
Abstract
Background:
Sjögren’s syndrome (SS) is a systemic autoimmune disorder characterized by chronic inflammation, oxidative stress, and progressive salivary gland dysfunction. Current therapies remain limited in efficacy.
Aim:
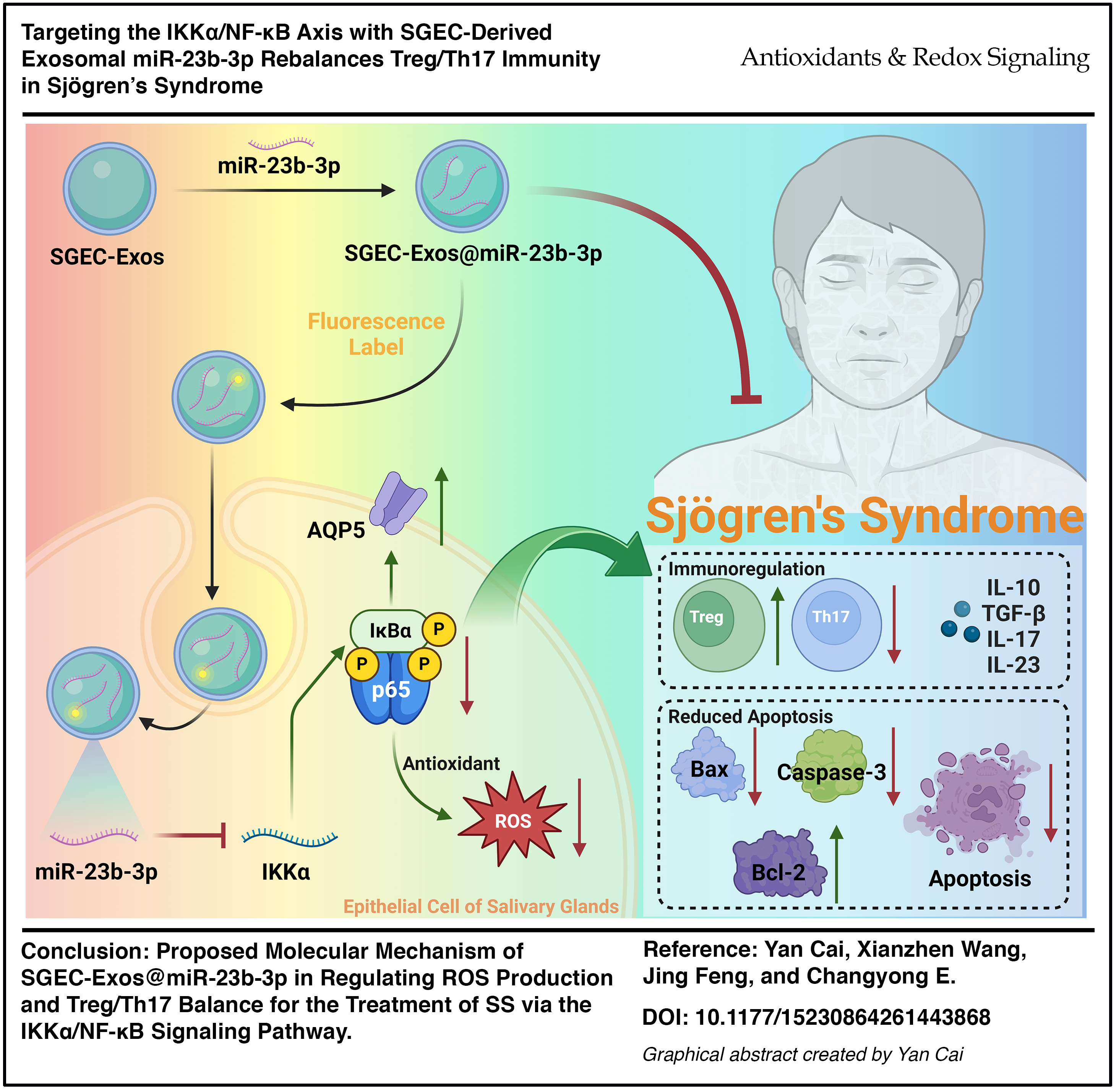
This study explored the regulatory effect of exosome (Exo)-transported miR-23b-3p on the IκB kinase alpha (IKKα)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling axis in SS.
Methods:
Key SS-related microRNAs (miRNAs) were identified by integrating weighted gene co-expression network analysis with machine-learning-based transcriptomic profiling. Target genes and functional pathways were analyzed by bioinformatics methods. The direct binding between miR-23b and IKKα (CHUK) was validated by a dual-luciferase reporter assay. An in vitro SS cell model was established to examine the effects of miR-23b-3p on the IKKα/NF-κB pathway, oxidative stress, inflammation, and apoptosis. miR-23b-3p was loaded into salivary gland epithelial cell-derived Exos (SGEC-Exos) via electroporation. In vitro coculture experiments assessed reactive oxygen species (ROS) levels, inflammatory cytokines, Treg/Th17 balance, and cell apoptosis. In vivo effects were evaluated in NOD/Ltj mice by measuring salivary flow rate, histopathology, and expression of salivary-function-related proteins AQP5 and GPER.
Results:
SGEC-Exos@miR-23b-3p significantly suppressed IKKα expression and NF-κB activation, reduced ROS production, and modulated immune responses by restoring the Treg/Th17 balance. It also inhibited apoptosis by decreasing Bax and caspase-3 expression and increasing Bcl-2 levels. These effects were partially reversed by reactivation of the IKKα/NF-κB pathway. In NOD/Ltj mice, SGEC-Exos@miR-23b-3p improved salivary flow, alleviated glandular pathology, and upregulated AQP5 and GPER expression.
Conclusions:
SGEC-Exos@miR-23b-3p offers a translational approach to address oxidative stress, immune imbalance, and glandular injury in SS, highlighting the potential of Exo-based miRNA therapy. Antioxid. Redox Signal. 00, 000–000.
Introduction
Sjögren’s syndrome (SS) is a chronic autoimmune disorder characterized by immune-mediated damage to exocrine glands, primarily the salivary and lacrimal glands, resulting in xerostomia and keratoconjunctivitis sicca (Bjordal et al., 2020; Negrini et al., 2022; Ogawa et al., 2021; Parisis et al., 2020; Rihab et al., 2023). Beyond glandular dysfunction, SS often presents with systemic involvement and is frequently associated with other autoimmune conditions. Pathologically, SS is characterized by acinar epithelial cell damage, lymphocytic infiltration, and persistent inflammation mediated by autoantibodies. Despite current interventions—including immunosuppressive therapy and symptomatic agents like artificial tears—most treatments are palliative and fail to halt or reverse glandular destruction (Cafaro et al., 2019; Wu et al., 2022). Given the clinical heterogeneity and limited therapeutic efficacy of existing options, there is a pressing need for innovative strategies with disease-modifying potential.
MicroRNAs (miRNAs) have emerged as important modulators of immune responses and inflammatory processes (Cao et al., 2019; Roszkowska et al., 2021; Ye et al., 2019). Among them, miR-23b-3p has gained attention for its regulatory role in autoimmune and inflammatory disorders (Burrows et al., 2023; Ortiz et al., 2023). It has been shown to influence immune signaling by targeting key components of the NF-κB pathway, particularly through suppression of IKKα (Sun et al., 2020). This modulation leads to decreased NF-κB activity and downstream inflammatory cytokine production. In addition, miR-23b-3p has been implicated in shaping T-cell subset differentiation by promoting Treg cell populations while inhibiting Th17 response cells (Wei et al., 2021; Yu et al., 2024), a balance essential for maintaining immune homeostasis in autoimmune disease settings.
Exosomes (Exos), nanosized extracellular vesicles secreted by diverse cell types, have recently garnered interest as natural carriers for therapeutic molecules due to their excellent biocompatibility and low immunogenicity (Ferreira et al., 2022; Santos et al., 2022; Wei et al., 2024). Compared with conventional drug delivery systems, Exos offer superior stability for RNA-based therapies, protecting miRNAs from enzymatic degradation and enhancing targeted delivery to recipient cells (Ortega et al., 2020; Suh et al., 2021; Wang et al., 2021). Increasing evidence supports the use of Exos for the effective transfer of functional miRNAs into diseased tissues, facilitating precise regulation of disease mechanisms (Zhang et al., 2024).
While miR-23b-3p has demonstrated anti-inflammatory effects in diseases such as rheumatoid arthritis and systemic lupus erythematosus (Lin et al., 2025), its function in SS remains largely unexplored. Current research has not sufficiently addressed how modulation of the IKKα/NF-κB axis by miR-23b-3p might impact SS progression, particularly in oxidative stress and programmed cell death. Moreover, the impact of this miRNA on Treg/Th17 dynamics in SS has yet to be fully characterized. These knowledge gaps underscore the therapeutic promise of delivering miR-23b-3p via Exos for immune modulation and tissue protection in SS.
