKI. Our findings revealed that SMAD4 exerts renoprotective effects by repressing the transcriptional activity of HO-1. Mechanistically, downregulation of SMAD4 leads to persistent HO-1 overexpression, thereby promoting iron accumulation, lipid peroxidation, and ferroptotic damage in tubular epithelial cells. Conversely, SMAD4 activation alleviates ferroptosis and attenuates renal injury by suppressing HO-1 expression. These results uncover a novel regulatory mechanism underlying the pathogenesis of SA-AKI and highlight the SMAD4/HO-1 axis as a promising therapeutic target for preventing sepsis-induced renal dysfunction.
Ferroptosis is a form of regulated cell death that is iron-dependent and characterized by excessive lipid peroxidation (C11) and disruption of redox homeostasis (Tang et al., 2021a). Unlike traditional forms of cell death such as apoptosis, necrosis, or autophagy, ferroptosis exhibits distinct features, including intracellular glutathione depletion, reduced glutathione peroxidase 4 (GPX4) activity, and accumulation of lipid reactive oxygen species (ROS) (Jiang et al., 2021). In recent years, increasing evidence has implicated ferroptosis in the pathogenesis of SA-AKI (Chen et al., 2021; Cross et al., 2019; Yang et al., 2024). However, the upstream molecular events that trigger ferroptosis in SA-AKI remain incompletely understood.
Heme oxygenase-1 (HO-1) is a stress-inducible enzyme that catalyzes the degradation of heme into carbon monoxide (CO), biliverdin, and free ferrous iron (Schipper et al., 2009). Under conditions of oxidative stress, HO-1 confers cytoprotective effects by converting pro-oxidant heme into metabolites with antioxidant properties. However, its by-products—particularly free iron and CO—may exacerbate intracellular oxidative damage and trigger ferroptosis (Campbell et al., 2021). Thus, HO-1 exerts a context-dependent, “double-edged sword” effect, playing either protective or detrimental roles depending on the pathological state (Yuan et al., 2008). In SA-AKI, the precise role and regulatory mechanisms of HO-1 remain poorly understood and warrant further investigation.
SMAD4, a central mediator of the transforming growth factor beta (TGF-β) signaling pathway, plays a critical role in regulating the transcription of genes involved in inflammation, fibrosis, and cellular stress responses (Lan, 2011). Recent studies have demonstrated that SMAD4 can function as a transcription factor under specific pathological conditions (Deng et al., 2019; Du et al., 2020; Liang et al., 2020; Shi et al., 2023). In highly invasive pancreatic cancer cells induced by TGF-β1, SMAD4 has been shown to act as a transcriptional repressor of GPX4, thereby promoting ferroptosis (Chen et al., 2024), suggesting its potential involvement in the regulation of cell death pathways. However, direct evidence linking SMAD4 to ferroptosis in the context of inflammation-induced acute kidney injury remains lacking.
This study focuses on the transcriptional regulation of HO-1—a key enzyme in ferroptosis—by SMAD4, aiming to uncover its role in mediating ferroptosis during SA-AKI. By integrating in vivo and in vitro models, spatial proteomics, and molecular mechanistic approaches, we seek to elucidate the SMAD4/HO-1 signaling axis in the regulation of ferroptosis during SA-AKI. These findings may offer novel insights into the pathogenesis of SA-AKI and identify potential therapeutic targets.
Result
Cecal ligation and puncture induces SA-AKI and activates ferroptosis in vivo
We first established a cecal ligation and puncture (CLP) model to induce SA-AKI in mice. Histopathological analysis, including hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) staining, revealed significant renal tubular epithelial cell damage after CLP. This was characterized by tubular dilation, epithelial flattening, cell detachment, and extensive coagulative necrosis, with representative injured areas locally magnified and indicated by red arrows (Fig. 1A). Kaplan–Meier survival analysis showed that CLP significantly reduced the survival rate compared with sham controls (Fig. 1B). Serum creatinine (Scr) and blood urea nitrogen (BUN) levels were elevated following CLP, indicating impaired renal function (Fig. 1C, D). Similarly, KIM-1 and NGAL, recognized as sensitive and specific biomarkers for AKI, were markedly upregulated in CLP-treated mice compared with sham controls (Fig. 1A, E–G). Inflammatory cytokines, including TNF-α, IL-6, and IL-1β, were also significantly increased in the CLP group (Fig. 1H–M). These results demonstrate that CLP successfully induces a typical SA-AKI phenotype accompanied by a pronounced inflammatory response.

To further investigate the underlying mechanisms, spatial proteomics analysis was performed on microdissected proximal tubules. Proximal tubular regions were precisely isolated by laser capture microdissection, thereby minimizing signal interference from different nephron segments and ensuring the spatial specificity and reliability of the proteomic data. Kyoto Encyclopedia of Genes and Genomes pathway enrichment and gene set enrichment analysis identified ferroptosis as a significantly activated pathway in SA-AKI (Fig. 1N, O). In addition, multiple infection- and immunity-related pathways also exhibited a tendency toward positive enrichment. These findings suggest that ferroptosis may represent a key mechanism driving tubular epithelial cell injury in the context of SA-AKI. Based on the proteomics results, key regulators of ferroptosis were further examined. Western blotting confirmed that CLP enhanced the expression of the ferroptosis-associated protein ACSL4 and reduced the expression of GPX4 (Fig. 1P). Collectively, spatial proteomics analysis combined with functional validation systematically reveals robust activation of the ferroptosis pathway in SA-AKI from both the spatial and molecular functional perspectives.
Ferrostatin-1 alleviates ferroptosis and renal injury
To investigate the role of ferroptosis in SA-AKI, we employed ferrostatin-1 (Fer-1), a specific ferroptosis inhibitor. Transmission electron microscopy revealed pronounced mitochondrial shrinkage and cristae loss in the CLP group. In contrast, Fer-1 treatment mitigated mitochondrial atrophy and restored cristae structure (Fig. 2A). Assessment of oxidative stress-related parameters showed that renal levels of Oxidized Glutathione (Glutathione disulfide) and Malondialdehyde (MDA) were significantly elevated in the CLP group, reflecting a pronounced oxidative stress response, but were markedly reduced upon Fer-1 administration (Fig. 2B,D). Moreover, Fer-1 treatment restored the depleted Reduced Glutathione (GSH) levels induced by CLP (Fig. 2C), indicating a potent antioxidant effect. Tissue ferrous iron, measured using a specific detection kit, was increased in the CLP group, whereas Fer-1 markedly suppressed this elevation (Fig. 2E). Western blot analysis showed that CLP induced a marked upregulation of ACSL4 and significant downregulation of GPX4, whereas these changes were reversed by Fer-1 treatment, suggesting that Fer-1 effectively inhibits key regulatory components of the ferroptosis pathway (Fig. 2F). Consistently, immunohistochemical analysis confirmed reduced expression of C11 marker 4-HNE following Fer-1 treatment, indicating a significant reduction in C11 damage (Fig. 2G). Collectively, these findings demonstrate that Fer-1 effectively suppresses CLP-induced renal ferroptosis by alleviating mitochondrial damage and inhibiting oxidative stress and iron accumulation, thereby further supporting a critical role of ferroptosis in the pathogenesis of SA-AKI.
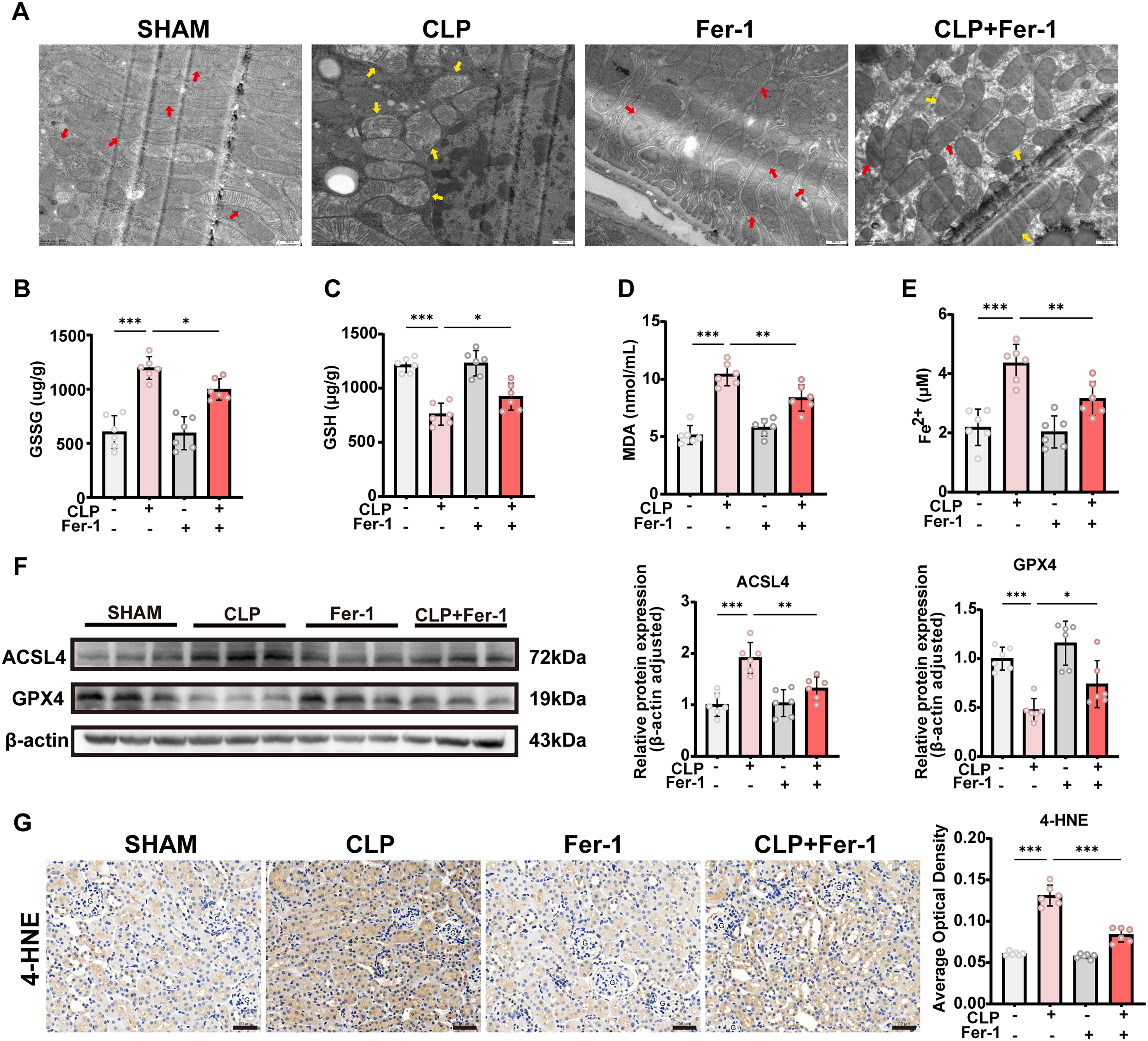
We further assessed the impact of Fer-1 on renal injury in CLP-treated mice. Fer-1 partially alleviated tubular dilation and structural damage (Fig. 3A). Renal function analysis showed that serum Scr and BUN levels were significantly elevated in CLP-treated mice, whereas Fer-1 treatment markedly reduced both parameters, indicating a protective effect on renal function (Fig. 3B, C). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis showed that multiple inflammatory cytokines, including IL-6, IL-1β, and TNF-α, were significantly increased in renal tissues after CLP, whereas their messenger ribonucleic acid (mRNA) levels were markedly decreased following Fer-1 intervention, suggesting an inhibitory effect of Fer-1 on the inflammatory response (Fig. 3D–F). Serum cytokine measurements were consistent with these findings and further confirmed the anti-inflammatory effect of Fer-1 (Fig. 3G–I). Moreover, RT-qPCR analysis demonstrated that mRNA levels of the renal injury markers KIM-1 and NGAL were significantly upregulated after CLP, whereas immunohistochemistry (IHC) and Western blot analyses showed corresponding increase at the protein level. Notably, these changes were effectively reversed by Fer-1 treatment, indicating attenuation of tubular injury (Fig. 3J–M). Collectively, these findings indicate that, by inhibiting ferroptosis, Fer-1 ameliorates CLP-induced SA-AKI by improving renal histological structure, restoring renal function, and suppressing inflammatory responses.