Herein, our study focused on the therapeutic potential and underlying mechanisms of miR-23b-3p delivered by salivary gland epithelial-cell-derived Exos (SGEC-Exos@miR-23b-3p) in a model of SS. We hypothesized that this exosomal delivery system would downregulate IKKα expression, suppress NF-κB signaling, and mitigate oxidative damage, thereby improving glandular function. In parallel, we expected SGEC-Exos@miR-23b-3p to rebalance Treg/Th17 cell populations, reduce inflammation, and protect epithelial cells by regulating apoptosis-related markers. Through this investigation, we sought not only to clarify the role of miR-23b-3p and its delivery via Exos in SS, but also to provide preclinical evidence supporting Exo-based miRNA therapies as a viable approach for autoimmune disease treatment.
Innovation
This study identifies an exosome (Exo)-mediated redox–immune regulatory mechanism in Sjögren’s syndrome (SS) by targeting the IKKα/NF-κB axis with salivary gland epithelial cell (SGEC)-derived exosomal miR-23b-3p. Unlike conventional anti-inflammatory approaches, this work integrates oxidative stress regulation, immune homeostasis, and epithelial cell survival within a single therapeutic framework.
First, we demonstrate that miR-23b-3p directly suppresses IKKα, thereby attenuating NF-κB-driven oxidative stress and inflammatory signaling in SGECs. This establishes a mechanistic link between microRNA (miRNA) regulation and redox-sensitive NF-κB activation in SS.
Second, we reveal that exosomal delivery of miR-23b-3p simultaneously restores Treg/Th17 immune balance and reduces reactive oxygen species accumulation, highlighting a coordinated redox–immune modulation that has not previously been demonstrated in this disease context.
Third, by using SGEC-derived Exos as a targeted delivery platform, this study provides a cell-type-specific strategy to enhance miRNA stability, tissue targeting, and biological efficacy, thereby overcoming key limitations of conventional miRNA-based therapies.
Collectively, this work introduces a redox-centered, Exo-based miRNA therapeutic paradigm that addresses oxidative stress, immune dysregulation, and glandular injury in SS, offering a mechanistically grounded and translationally relevant approach for autoimmune disease intervention.
Results
Identification of miR-23b-3p as a key regulator in SS via bioinformatics analysis
To uncover miRNAs potentially involved in SS progression, we analyzed miRNA expression profiles from salivary gland tissues of SS model mice. Differential expression analysis revealed 239 upregulated and 214 downregulated miRNAs in the SS group relative to controls (Fig. 1A). To identify functionally relevant miRNAs, we performed weighted gene co-expression network analysis (WGCNA). A soft-thresholding power of β = 9 was chosen to meet scale-free topology criteria (Fig. 1B). Module detection was performed using dynamic tree cutting (minimum module size = 10; module merging threshold = 0.6), resulting in three distinct modules: MEblue, MEbrown, and MEgrey (Fig. 1C). Among these, the MEbrown module showed the strongest association with SS (r = 0.98, p < 0.001; Fig. 1D, E).
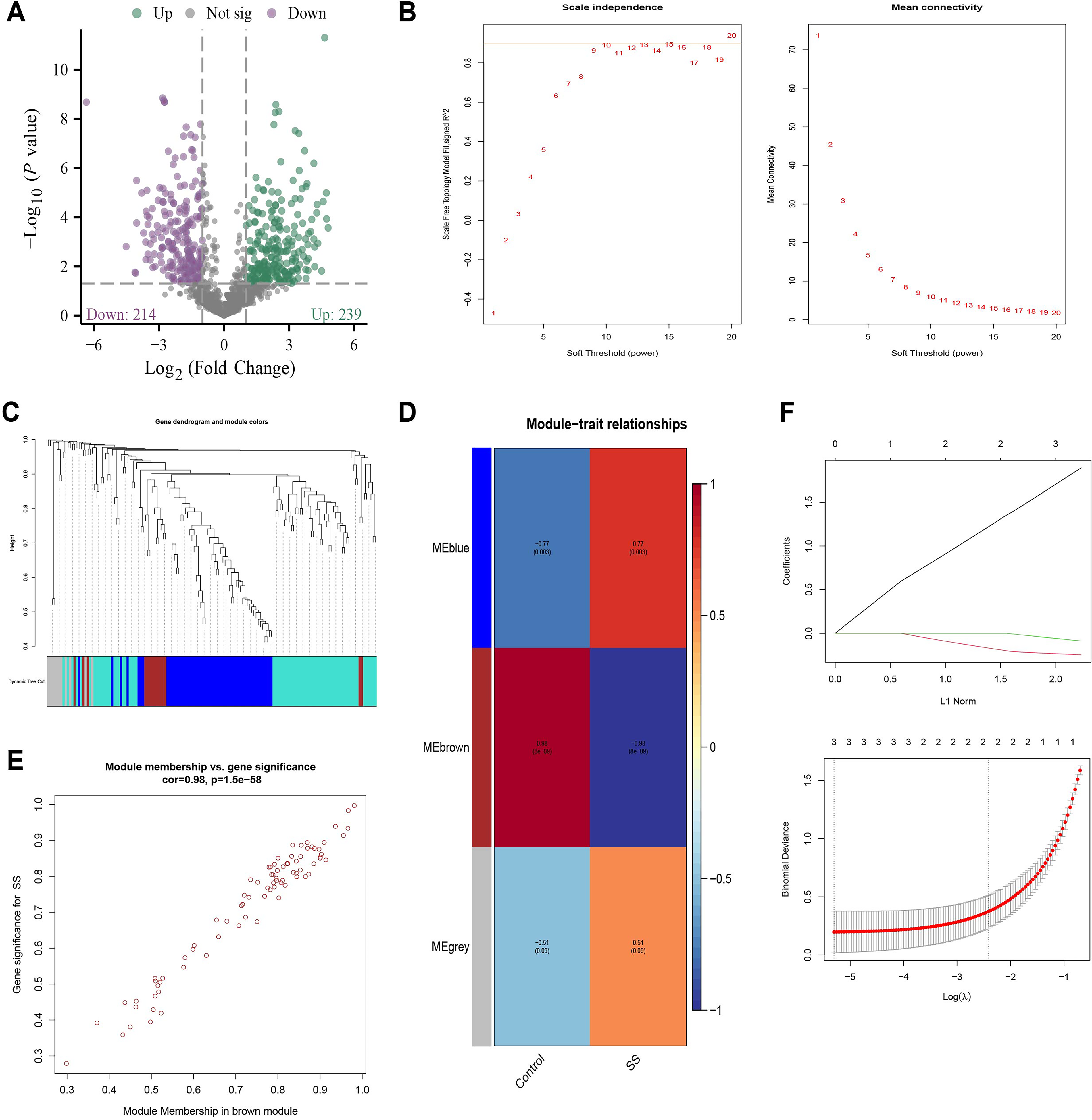
To further refine the selection of candidate miRNAs, Least Absolute Shrinkage and Selection Operator (LASSO) regression was performed on genes within the MEbrown module using the glmnet package in R. Through cross-validation, we identified three feature miRNAs with nonzero coefficients at the optimal lambda value: miR-544-5p, miR-7019-3p, and miR-23b-3p (Fig. 1F). While the biological relevance of miR-544-5p and miR-7019-3p remains unclear, miR-23b-3p has been extensively reported in inflammation-related processes (Romero-López et al., 2022) and is notably downregulated in SS model mice (Supplementary Fig. S1A). Its involvement in immune homeostasis and epithelial dysfunction suggests a potential pathogenic role in SS (Fox, 2005).
To elucidate the regulatory network of miR-23b-3p, we integrated predicted target genes from ENCORI, miRTarBase, and miRWalk, yielding 104 consensus targets (Supplementary Fig. S1B). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed that these targets were significantly enriched in immune-related pathways, including Th1/Th2 differentiation, interleukin (IL)-17 signaling, apoptosis, and notably the NF-κB pathway (Supplementary Fig. S1C), all of which are central to SS pathogenesis.
To prioritize potential mediators of SS pathology, we intersected these predicted targets with SS-associated genes from the GeneCards database, identifying eight shared genes: TNFAIP3, CHUK, CA2, PTEN, TNRC6A, NOTCH2, BTLA, and HMGB2 (Supplementary Fig. S1D). Among them, CHUK (IKKα) and TNFAIP3 (A20) are critical regulators of the NF-κB signaling cascade. TNFAIP3 limits inflammatory responses via negative regulation of NF-κB (Zhou et al., 2016), while CHUK encodes IKKα, a pivotal upstream kinase in NF-κB activation (Karin and Ben-Neriah, 2000). In recent years, multiple studies have confirmed that the transcription factor NF-κB plays a central role in regulating immune-inflammatory responses, and its aberrant activation contributes to SS pathogenesis (Sisto et al., 2020).