Lipopolysaccharide induces ferroptosis in HK-2 cells in vitro
Building on our in vivo findings that ferroptosis is activated during SA-AKI and that its inhibition alleviates renal injury, we next explored the cellular mechanisms underlying ferroptosis using an in vitro model. HK-2 cells were treated with lipopolysaccharide (LPS) at varying concentrations (0, 5, 10, 15, 20, and 25 μg/mL) to determine the optimal dose, and 15 μg/mL was selected for subsequent experiments based on cell viability and injury markers. CCK-8 assay results demonstrated a dose-dependent decline in cell viability, with ∼60% viability observed at 15 μg/mL, indicating significant cellular damage at this concentration (Fig. 4A). Consistently, Lactate Dehydrogenase (LDH) release increased dose-dependently, reaching about 50% at 15 μg/mL, reflecting substantial membrane damage (Fig. 4B). Lipid peroxidation, measured by MDA levels, also rose with increasing LPS doses, reaching ∼15 nmol·mg−1 protein at 15 μg/mL (Fig. 4C). Time-course experiments were performed with 15 μg/mL LPS treatment at 0, 6, 12, 24, and 48 h. Western blot analysis showed that the ferroptosis marker ACSL4 began to increase at 12 h and peaked at 24 h, whereas GPX4 levels declined to their lowest at 24 h and remained similarly low at 48 h (Fig. 4D). Thus, 24 h was selected as the optimal stimulation duration for downstream studies. FerroOrange staining revealed a significant increase in intracellular ferrous iron accumulation in LPS-treated cells, further confirming the activation of ferroptosis (Fig. 4E). ROS levels, assessed using fluorescent probes, showed enhanced green fluorescence following LPS stimulation, indicating elevated oxidative stress (Fig. 4F). Mitochondrial membrane potential was evaluated using the JC-1 probe, which demonstrated a shift in fluorescence from red to green upon LPS treatment, reflecting loss of mitochondrial membrane potential (Fig. 4F). Similarly, C11 levels, detected using the C11-BODIPY probe, were markedly increased in LPS-treated cells, as evidenced by a shift from red to green fluorescence (Fig. 4G). Together, these findings demonstrate that LPS induces ferroptosis in HK-2 cells in a dose- and time-dependent manner, supporting the in vivo observations and providing further insight into the cellular mechanisms involved in SA-AKI.

HO-1 is upregulated in SA-AKI and enriched in ferroptosis pathway
We have confirmed that ferroptosis is activated in both in vivo and in vitro models of SA-AKI. To further explore the underlying regulatory mechanisms, we reanalyzed the previously obtained spatial proteomic data and found that the ferroptosis-related gene HO-1 was significantly upregulated in the CLP group and enriched in the ferroptosis pathway (Fig. 5A and Supplementary Fig. S1B). As a heme oxygenase, HO-1 catalyzes the degradation of heme, releasing free ferrous iron, which promotes intracellular iron accumulation. Combined with the observed significant increase in ferrous iron levels in both tissue and cellular experiments, these results suggest that HO-1 may serve as a critical upstream factor inducing ferroptosis. Further analyses by Western blotting, RT-qPCR, and IHC revealed marked upregulation of HO-1 at both the mRNA and protein levels in tissues and cells (Fig. 5B–F). Notably, the consistent increase in HO-1 expression at both the transcriptional and protein levels indicates that its regulation primarily occurs at the transcriptional level. Based on these findings, we hypothesize that HO-1 is regulated by specific transcription factors and plays a key role in mediating ferroptosis in SA-AKI.
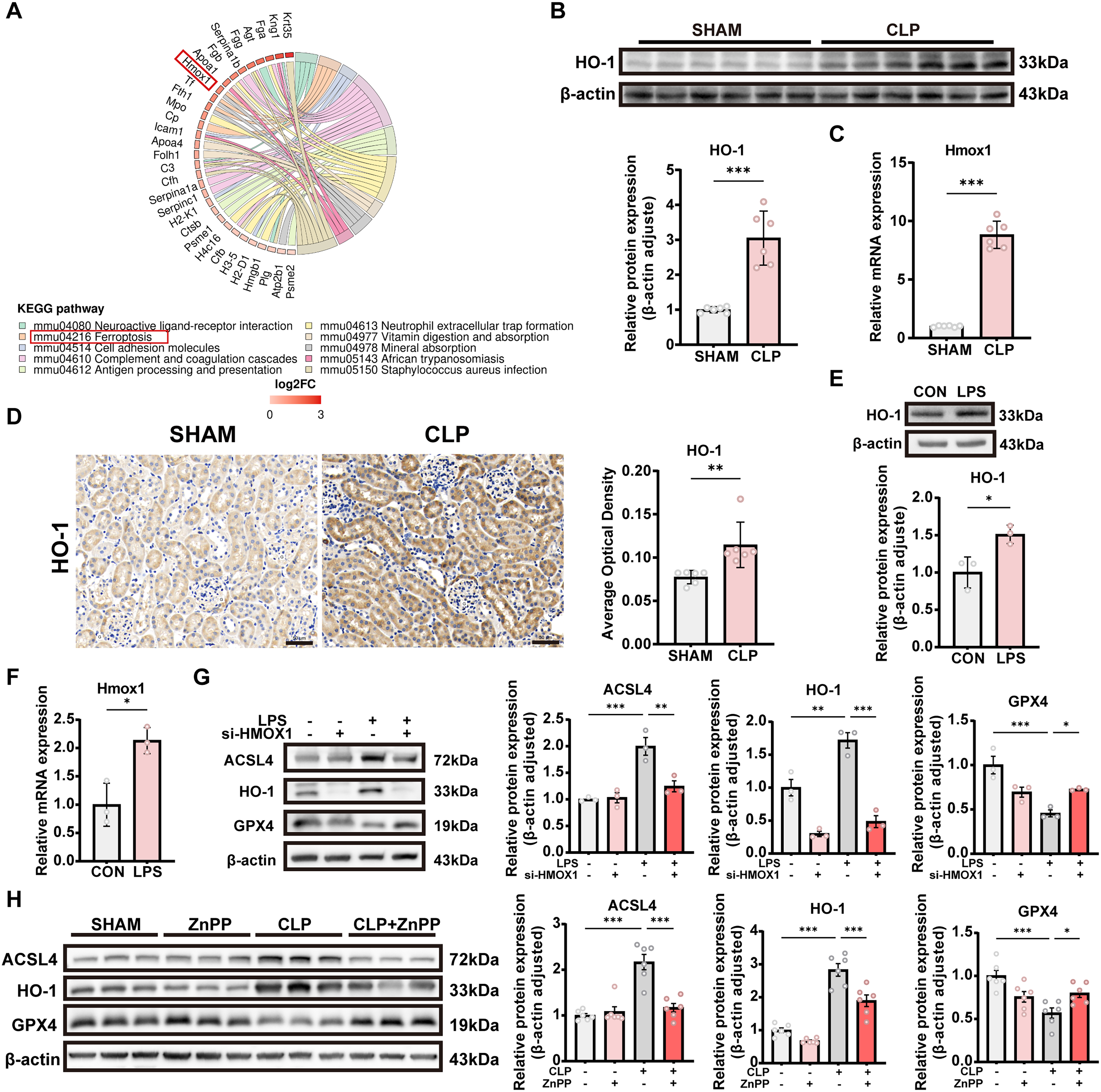
To further investigate the functional role of HO-1 in regulating ferroptosis in SA-AKI, we inhibited HO-1 expression in both in vivo and in vitro models and examined changes in key ferroptosis-related proteins. In the cellular model, HK-2 cells were transfected with HO-1–specific small interfering RNA (si-HO-1) to reduce HO-1 expression. Western blot analysis showed that, compared with the LPS-treated group, HO-1 knockdown decreased ACSL4 expression and increased GPX4 expression (Fig. 5G), consistent with the in vivo findings. In the animal experiments, the HO-1–specific inhibitor zinc protoporphyrin (ZnPP) was administered intraperitoneally to suppress HO-1 expression. Western blot analysis showed that, in the CLP model, suppression of HO-1 markedly attenuated the elevated expression of ACSL4 and partially restored the reduced expression of GPX4 (Fig. 5H), indicating that HO-1 upregulation directly contributes to the induction of ferroptosis in vivo. Together, these results demonstrate that inhibition of HO-1 in both in vivo and in vitro models partially reverses the abnormal changes in ferroptosis-related proteins, further supporting a critical pro-ferroptotic role of HO-1 in SA-AKI rather than a merely passive association.
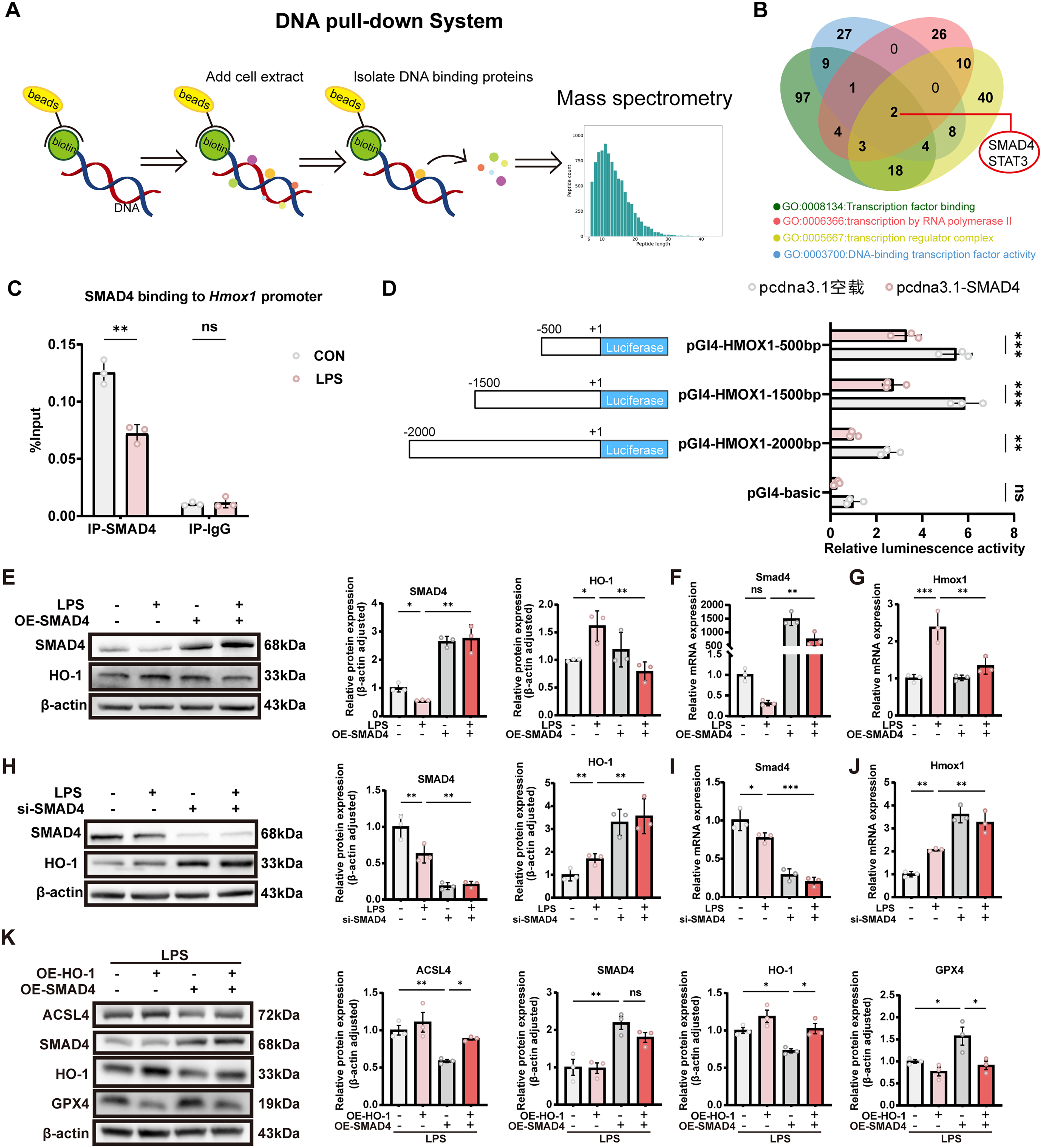
Identification of SMAD4 as a transcriptional repressor of HO-1
To identify potential regulators of HO-1, we performed a DNA pull-down assay using a biotin-labeled HMOX1 promoter sequence to capture nuclear proteins from HK-2 cells, followed by quantitative mass spectrometry (MS) analysis (Fig. 6A). Cross-referencing with transcription-related Gene Ontology enrichment analysis revealed SMAD4 and STAT3 as candidate transcription factors potentially regulating HMOX1 expression (Fig. 6B). Among these, SMAD4 exhibited more pronounced differential expression compared to STAT3 and was therefore selected for further investigation. Chromatin immunoprecipitation (ChIP)-qPCR confirmed binding of SMAD4 to the HMOX1 promoter region, which was diminished upon LPS treatment (Fig. 6C). Furthermore, luciferase reporter assays performed in HEK293T cells transfected with SMAD4 overexpression plasmids demonstrated that SMAD4 suppressed activation of HMOX1 promoter constructs spanning 2000 bp, 1500 bp, and 500 bp upstream regions, suggesting that the SMAD4 binding site is likely located within the proximal 500 bp of the promoter (Fig. 6D). Subsequently, HK-2 cells treated with LPS were transfected with either an empty vector or SMAD4 overexpression plasmids. qPCR and Western blot analyses showed that SMAD4 overexpression led to decreased HO-1 expression (Fig. 6E–G), indicating a negative regulatory role of SMAD4 on HO-1. Conversely, knockdown of SMAD4 via siRNA resulted in derepression of HO-1 expression, as confirmed by qPCR and Western blotting (Fig. 6H–J). In rescue experiments, HO-1 overexpression reversed the alterations in ACSL4 and GPX4 levels induced by SMAD4 overexpression. Conversely, silencing HO-1 markedly attenuated the ferroptotic changes triggered by SMAD4 knockdown, further supporting that HO-1 functions as a key downstream effector of SMAD4 in the regulation of ferroptosis (Fig. 6K and Supplementary Fig. S1D). Collectively, these findings demonstrate that SMAD4 acts as a transcriptional repressor of HO-1, and that the SMAD4–HO-1 axis plays a critical regulatory role in ferroptosis during SA-AKI.