Considering that miRNAs primarily suppress gene expression, CHUK/IKKα is a likely direct target through which miR-23b-3p modulates inflammatory signaling. Collectively, miR-23b-3p may mitigate SS progression by targeting IKKα, thereby inhibiting NF-κB signaling and subsequent immune activation.
miR-23b-3p directly targets IKKα and suppresses NF-κB pathway activation
To determine whether miR-23b-3p directly regulates IKKα expression, a luciferase reporter assay was performed in HEK293 cells. We first assessed the expression of miR-23b-3p and IKKα in salivary gland tissues from SS model mice (female NOD/Ltj mice). miR-23b-3p expression was significantly reduced, whereas IKKα expression was markedly elevated in SS mice (Fig. 2A). Co-transfection with miR-23b-3p mimics notably suppressed luciferase activity driven by the IKKα 3′-untranslated region (UTR), indicating that miR-23b-3p directly binds and represses IKKα expression (Fig. 2B, C). In addition, RNA immunoprecipitation (RIP) using an anti-AGO2 antibody revealed substantial enrichment of IKKα mRNA in miR-23b-3p-overexpressing SGECs, further confirming its direct interaction via the RNA-induced silencing complex (RISC) (Fig. 2D).
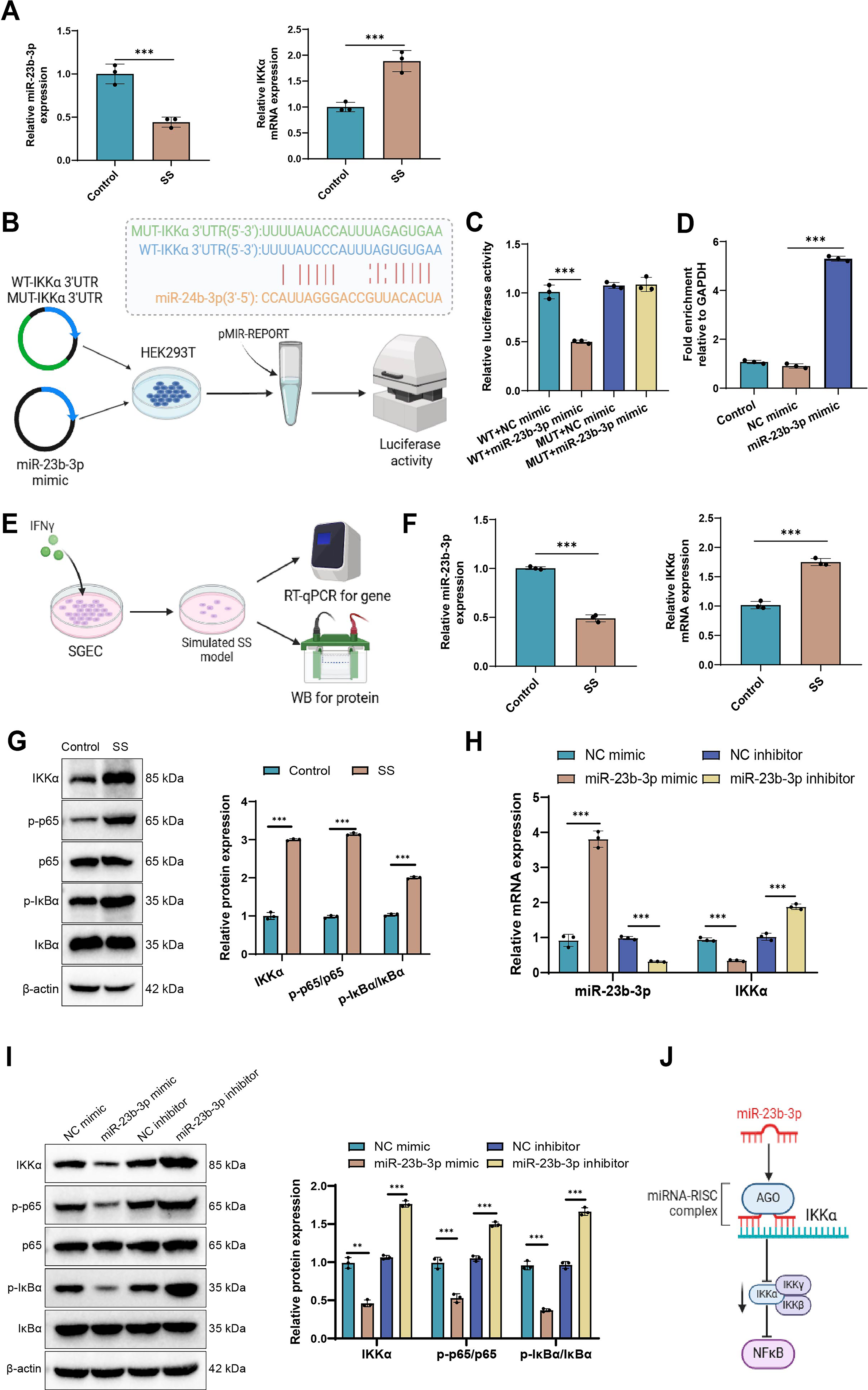
To explore this regulatory axis under pathological conditions, we established an in vitro SS-like cell model using interferon-γ (IFN-γ)-stimulated A253 cells, a validated SGEC model (Mougeot et al., 2024). Western blot (WB) and immunofluorescence analyses demonstrated increased expression of the pro-inflammatory cytokines IL-6 and tumor necrosis factor-alpha (TNF-α), accompanied by reduced AQP5 levels, thereby confirming the inflammatory phenotype (Fig. 2E, Supplementary Fig. S2A–C).
Expression profiling by reverse transcription quantitative polymerase chain reaction (PCR) (RT-qPCR) and WB revealed that miR-23b-3p was diminished, while IKKα and phosphorylated NF-κB pathway proteins (p-p65 and p-IκBα) were markedly upregulated in the IFN-γ-treated model group (Fig. 2F, G), highlighting a potential inverse relationship between miR-23b-3p and IKKα/NF-κB activity.
To further test this regulatory interaction, A253 cells were transfected with miR-23b-3p mimics or inhibitors. Overexpression of miR-23b-3p led to reduced IKKα mRNA and protein levels, along with decreased p-p65 and p-IκBα expression. In contrast, miR-23b-3p inhibition led to an elevation in IKKα expression and an enhancement in NF-κB pathway activation (Fig. 2H, I). In addition, we observed that IKKα protein levels gradually increased with prolonged IFN-γ stimulation, showing a significant elevation as early as 2 h and reaching a peak at ∼16 h. Meanwhile, reactive oxygen species (ROS) levels also increased with the duration of IFN-γ treatment; however, the elevation in ROS became evident only around 4 h after stimulation. These findings suggest that the increase in ROS levels may be associated with the upregulation of IKKα protein (Supplementary Fig. S2D, E).
Collectively, these results indicate that miR-23b-3p suppresses IKKα at both the mRNA and protein levels, thereby restraining NF-κB signaling. Loss of miR-23b-3p in SS may exacerbate inflammatory activation through IKKα-dependent NF-κB pathway upregulation. The proposed mechanism is illustrated in Figure 2J.
Construction and characterization of SGEC-derived Exos loaded with miR-23b-3p
Exos were successfully isolated from cultured human SGECs via differential ultracentrifugation followed by immunoaffinity capture using anti-CD63 magnetic beads. miR-23b-3p was subsequently loaded into these vesicles using an optimized electroporation protocol, yielding the SGEC-Exos@miR-23b-3p complex (abbreviated as SGEC-Exos@miR) (Fig. 3A). Transmission electron microscopy (TEM) revealed that the Exo structure remained intact after miRNA loading, retaining the characteristic cup-shaped morphology (Fig. 3B). Dynamic light scattering (DLS) analysis indicated that the native SGEC-Exos had a mean particle diameter of 105 ± 1.2 nm and a zeta potential of −20.5 ± 0.6 mV. Following miR-23b-3p incorporation, a slight increase in size to 123 ± 2.3 nm and a marginal shift in surface charge to −19.8 ± 0.4 mV were observed (Fig. 3C, D), indicating successful loading without major structural disruption. WB analysis verified the expression of canonical exosomal markers CD9 and TSG101, while the absence of calnexin confirmed vesicle purity and excluded contamination from cellular organelles (Fig. 3E). RT-qPCR analysis further confirmed that miR-23b-3p was efficiently enriched in SGEC-Exos@miR compared with unloaded Exos (Fig. 3F).
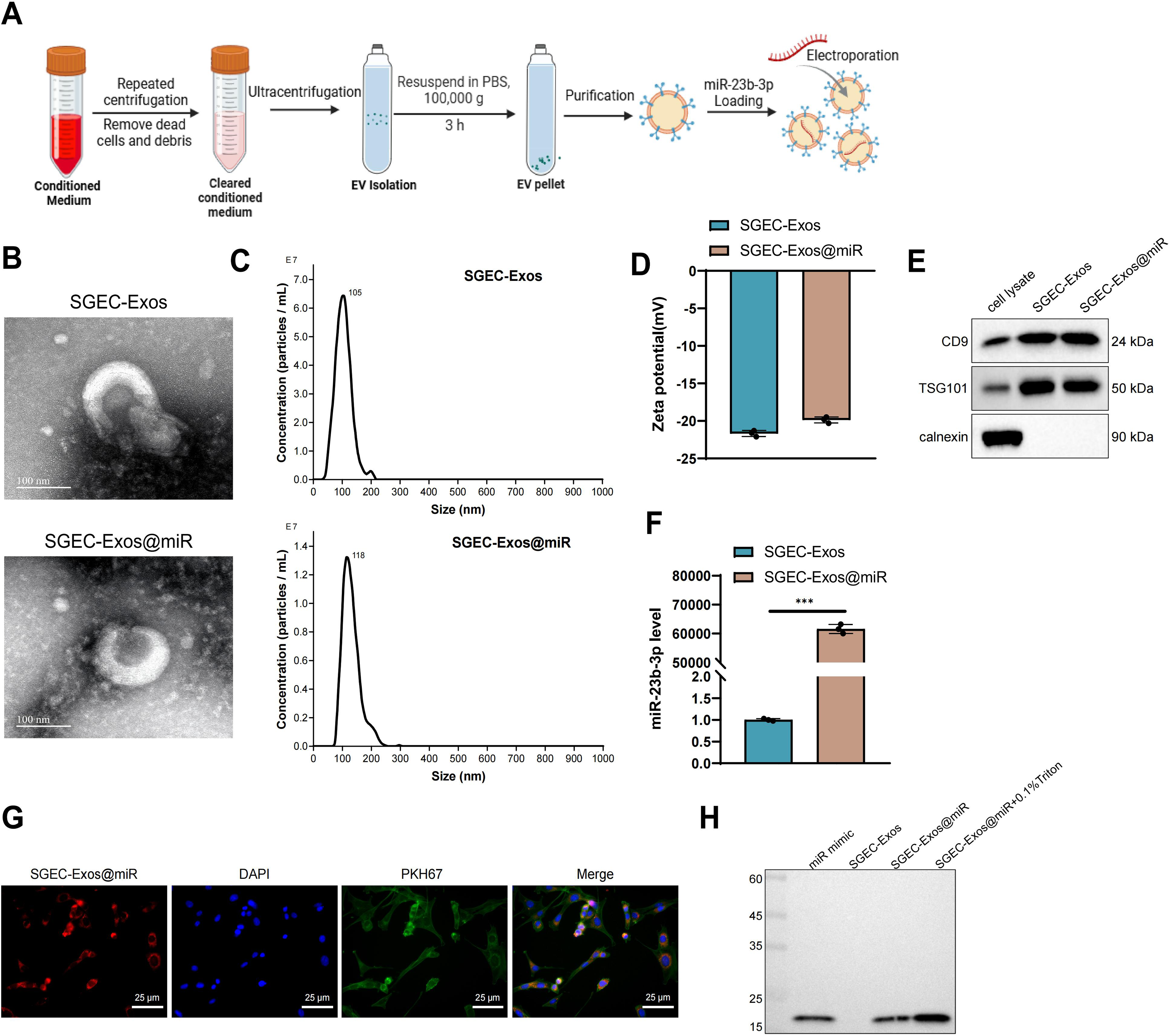
To assess cellular uptake, SGEC-Exos@miR was labeled with Dil (red fluorescence) and incubated with SGECs prestained with PKH67 (green membrane dye). Confocal laser scanning microscopy revealed strong intracellular colocalization signals, confirming successful internalization of the engineered Exos by recipient SGECs (Fig. 3G). In addition, we used Northern blot to determine whether free miRNA contamination was present after miR-23b-3p loading. The results showed that, when using a DIG-labeled miR-23b-3p probe, the SGEC-Exos@miR group displayed a specific band at ∼22 nt, whereas no such band was observed in the unloaded Exos or free miRNA control groups, indicating the absence of free miRNA contamination (Fig. 3H).
These results demonstrate that miR-23b-3p was effectively incorporated into SGEC-derived Exos and that the engineered vesicles retained structural integrity, molecular markers, and efficient cellular delivery capacity.
SGEC-Exos@miR attenuates ROS generation and inflammation while enhancing AQP5 expression via the IKKα/NF-κB pathway
Diminished AQP5 expression is closely linked to reduced salivary secretion in SS. Previous studies demonstrated that AQP5 protein expression is significantly downregulated in salivary gland tissues of SS patients and that TNF-α and IFN-γ activate NF-κB signaling, directly suppressing AQP5 promoter activity and exacerbating glandular dysfunction (Chivasso et al., 2023). In an SS dry-eye model, AQP5 downregulation was also observed in lacrimal gland epithelial cells, suggesting that AQP5 is a key node in inflammation-mediated exocrine gland dysfunction (Fu et al., 2024). Furthermore, ∼42% of primary SS patients have detectable serum autoantibodies against AQP5. Passive transfer of these antibodies induces salivary gland inflammation and downregulation of AQP5 expression, indicating that AQP5 is not only a target of autoimmune attack but also a critical effector molecule in SS pathogenesis (Wang et al., 2024). NF-κB activation suppresses AQP5 transcription (Chang et al., 2017; Yao et al., 2010). To determine whether SGEC-Exos@miR exerts its therapeutic effects through modulation of the IKKα/NF-κB signaling cascade, we examined AQP5 expression and pathway activity in an in vitro SS cell model (Fig. 4A).
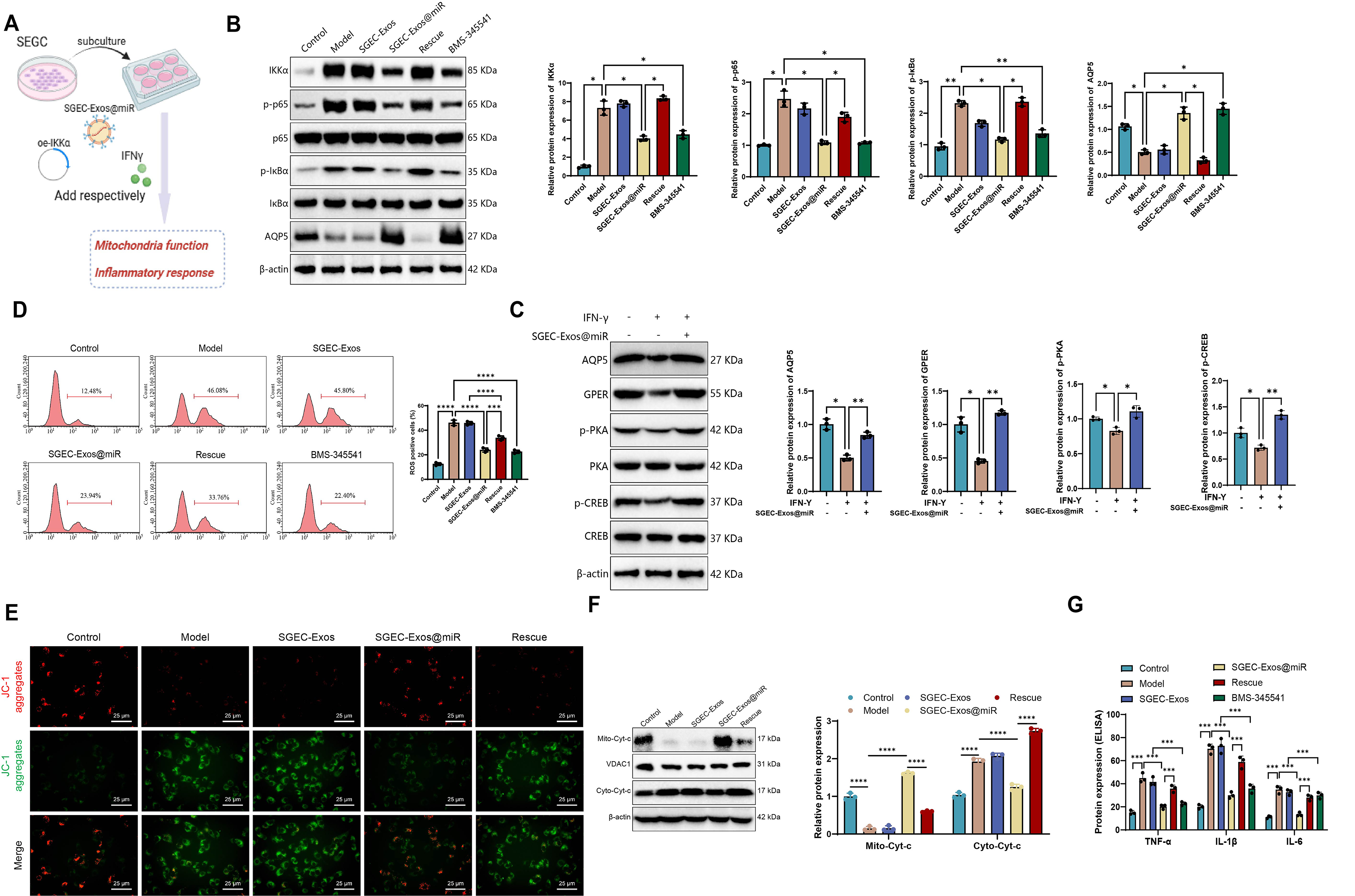
WB analysis showed that, relative to the model group, SGEC-Exos treatment induced no significant changes, whereas BMS-345541 or SGEC-Exos@miR treatment markedly reduced IKKα, p-p65, and p-IκBα expression, accompanied by a pronounced restoration of AQP5 levels. In contrast, the Rescue group (SGEC-Exos@miR + oe-IKKα) exhibited restored pathway activation and a partial reversal of AQP5 upregulation (Fig. 4B), suggesting that miR-23b-3p regulates AQP5 via the IKKα/NF-κB axis. In addition, we examined the expression of AQP5-associated proteins, including GPER, PKA (p-PKA), and CREB (p-CREB). The SGEC-Exos@miR group displayed significantly increased expression of GPER, PKA (p-PKA), and CREB (p-CREB) compared with the model group (Fig. 4C).