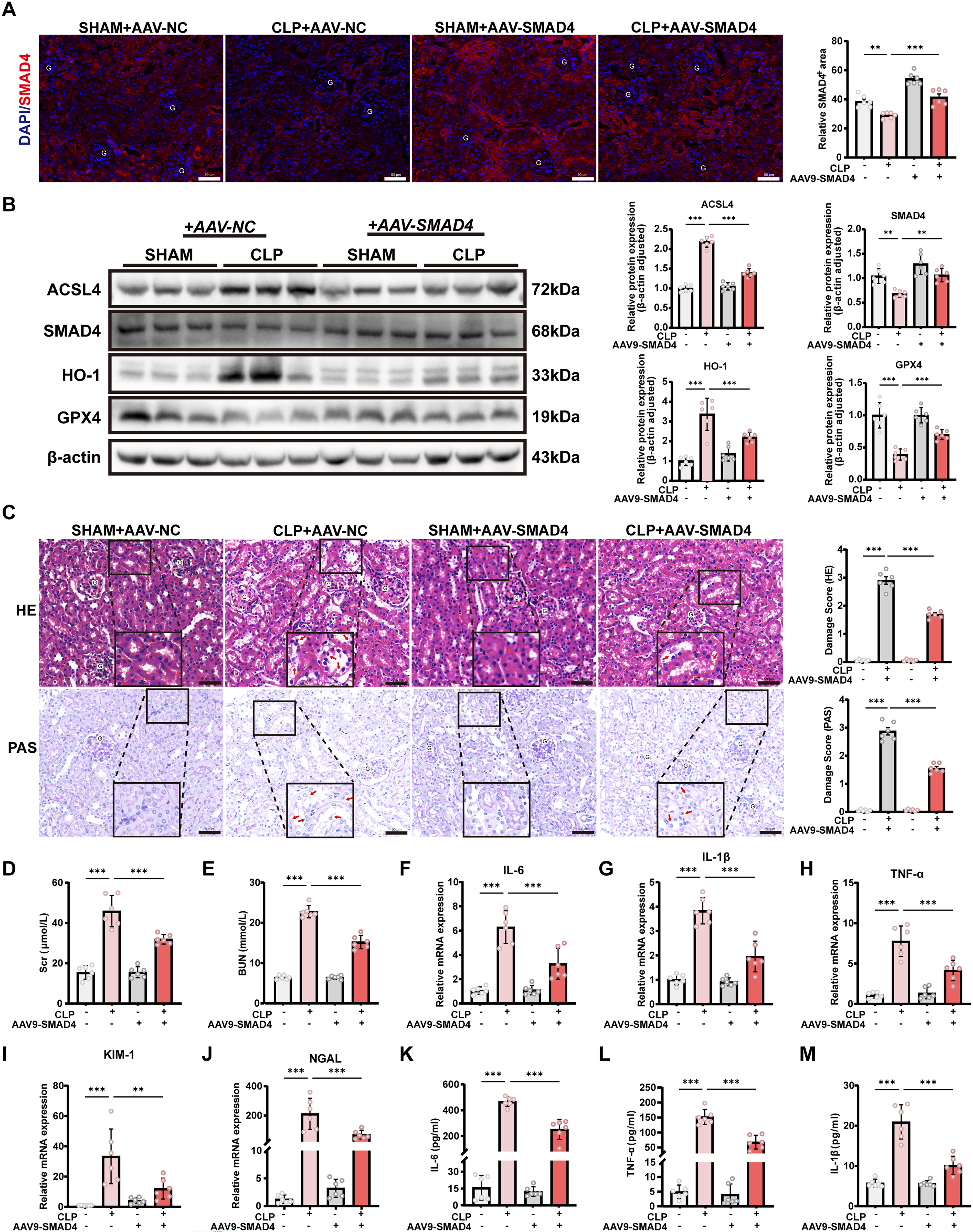
AAV-mediated SMAD4 overexpression alleviates ferroptosis and renal injury
Building on the regulatory role of SMAD4 in HO-1 transcription demonstrated in vitro, we next investigated whether SMAD4 overexpression could alleviate ferroptosis and renal injury in the CLP-induced SA-AKI mouse model. To genetically activate SMAD4 in vivo, mice were systemically injected with AAV9 encoding SMAD4 (AAV-SMAD4) or negative control (AAV-NC); the structure of the AAV construct is shown in Supplementary Figure S1E. Robust GFP fluorescence confirmed efficient renal transduction in both AAV-NC- and AAV-SMAD4–treated mice (Supplementary Fig. S1F). Immunofluorescence staining revealed that SMAD4 expression was markedly reduced in renal tubular cells following CLP surgery, whereas AAV-SMAD4 administration effectively restored SMAD4 expression (Fig. 7A), which was further confirmed by Western blot analysis (Fig. 7B). Notably, SMAD4 overexpression significantly suppressed HO-1 expression in CLP-injured kidneys and attenuated ferroptosis, as evidenced by increased GPX4 and decreased ACSL4 levels (Fig. 7B). Histological analysis using H&E and PAS staining demonstrated that AAV-SMAD4 markedly alleviated CLP-induced tubular injury compared with AAV-NC–treated mice (Fig. 7C). Consistently, renal function was improved, as reflected by significantly reduced Scr and BUN levels in the AAV-SMAD4 group (Fig. 7D, E). In addition, AAV-SMAD4 significantly reduced renal inflammation and tubular injury, as shown by decreased mRNA expression of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β (Fig. 7F–H), as well as reduced expression of KIM-1 and NGAL (Fig. 7I, J). Serum levels of TNF-α, IL-6, and IL-1β were also markedly decreased in AAV-SMAD4–treated mice compared with AAV-NC controls (Fig. 7K–M). Collectively, these results demonstrate that AAV-mediated SMAD4 overexpression suppresses HO-1–dependent ferroptosis, attenuates renal inflammation, and protects against renal injury in CLP-induced SA-AKI.