As oxidative stress contributes to glandular injury in SS (Zhou et al., 2024), intracellular ROS levels were assessed by flow cytometry. Compared with the model group, SGEC-Exos treatment produced no significant effect, whereas BMS-345541 or SGEC-Exos@miR markedly reduced ROS accumulation in SS model cells. This antioxidant effect was largely abolished in the Rescue group, indicating that suppression of ROS by SGEC-Exos@miR is primarily mediated through inhibition of the IKKα/NF-κB pathway (Fig. 4D).
To evaluate mitochondrial function, we monitored Δψm using JC-1 staining. Compared with the model group, SGEC-Exos treatment produced no significant effect, whereas SGEC-Exos@miR treatment markedly increased the red-to-green fluorescence ratio, indicating preservation of mitochondrial membrane potential and integrity. This protective effect was attenuated upon IKKα re-expression (Fig. 4E). Moreover, WB analysis showed that compared with the model group, no significant changes were observed in the SGEC-Exos group, while reduced Cyt-c and enhanced mitochondrial retention of Cyt-c were seen in the SGEC-Exos@miR group, suggesting effective prevention of mitochondrial outer membrane permeabilization. These effects were reversed in the Rescue group, where increased Cyt-c release was observed (Fig. 4F).
To further assess the anti-inflammatory effects of SGEC-Exos@miR, pro-inflammatory cytokine levels were quantified by enzyme-linked immunosorbent assay (ELISA). No significant differences were observed between the SGEC-Exos group and the model group. Cytokine levels were diminished following treatment with BMS-345541 or SGEC-Exos@miR, whereas reactivation of IKKα restored cytokine production, supporting a mechanistic link between NF-κB inhibition and anti-inflammatory effects (Fig. 4G).
In summary, these findings indicate that unloaded SGEC-Exos had no impact on the relevant indicators. SGEC-Exos@miR effectively downregulates the IKKα/NF-κB axis, leading to reduced oxidative stress, suppressed inflammatory responses, and restored AQP5 expression. Rescue experiments further confirm that these protective effects are dependent on miR-23b-3p-mediated inhibition of IKKα, highlighting a critical regulatory axis in SS pathogenesis.
SGEC-Exos@miR regulates Treg/Th17 imbalance via IKKα/NF-κB pathway in SS
To investigate the immunomodulatory effect of SGEC-Exos@miR on T-cell polarization, a coculture system was established using SGECs and Jurkat T cells (Fig. 5A). Flow cytometric analysis showed that SGEC-Exos treatment did not significantly alter immune cell proportions compared with the model group, while BMS-345541 or SGEC-Exos@miR notably increased the proportion of Tregs (CD4+CD25+Foxp3+) and decreased the proportion of Th17 cells (CD4+IL-17A+), indicating enhanced immune tolerance and attenuated inflammatory potential. In contrast, IKKα overexpression in the Rescue group reversed these effects, reducing Tregs and increasing Th17 cell populations, highlighting the pivotal role of the IKKα/NF-κB axis in regulating this immune balance (Fig. 5B, C).
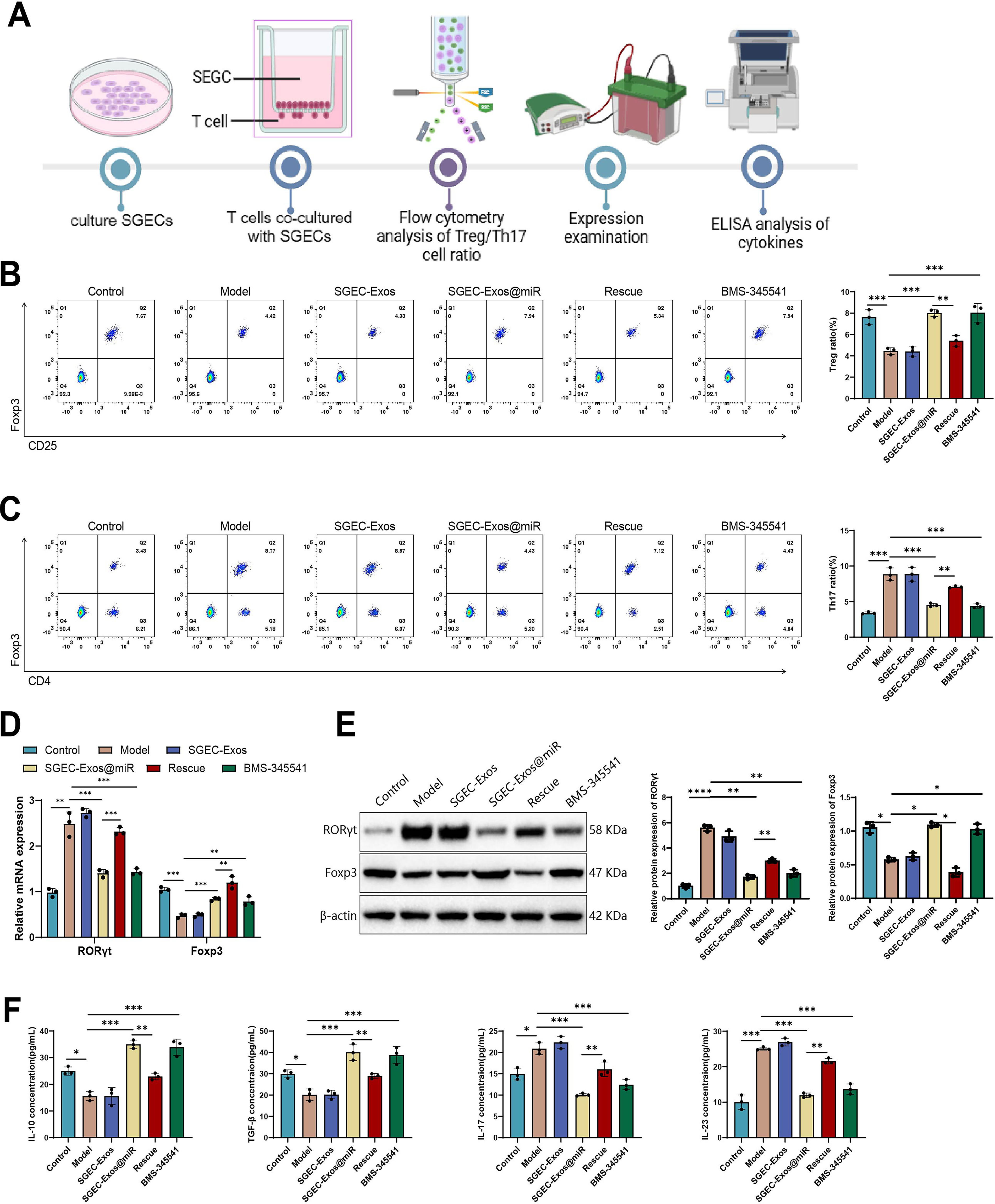
Using qRT-PCR and WB, we examined the expression of Foxp3, a Treg-cell-specific transcription factor, and RORγt, a Th17-cell-specific transcription factor. The results showed that, compared with the model group, no significant changes were observed in the SGEC-Exos group. In contrast, BMS-345541 or SGEC-Exos@miR treatment markedly upregulated Foxp3 expression while significantly downregulating RORγt expression, further supporting its role in promoting Treg cell differentiation and suppressing Th17 cell differentiation. In the Rescue group, both the upregulation of Foxp3 and the downregulation of RORγt were significantly inhibited, indicating that restored IKKα/NF-κB expression exerts a strong counter-regulatory effect on the Treg/Th17 balance modulated by SGEC-Exos@miR (Fig. 5D, E).