Discussion
SA-AKI remains a major challenge in critical care medicine, with complex pathophysiology and limited effective therapeutic strategies. It is generally recognized that the predominant pathological feature of SA-AKI is tubular epithelial injury rather than glomerular dysfunction (Aslan et al., 2018). Therefore, preserving tubular epithelial cell integrity is crucial for the treatment and prevention of this condition. In recent years, ferroptosis—a newly characterized form of regulated cell death driven by iron-dependent lipid peroxidation—has been increasingly implicated in the pathogenesis of AKI, including SA-AKI (Huang et al., 2024; Li et al., 2023; Yang et al., 2024).
In the present study, we identify a transcriptional mechanism by which SMAD4 restrains ferroptosis in SA-AKI via repression of HO-1. As schematically illustrated in Figure 8, the loss of SMAD4 under septic conditions results in aberrant HO-1 upregulation, iron accumulation, and lipid peroxidation, thereby promoting ferroptotic tubular injury. These findings offer new mechanistic insights into the progression of SA-AKI. We first confirmed that the CLP-induced SA-AKI model presents classical tubular damage accompanied by hallmark features of ferroptosis, including downregulation of GPX4, upregulation of ACSL4, increased MDA accumulation, and mitochondrial injury. Treatment with Fer-1, a ferroptosis inhibitor, significantly attenuated these phenotypes, supporting a causal and pathogenic role for ferroptosis rather than it being a mere secondary consequence in SA-AKI.

Further mechanistic investigation revealed that HO-1 expression was persistently upregulated in the SA-AKI model and enriched in the ferroptosis pathway based on spatial proteomics analysis. As a heme oxygenase, HO-1 exerts dual, context-dependent roles. On one hand, its enzymatic activity produces free Fe2+, which may catalyze Fenton reactions and amplify lipid peroxidation, thereby promoting ferroptosis (de Oliveira et al., 2022; Ryter, 2021; Wu et al., 2022). On the other hand, its by-products, such as biliverdin and CO, possess antioxidant and cytoprotective properties (An et al., 2021; Asghar et al., 2025), and in some disease contexts, HO-1 is suggested to exert anti-ferroptotic effects (Wang et al., 2024). However, recent studies across various disease models—including colorectal cancer, retinal pigment epithelial degeneration, and cisplatin-induced AKI—have confirmed that sustained or excessive HO-1 expression can drive ferroptosis and exacerbate tissue injury (Lin et al., 2023; Tang et al., 2021b; Wei et al., 2021). Consistent with these findings, our data show that HO-1 expression increases in parallel with ferroptosis severity in both CLP-induced kidneys and LPS-treated cells, supporting its role as a pro-ferroptotic factor in SA-AKI. Although HO-1 protein expression does not necessarily equate to its enzymatic activity, the stable association between HO-1 upregulation and ferroptotic phenotypes, together with functional perturbation data, supports a biologically relevant role of HO-1 in this pathological context. Notably, our previous work also demonstrated the pro-ferroptotic effect of HO-1: In LPS- or erastin-induced cell injury models, pharmacological inhibition of HO-1 by ZnPP significantly reduced C11 and cellular injury and decreased ferroptosis markers (Li et al., 2025). In the present study, we further suppressed HO-1 expression both in vivo and in vitro and observed a marked alleviation of ferroptosis in SA-AKI models following HO-1 inhibition, thereby providing additional functional evidence that HO-1 actively promotes ferroptotic cell death in this disease setting. Notably, the concordant changes in HO-1 at both the mRNA and protein levels suggest that its pathological upregulation is primarily regulated at the transcriptional level.
The major novelty of this study lies in the identification of a previously unrecognized transcriptional regulatory mechanism of HO-1. Through DNA pull-down assays, ChIP-qPCR, and dual-luciferase reporter assays, we demonstrate for the first time that SMAD4 directly binds to the HO-1 promoter and suppresses its transcriptional activity. Although traditionally viewed as a transcriptional activator in the canonical TGF-β pathway, growing evidence suggests that SMAD4 can also act as a transcriptional repressor for specific target genes (Chen et al., 2024; Deng et al., 2019; Wang et al., 2021). In our model, SMAD4 expression was downregulated under LPS stimulation, coinciding with increased HO-1 expression. Conversely, overexpression of SMAD4 reduced HO-1 mRNA and protein levels and mitigated the ferroptotic phenotype. Importantly, genetic restoration of SMAD4 expression in vivo using AAV-mediated overexpression reproduced these protective effects, leading to suppression of HO-1 expression, attenuation of ferroptosis, and marked improvement in renal injury in CLP-induced SA-AKI. Together, these findings establish SMAD4 as a previously unrecognized transcriptional repressor of HO-1 and highlight the SMAD4/HO-1 axis as a critical regulator of ferroptosis in SA-AKI.
Interestingly, this inverse relationship between HO-1 and SMAD4 was also observed in a diabetic nephropathy model, in which kidney tissues exhibited elevated HO-1 and reduced SMAD4 protein expression (Xu et al., 2025), suggesting a potentially conserved regulatory axis across diseases. Beyond HO-1, SMAD4 may modulate ferroptosis through other pathways as well. For instance, in pancreatic cancer, SMAD4 suppresses GPX4 transcription, thereby enhancing ferroptosis susceptibility (Chen et al., 2024), whereas in ovalbumin-induced asthma models, SMAD4 silencing alleviated ferroptosis-associated damage (Rao et al., 2024). These studies reinforce the notion that SMAD4 serves as a multifaceted regulator of ferroptosis.
From a broader perspective, cellular redox regulation can be viewed as a balance between adaptive stress responses and maladaptive injury pathways. While the Nrf2-associated resilience network, including vitagenes, has been proposed to mediate cytoprotection under mild stress conditions (Flanagan et al., 2020), our findings highlight a contrasting pathological setting in which sustained inflammatory stress drives ferroptosis and tissue damage in SA-AKI. This study has several limitations. Although we demonstrated that SMAD4 binds to the HO-1 promoter and represses its transcription, we did not further characterize the specific binding sites or potential co-repressors (e.g., HDACs) involved. Future studies employing ChIP-seq or CUT&Tag techniques will be necessary to precisely map the regulatory elements and cofactors that mediate SMAD4-driven transcriptional repression. In conclusion, we uncovered a novel SMAD4/HO-1 axis that regulates ferroptosis in SA-AKI. Modulating SMAD4 expression or its downstream targets may represent a promising therapeutic strategy for sepsis-associated kidney injury.