Cytokines associated with Treg and Th17 function were quantified in culture supernatants by ELISA. No significant differences were observed between the SGEC-Exos group and the model group. BMS-345541 or SGEC-Exos@miR treatment increased levels of immunosuppressive cytokines IL-10 and transforming growth factor (TGF)-β while decreasing IL-17 and IL-23. Restoration of IKKα in the Rescue group reversed these trends, further supporting the role of SGEC-Exos@miR in immune regulation via inhibition of the IKKα/NF-κB pathway (Fig. 5F).
Collectively, these results demonstrate that unloaded SGEC-Exos had no impact on the relevant indicators. SGEC-Exos@miR reestablishes immune homeostasis in SS by shifting the Treg/Th17 balance toward immune tolerance. This effect is mediated through downregulation of IKKα/NF-κB signaling, highlighting a potential mechanism through which SGEC-Exos@miR exerts its anti-inflammatory and immunoregulatory functions.
SGEC-Exos@miR attenuates apoptosis via suppression of the IKKα/NF-κB pathway in SS-like cells
To assess the protective effects of SGEC-Exos@miR on SGEC survival under SS-like conditions, we examined cell viability and apoptosis-related markers. Cell Counting Kit-8 (CCK-8) assays showed that IFN-γ-induced injury markedly reduced SGEC viability. Compared with the model group, SGEC-Exos treatment produced no significant effect, whereas treatment with BMS-345541 or SGEC-Exos@miR substantially restored cell proliferation. However, this restorative effect was partially reversed upon IKKα overexpression, suggesting that suppression of IKKα is critical to the cytoprotective role of SGEC-Exos@miR (Fig. 6A). Caspase-3 activity, a key indicator of apoptosis, was markedly increased in the SS model group. In contrast, no significant change was observed in the SGEC-Exos group compared with the model group. BMS-345541 or SGEC-Exos@miR treatment significantly inhibited caspase-3 activation, while IKKα re-expression abolished this suppression, further confirming the dependence of this antiapoptotic effect on IKKα downregulation (Fig. 6B).
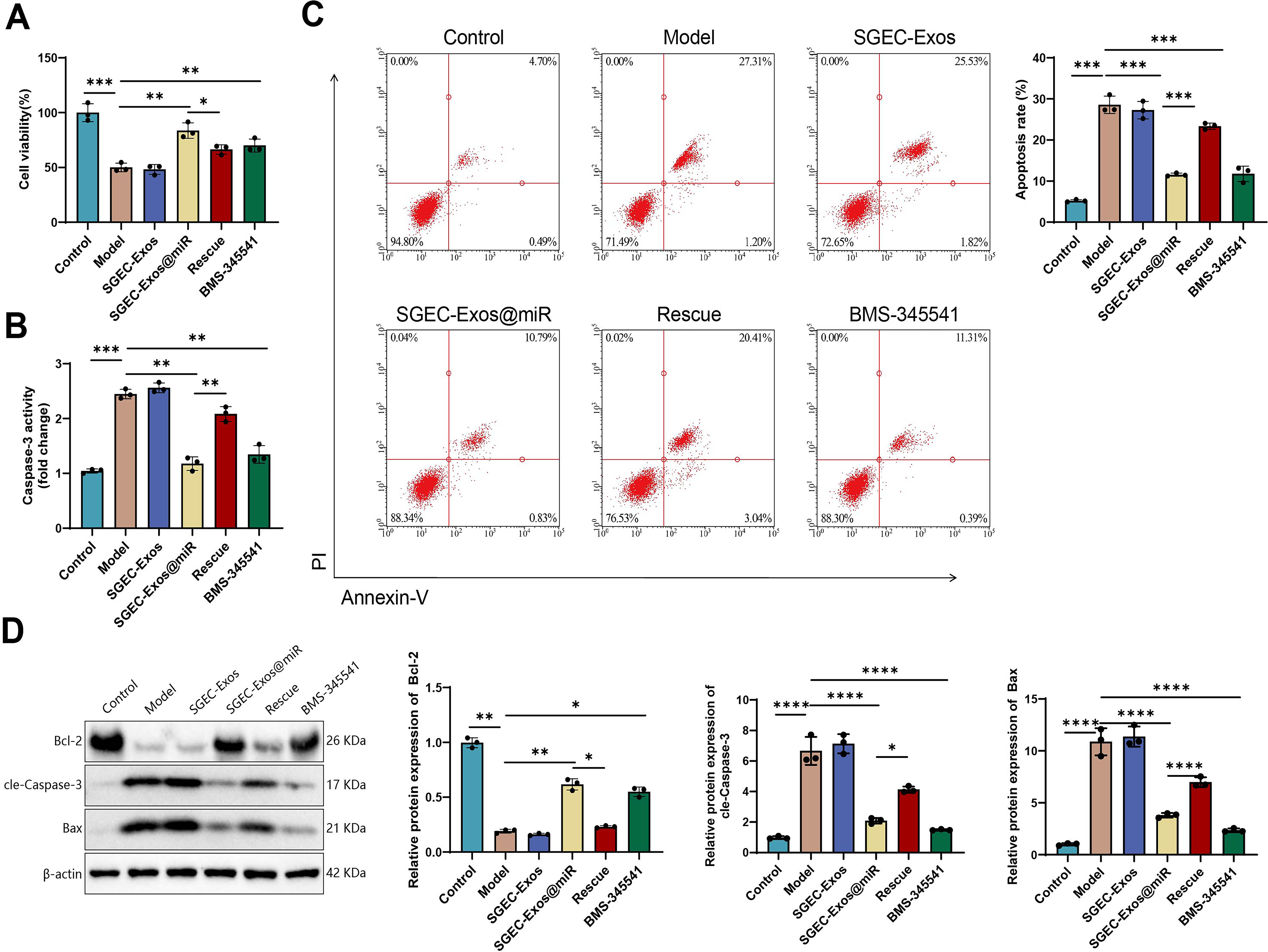
Annexin V/PI dual staining with flow cytometric analysis further corroborated these findings, with no significant differences observed between the SGEC-Exos group and the model group. The SS model group exhibited a higher proportion of apoptotic cells, which was notably reduced after BMS-345541 or SGEC-Exos@miR treatment. In contrast, reactivation of IKKα expression reversed the reduction in apoptosis, underscoring the pivotal role of the IKKα/NF-κB axis in regulating cell death (Fig. 6C). Consistently, WB analysis showed increased Bax and cleaved caspase-3 levels, together with reduced Bcl-2 expression, in the model group. No significant differences were observed in the SGEC-Exos group compared with the model group. Treatment with BMS-345541 or SGEC-Exos@miR reversed these trends by suppressing Bax and caspase-3 expression and enhancing Bcl-2 levels. These effects were largely negated in the Rescue group, in which IKKα expression was restored (Fig. 6D).
In summary, these findings indicate that unloaded SGEC-Exos had no impact on the relevant indicators. SGEC-Exos@miR exerts potent antiapoptotic effects on SGECs by modulating the IKKα/NF-κB axis.
In vivo functional validation of SGEC-Exos@miR
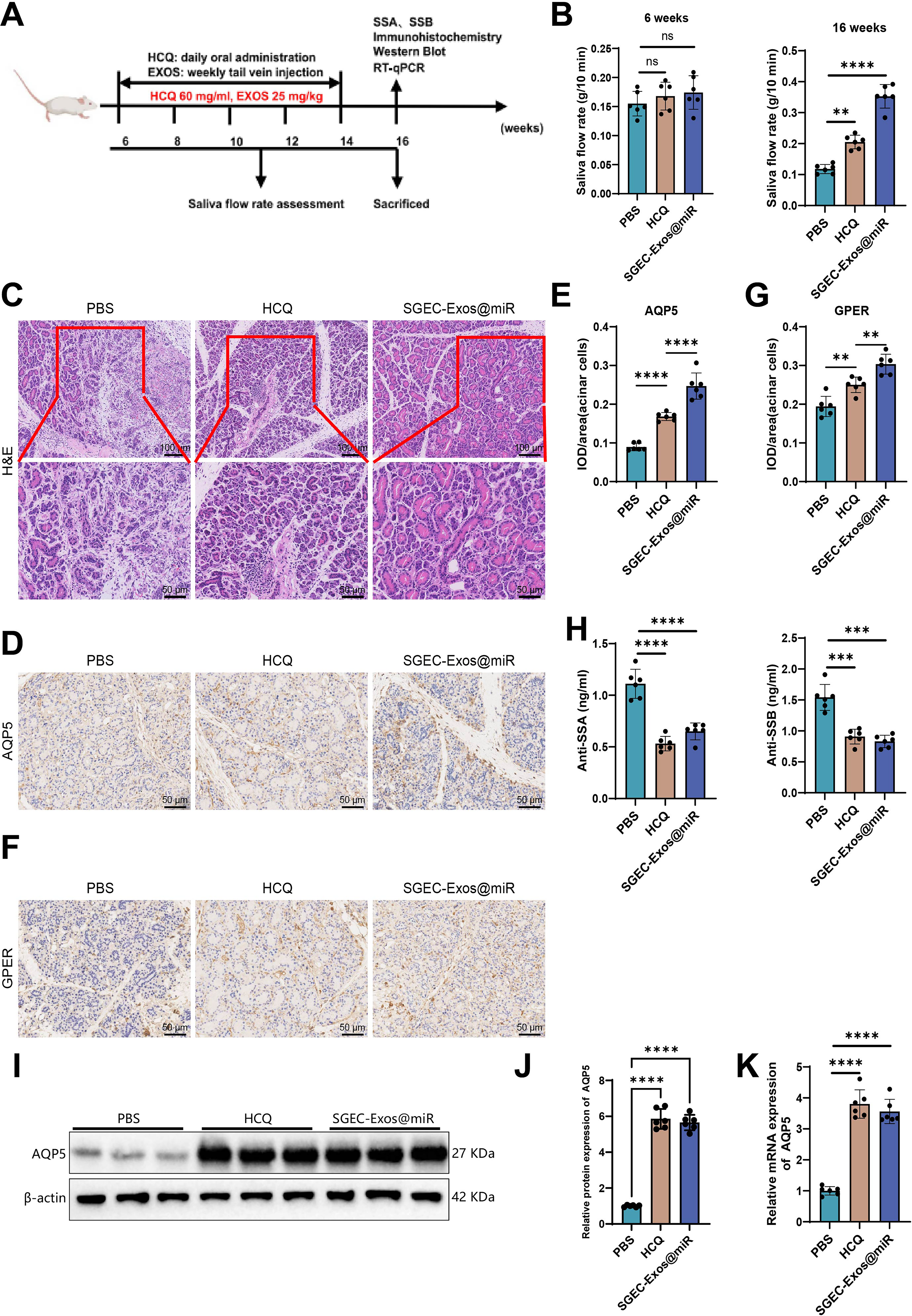
NOD/Ltj mice were employed as a primary SS model, characterized by progressive salivary gland dysfunction (Scuron et al., 2019). The experimental design is illustrated in Figure 7A. The salivary flow rate of 16-week-old NOD/Ltj mice was significantly lower than that of 6-week-old NOD/Ltj mice. After 10 weeks of SGEC-Exos@miR treatment, salivary flow rates in NOD/Ltj mice were significantly increased (Fig. 7B). Notably, the improvement in salivary secretion induced by SGEC-Exos@miR was more pronounced than that achieved with the positive control hydroxychloroquine (HCQ). Hematoxylin and eosin (H&E) staining revealed pronounced periductal acinar vacuolization and severe glandular atrophy in the salivary glands of NOD/Ltj mice. Compared with phosphate-buffered saline (PBS)-treated controls, both HCQ- and SGEC-Exos@miR-treated mice showed a marked reduction in the number and area of lymphocytic infiltration foci within the salivary glands (Fig. 7C). Immunohistochemical (IHM) analyses (Fig. 7D, E) demonstrated that, compared with the PBS group, AQP5 immunoreactivity was markedly enhanced in the SGEC-Exos@miR group, particularly at the apical membrane of acinar cells. In addition, GPER expression was also significantly upregulated in SGEC-Exos@miR-treated mice relative to PBS-treated controls (Fig. 7F, G). Furthermore, both SGEC-Exos@miR and HCQ treatments significantly decreased serum anti-Sjögren’s syndrome antigen A (SSA)/Ro and anti-Sjögren’s syndrome antigen B (SSB)/La antibody levels in NOD/Ltj mice (Fig. 7H). WB analyses (Fig. 7I, J) and RT-qPCR assays (Fig. 7K) were performed to evaluate AQP5 expression in the submandibular glands. Compared with the PBS group, both SGEC-Exos@miR and HCQ groups showed increased AQP5 mRNA and protein expression levels. Collectively, these findings demonstrate that SGEC-Exos@miR treatment effectively improves salivary gland function in NOD/Ltj mice.
Discussion
SS is a chronic systemic autoimmune disease that predominantly impairs exocrine gland function, especially in the salivary and lacrimal glands, leading to xerostomia and keratoconjunctivitis sicca and markedly reducing quality of life. In addition, SS is associated with an increased risk of severe complications, including lymphoma (Argyropoulou et al., 2018). Compared with chemically induced SS models, NOD mice offer a closer approximation of the human disease. These animals spontaneously develop progressive lymphocytic infiltration and secretory gland dysfunction without the need for external stimulation, thereby more accurately reflecting the underlying autoimmune mechanisms (Peck and Nguyen, 2017). Although the pathological process in NOD mice advances slowly, their reproducible glandular damage and immune profile make them highly suitable for mechanistic studies on SS (Gao et al., 2006; Shi et al., 2014). In the present study, both the in vivo NOD model and the IFN-γ-induced A253 cell model were utilized. Although A253 cells are derived from salivary gland tumors and thus may not fully replicate the behavior of primary epithelial cells, consistency between these in vitro results, our in vivo findings, and published data validates the use of this system for mechanistic exploration. While current therapies—including immunosuppressants and anti-inflammatory agents—provide symptomatic relief (Ibáñez-Cabellos et al., 2019), they do not reverse glandular atrophy or alter long-term disease trajectory (Chen et al., 2016). This limitation underscores the urgent need for targeted and disease-modifying treatments.
Recent research has increasingly focused on miRNAs as post-transcriptional regulators with potential therapeutic value in autoimmune diseases (Chen et al., 2017). In this context, WGCNA was applied to identify miRNA expression modules correlated with the SS phenotype. Rather than constructing conventional miRNA–mRNA regulatory networks, WGCNA was used to detect hub miRNAs within co-expression modules. The MEbrown module demonstrated the strongest association with SS pathology, and from this, miR-23b-3p emerged as a key regulator. To investigate its therapeutic relevance, we developed a targeted delivery system using SGEC-Exos to encapsulate miR-23b-3p. This strategy enabled efficient transfer of miR-23b-3p into SGECs and successfully inhibited IKKα/NF-κB signaling, which is critical in inflammation and immune activation. Notably, this approach not only reduced oxidative stress and inflammatory cytokine production but also restored immune homeostasis by shifting the Treg/Th17 cell balance. In contrast to prior studies that focused solely on miRNA expression changes or indirect modulation of gene targets, our work provides direct evidence of functional miRNA delivery using Exo-based systems. This not only validates the therapeutic relevance of miR-23b-3p but also highlights SGEC-derived Exos as a promising vehicle for targeted RNA delivery in autoimmune disorders such as SS.
Exos are emerging as biocompatible nanoscale delivery vehicles capable of shielding miRNAs from enzymatic degradation while inherently guiding them to injured tissues, owing to the homing characteristics conferred by their parent cells (Jang et al., 2023). In this study, Exos derived from SGEC-Exos were employed as miR-23b-3p carriers, leveraging their natural targeting capacity toward damaged salivary tissue to improve therapeutic precision and efficacy. Unlike conventional viral vectors, Exos exhibit minimal immunogenicity, making them well-suited for repeated or long-term administration. Compared with traditional delivery systems such as liposomes or direct nucleic acid injection, the SGEC-Exos-based strategy demonstrated enhanced cellular uptake specificity and favorable biosafety, thereby optimizing the therapeutic utility of miR-23b-3p. Nonetheless, several translational challenges persist. Targeting accuracy remains influenced by the donor cell phenotype and the recipient’s microenvironment, potentially affecting biodistribution and efficacy in vivo (Shao et al., 2020). Furthermore, large-scale manufacturing and long-term preservation of Exos are still under development. Although freeze–drying (lyophilization) has been explored to improve Exo stability (Charoenviriyakul et al., 2018), it may compromise drug-loading efficiency and bioactivity. In addition, the pharmacokinetics, clearance routes, and long-term biosafety profiles of Exo-based therapeutics remain inadequately characterized. While this study demonstrates that SGEC-Exos@miR-23b-3p effectively alleviates salivary gland dysfunction in NOD/Ltj mice, its long-term toxicity warrants further investigation using metabolic tracking approaches.