Materials and Methods
Animal model
All animal experiments were conducted using male C57BL/6J mice (7–8 weeks old, 20–22 g). Prior to the experiments, mice were housed under a 12-h light/dark cycle for at least 3 days to acclimate to the environment. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Harbin Medical University (YJSDW2024-202). All experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
SA-AKI was induced in mice using the CLP model. Briefly, a 2 cm midline abdominal incision was made to expose the cecum, which was then ligated with a 4-0 silk suture and punctured once with a 22 G needle to extrude a small amount of fecal content. The abdominal cavity was closed in two layers. Mice in the sham group underwent identical surgical procedures without cecal ligation or puncture. In the CLP + Fer-1 group, mice received a tail vein injection of Fer-1 (0.8 mL/kg; MCE, USA) 1 h after CLP surgery. In the CLP + AAV-SMAD4 group, mice received a single renal pedicle injection of AAV9-SMAD4 (1 × 1013 viral genomes per mouse) 30 days prior to CLP surgery. To assess the independent effects of each treatment, a Fer-1 group and an AAV-SMAD4 group were included. These mice underwent the same sham surgical procedures as the sham group (without cecal ligation or puncture) and received the respective treatments using the same dosing regimens described above. All mice received a single subcutaneous injection of 0.9% (w/v) saline (50 mL/kg) for fluid resuscitation immediately after surgery. Kidney tissues were collected 24 h post procedure for further analysis.
Spatial proteomics
Paraffin-embedded kidney tissues were sectioned at a thickness of 7 μm and stained with H&E. Specific regions were microdissected using a PALM laser capture system (Zeiss) at 20× magnification (cut energy: 39; LPC energy: 55; cut speed: 20), and stored at −80°C. Proteins were extracted, quantified, and reduced with 5 mM dithiothreitol at 56°C for 30 min, followed by alkylation with 11 mM iodoacetamide at room temperature in the dark for 15 min. After dilution of urea to <2 M, samples were digested with trypsin (1:50) overnight at 37°C and further digested for 4 h at a 1:100 ratio. Peptides were resuspended in 2% (v/v) acetonitrile with 0.1% (v/v) formic acid and separated using a NanoElute UPLC system (flow rate: 100 nL/min; gradient: 7–16–26–40–80% B). MS was performed on a timsTOF Pro (Bruker) in dia-PASEF mode, with MS1 scans over 100–1700 m/z, followed by three PASEF MS/MS scans (400–1200 m/z, 25 m/z windows).
Cell culture and treatment
HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Pricella, China) supplemented with 10% (v/v) fetal bovine serum (FBS; Pricella, China) and 1% (v/v) penicillin–streptomycin (Solarbio, China). HK-2 cells were maintained in DMEM/F12 medium (Meilunbio, China) supplemented with 10% (v/v) FBS and 1% (v/v) penicillin–streptomycin. All cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. For drug treatments, cells were seeded into appropriate culture plates and grown to ∼70%–80% confluence before stimulation. To simulate septic conditions, lipopolysaccharide (LPS, 15 μg/mL; Sigma, USA) was added to the culture medium, and cells were incubated for 24 h.
Transfection
Cells were cultured to ∼70% confluence prior to transfection. For transient transfection, small interfering RNAs targeting SMAD4 (si-SMAD4) or HMOX1 (si-HMOX1), overexpression plasmids for SMAD4 (OE-SMAD4) or HMOX1 (OE-HMOX1), along with their corresponding negative controls (NCs) and other indicated plasmids, were diluted in serum-free DMEM and transfected using GP-transfect-mate reagent (GenePharma, China) according to the manufacturer’s instructions. The final concentration of siRNAs was 20 μM. Among the four tested siRNA sequences targeting SMAD4, the one used in this study achieved a knockdown efficiency of 94.33% at the mRNA level, whereas the selected siRNA against HMOX1 achieved a knockdown efficiency of 90.57%, as determined by RT-qPCR (Supplementary Fig. S1A, C). The siRNA sequences used in this study were as follows: NC siRNA, 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense); si-HMOX1, 5′-CCAGCAACAAAGUGCAAGATT-3′ (sense) and 5′-UCUUGCACUUUGUUGCUGGTT-3′ (antisense); and si-SMAD4, 5′-CCAGCAUCCACCAAGUAAUTT-3′ (sense) and 5′-AUUACUUGGUGGUGGAUGCUGGTT-3′ (antisense). All siRNAs and plasmids used in this study were synthesized by GenePharma.
Histological and immunohistochemical analysis
Kidney samples were dissected and fixed in 4% paraformaldehyde overnight, then dehydrated and embedded in paraffin. The paraffin-embedded tissues were sectioned into 4-μm thick slices for H&E, PAS, and IHC staining. All histological fields analyzed and presented in this study were obtained from the renal cortex. Tubular injury was evaluated in a blinded manner using a semiquantitative scoring system, as described previously: 0, all tubules normal (no injury); 1, <10% of tubules affected (minimal injury); 2, 10%–25% of tubules affected (mild injury); 3, 25%–75% of tubules affected (moderate injury); and 4, >75% of tubules affected (severe injury). For H&E, injury criteria included tubular cell swelling, vacuolization, brush border loss, luminal debris, and necrosis. For PAS staining, injury criteria included basement membrane thickening, tubular glycoprotein deposition, and overall tubular architecture disruption. For IHC, tissue sections were deparaffinized in xylene and rehydrated through a graded ethanol series. Antigen retrieval was performed by heating slides in a pressure cooker with 10 mM sodium citrate buffer (pH 6.0) for 3 min at ∼120°C, followed by natural cooling to room temperature. Endogenous peroxidase activity was blocked with 3% (v/v) hydrogen peroxide in methanol for 20 min at room temperature, and nonspecific binding sites were blocked with 10% (v/v) normal goat serum in phosphate-buffered saline for 30 min at 37°C. Sections were incubated overnight at 4°C with primary antibodies against KIM-1 (1:400), HO-1 (1:200), and 4-HNE (1:100). After washing, sections were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse secondary antibodies for 1 h at 37°C. Immunoreactivity was visualized using a diaminobenzidine substrate kit, with development time microscopically controlled to 1–2 min. Finally, sections were counterstained with hematoxylin for 3 min, dehydrated, cleared in xylene, and mounted with neutral balsam.
Serum biochemical and inflammatory cytokine analysis
Mouse blood samples were collected and allowed to clot at room temperature for 30 min, followed by centrifugation at 3000 rpm for 10 min at 4°C to obtain serum. Scr and BUN levels were measured using commercial assay kits (Jianchengbio Bio, China) according to the manufacturer’s instructions. To evaluate systemic inflammation, the concentrations of IL-6, IL-1β, and TNF-α in serum were determined using ELISA kits specific for mouse cytokines (Elabscience, China). Briefly, standards and samples were added to 96-well plates pre-coated with capture antibodies, followed by incubation with biotinylated detection antibodies and HRP-conjugated streptavidin. Color was developed using a TMB substrate and stopped with a stop solution. Absorbance was measured at 450 nm, and concentrations were calculated from standard curves.
Western blotting
Protein samples were denatured by boiling at 100°C for 5 min, separated on 10% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and subsequently transferred onto polyvinylidene difluoride membranes (Millipore, USA). Membranes were blocked with 5% (w/v) nonfat milk in Tris-buffered saline containing 0.1% (v/v) Tween-20 (TBST) for 1 h at room temperature and then incubated overnight at 4°C with primary antibodies. The primary antibodies used were as follows: anti-glutathione peroxidase 4 (Abcam, USA), anti-heme oxygenase 1 (PTMBIO, China), anti-SMAD4 (Proteintech, China or Engibody, China), anti-β-actin (Servicebio, China), and anti-ACSL4 (Abcam, USA). After three washes with TBST, membranes were incubated with HRP-conjugated secondary antibodies (Proteintech, China) for 1 h at room temperature. Protein bands were visualized using enhanced chemiluminescence reagents (Meilunbio, China) according to the manufacturer’s protocol. Signal intensities were captured using a Sage imaging system and quantified using ImageJ software.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from kidney tissues or cultured cells using TRIzol reagent (TransGen Biotech, China) following the manufacturer’s protocol. RNA purity was assessed by the A260/A280 ratio using a NanoDrop spectrophotometer, with values between 1.80 and 2.00 considered acceptable. Complementary DNA (cDNA) was synthesized using the All-in-One First-Strand cDNA Synthesis SuperMix (TransGen Biotech, China). qPCR was performed with gene-specific primers, using GAPDH as an internal control. Relative gene expression was calculated by the 2–ΔΔCt method. Primer sequences are listed in Table 1.
Primers That Were Used in This Study

Live-cell fluorescent probe staining
To evaluate oxidative stress, lipid peroxidation, mitochondrial membrane potential, and intracellular ferrous ion (Fe2+) levels, live-cell fluorescent staining was performed using specific probes. Cells were incubated with DCFH-DA (Solarbio, China) for 30 min at 37°C in the dark to detect ROS. C11 was assessed using BODIPY™ 581/591 C11 (Dojindo, Japan), with cells incubated in working solution for 30 min at 37°C. Mitochondrial membrane potential was measured using JC-1 (Boxbio, China) following 20 min staining at 37°C. Intracellular Fe2+ levels were detected using FerroOrange (Dojindo, Japan) after a 30 min incubation in Hank’s Balanced Salt Solution at 37°C. After staining, cells were washed appropriately, and fluorescence images were acquired using a confocal microscope with the corresponding excitation and emission settings.
DNA pull-down and MS analysis
A DNA pull-down assay was performed using biotin-labeled oligonucleotides to identify nuclear proteins that bind to the HO-1 promoter region. The biotin-labeled probe was generated by annealing a 5′-biotinylated forward strand (5′-GTGCTGGGATTACAGGTGTGAGCCA-3′) with a complementary reverse strand (5′-GACTGCCGGAGCCGCGG-3′). A nonbiotinylated probe of the same sequence (cold probe) was used in competition assays to assess binding specificity. For each reaction, 1 μg of the double-stranded probe was incubated with 300 μg of nuclear extract at room temperature for 20 min. Streptavidin agarose beads (30 μL), pre-blocked with poly(dI–dC), were added and incubated at 4°C for 4 h with gentle rotation. After incubation, beads were collected and extensively washed with low- and high-salt buffers to remove nonspecific binding. The bound proteins were eluted, separated by SDS-PAGE, and analyzed by MS.
Chromatin immunoprecipitation assay
ChIP was performed using the SimpleChIP® Plus Sonication Chromatin IP Kit (Cell Signaling Technology, USA) according to the manufacturer’s protocol. Cells cultured in 15 cm dishes were cross-linked with 1% (v/v) formaldehyde for 10 min at room temperature, and the reaction was quenched with glycine. After washing, cells were scraped, collected by centrifugation, and lysed sequentially using cell lysis and nuclear lysis buffers. Chromatin was sheared to appropriate fragment sizes using a SCIENTZ-IID sonicator (SCIENTZ, China). Equal amounts of chromatin were incubated overnight at 4°C with 8 μg of anti-SMAD4 antibody (Proteintech, Cat. no. 10231-1-AP) or normal IgG as a negative control, with gentle rotation. The next day, ChIP-Grade Protein G magnetic beads were added and incubated for 2 h at 4°C to capture the immune complexes. After washing, the complexes were reverse cross-linked at 65°C for 2 h in the presence of 5 M NaCl and Proteinase K. DNA was purified using spin columns provided in the kit and subjected to ChIP-qPCR analysis.
Dual-luciferase reporter assay
To investigate the regulatory effect of SMAD4 on HO-1 promoter activity, HEK293T cells were co-transfected with a firefly luciferase reporter plasmid containing the human HO-1 promoter region (pGL3-HO-1-promoter), a Renilla luciferase plasmid (pRL-TK) as an internal control, and either an SMAD4 overexpression plasmid (OE-SMAD4) or an empty vector. After 48 h of incubation, cells were lysed, and luciferase activities were measured using the Dual-Luciferase Reporter Assay Kit (Yeasen Biotech, China) according to the manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity to account for variations in transfection efficiency.
Statistical analysis
Data are expressed as mean ± standard deviation. Prior to statistical analysis, data distribution was assessed for normality using the Shapiro–Wilk test. For datasets showing a normal distribution, two-group comparisons were performed using unpaired Student’s t-tests, and multiple-group comparisons were analyzed by one-way analysis of variance followed by appropriate post hoc tests. For data not meeting normality assumptions, nonparametric tests were applied as indicated. A p value < 0.05 was considered statistically significant. All analyses were performed using GraphPad Prism 9.5.
The MS proteomics data have been deposited in the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository under the dataset identifier IPX0008335000 and IPX0014197000.
An electronic laboratory notebook was not used.
Footnotes
Acknowledgments
Heartfelt thanks to the co-authors for their valuable contributions to the writing of this research. Their professional insights, collaborative spirit, and selfless support have been crucial in completing this research.
Authors’ Contributions
Z.H.: Conceptualization, methodology, investigation, writing—original draft. J.L.: Data curation, formal analysis, visualization, writing—review & editing. C.G.: Investigation, resources, validation. Z.Q.: Investigation, supervision. S.Z.: Methodology, data curation. Y.L.: Visualization. L.W.: Supervision,. J.B.: Investigation. J.Z.: Conceptualization, supervision, writing—review & editing. H.W.: Project administration, supervision, funding acquisition.
Data Availability
The data used to support the findings of this study are included in the article.
Consent for Publication
All the authors give consent for the publication of identifiable details, which can include the text, figures, and other materials in this article.
Author Disclosure Statement
The authors declare that there is no conflict of interest regarding the publication of this article.
Funding Information
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.