The IKKα/NF-κB signaling cascade is pivotal in modulating immune and inflammatory responses, and its dysregulation shows linkage with the pathogenesis of various autoimmune conditions. In this study, dual-luciferase reporter assays and RIP assays confirmed that IKKα is a downstream target of miR-23b-3p. Delivery of miR-23b-3p via SGEC-derived Exos specifically reduced IKKα expression, thereby suppressing NF-κB pathway activation. These findings indicate that IKKα represents a major functional target of SGEC-Exos@miR-23b-3p. Compared with conventional approaches involving small-molecule inhibitors or neutralizing antibodies, this Exo-mediated RNA delivery system achieved higher cellular specificity with a lower likelihood of off-target effects or systemic toxicity. This precise molecular targeting not only highlights the therapeutic potential of miR-23b-3p in autoimmune disease contexts but also supports the feasibility of miRNA-Exo platforms as a new generation of immunomodulatory therapeutics.
AQP5 is a transmembrane water channel protein that is predominantly expressed in SGECs and plays a critical role in salivary secretion (Hu et al., 2023). In SS, inflammatory injury to the salivary glands induces SGEC death and suppresses AQP5 expression, leading to reduced salivary secretion and xerostomia. Accordingly, multiple therapeutic approaches have been reported to restore AQP5 expression in SGECs during SS treatment. For example, apigenin alleviates xerostomia by activating AQP5 via estrogen receptor-α signaling (Wei et al., 2022), while the traditional Chinese medicine formula Shaoyao-Gancao Decoction improves SS symptoms by modulating the cAMP–PKA signaling pathway and enhancing AQP5 expression (Wang et al., 2020). This study demonstrated that SGEC-Exos@miR-23b-3p upregulated AQP5 expression in an SS cell model, indicating a protective effect on SGECs and highlighting the therapeutic potential of SGEC-Exos@miR-23b-3p for the treatment of SS.
Excessive ROS generation is a key driver of inflammatory and autoimmune pathologies. In this study, SGEC-Exos@miR-23b-3p effectively reduced ROS accumulation by modulating NF-κB signaling, thereby alleviating oxidative stress and inflammatory injury. In the SS cell model, inhibition of IKKα expression by BMS-345541 led to a reduction in ROS levels, whereas restoration of IKKα expression resulted in a subsequent increase in ROS production. Collectively, these findings indicate that ROS generation occurs downstream of IKKα activation. In addition, the intervention restored immune balance by increasing Treg cell frequency and reducing Th17 cell proportions—an effect that parallels outcomes observed with biological agents and cellular immunotherapies. However, by specifically targeting molecular mediators, our approach offers a potentially more precise and efficient alternative.
Moreover, SGEC-Exos@miR-23b-3p exerted marked cytoprotective effects by downregulating pro-apoptotic markers such as Bax and cleaved caspase-3, while upregulating Bcl-2. These changes were associated with enhanced cell survival and improved salivary gland function, directly addressing SS-related glandular dysfunction. While apoptosis modulation has been extensively investigated in oncology and neurodegenerative disorders, its application in autoimmune disease therapy remains underexplored. Our findings suggest that miRNA-based antiapoptotic strategies may represent a novel therapeutic direction for SS and related conditions.
The partial attenuation of therapeutic efficacy following IKKα re-expression underscores the central role of the IKKα/NF-κB axis in mediating the protective effects of SGEC-Exos@miR-23b-3p. These observations also suggest the importance of continuous or sustained intervention in potential clinical regimens. Furthermore, these findings highlight the relevance of IKKα and related molecules as biomarkers for treatment response, offering new avenues for patient stratification and disease monitoring.
In conclusion, this study identifies miR-23b-3p as a key mediator of SS pathophysiology and demonstrates that its delivery via SGEC-derived Exos yields substantial anti-inflammatory, antioxidant, and antiapoptotic effects. The use of SGEC-Exos as a delivery platform ensures high target specificity and low immunogenicity, supporting its promise for clinical translation as a precision therapy for SS.
This study has certain limitations. First, we used the A253 tumor-derived cell line as an in vitro model of SS, which may not fully recapitulate the biological behavior of primary SGECs. In future studies, mechanistic validation will be performed in NOD/Ltj mice and primary SGECs derived from patients with SS. These validation experiments will include (i) overexpression or inhibition of miR-23b-3p in primary SGECs to assess its effects on the IKKα/NF-κB axis; (ii) evaluation of the impact of SGEC-Exos@miR-23b-3p treatment on IKKα/NF-κB signaling, ROS generation, inflammatory cytokines (TNF-α, IL-1β, and IL-6), and AQP5 expression; and (iii) coculture with T cells to determine effects on the Treg/Th17 balance, expression of transcription factors Foxp3 and RORγt, and levels of cytokines IL-10, TGF-β, IL-17, and IL-23, thereby further validating the anti-inflammatory, antiapoptotic, and immunomodulatory effects of SGEC-Exos@miR-23b-3p. In parallel, we will investigate whether SGEC-Exos@miR can ameliorate salivary gland dysfunction in NOD/Ltj mice by assessing salivary flow rate, performing histopathological analysis of salivary gland tissues, and evaluating the expression of key proteins involved in salivary secretion, including AQP5 and GPER, in the submandibular glands, thereby further validating its in vivo efficacy. Although the present study demonstrates significant effects of SGEC-Exos@miR-23b-3p in in vitro models, the correlation between miR-23b-3p and IKKα expression has not yet been validated in clinical samples from patients with SS. Future investigations will examine miR-23b-3p and IKKα levels in salivary gland tissues and peripheral blood from SS patients, with healthy individuals serving as controls, to confirm their inverse association and to determine its relevance to SS disease activity. In addition, long-term in vivo safety data for SGEC-Exos@miR-23b-3p are currently lacking. Future investigations will focus on evaluating its safety profile following repeated administration, including biodistribution analysis (using fluorescence imaging to track DiR-labeled SGEC-Exos@miR-23b-3p after tail vein injection in female NOD/Ltj mice), chronic toxicity assessment (monitoring body weight and measuring serum levels of creatine kinase (CK), lactate dehydrogenase (LDH), alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CREA), and uric acid (UA) using an automated biochemical analyzer), and histopathological evaluation (H&E staining of major organs, including the heart, liver, spleen, lungs, and kidneys). Finally, additional studies are needed to clarify the involvement of miR-23b-3p in regulating other inflammatory signaling pathways, which will be essential for advancing this therapeutic strategy toward clinical translation.
This study revealed the therapeutic potential of SGEC-Exos@miR-23b-3p in SS by targeting the IKKα/NF-κB pathway. Functional assays demonstrated that SGEC-Exos@miR-23b-3p effectively downregulated IKKα, p-p65, and p-IκBα, thereby suppressing NF-κB activation. This led to reduced oxidative stress, restoration of AQP5 expression, and improvement in salivary gland function. In addition, SGEC-Exos@miR-23b-3p restored the Treg/Th17 balance, enhancing immune regulation and mitigating inflammatory responses. The treatment also inhibited apoptosis, further contributing to cellular protection. Collectively, these findings validate that SGEC-Exos@miR-23b-3p exerts a multifaceted therapeutic effect in SS by simultaneously modulating inflammation, immune dysregulation, and cell death, offering a promising strategy for targeted gene therapy in autoimmune disorders.
Materials and Methods
SS animal model details
Female NOD/Ltj mice (10 weeks old, 20–22 g) and age-matched female ICR mice were obtained from HFK Bioscience Co., Ltd. (Beijing, China). Given that NOD mice are derived from the ICR lineage, ICR mice served as healthy controls. All animals were maintained under specific-pathogen-free conditions with a 12-h light/dark cycle (lights on from 7:00 AM to 7:00 PM), controlled temperature (23 ± 1°C), and relative humidity (55 ± 5%), with ad libitum access to standard chow and water. At the endpoint, mice were deeply anesthetized with isoflurane and euthanized by cervical dislocation in accordance with established protocols (Pu et al., 2022).
RNA extraction and sequencing
Salivary gland tissues from SS model and control mice (n = 6 per group) were collected for total RNA isolation using TRIzol reagent (15596026, Invitrogen, USA). RNA concentration and purity were assessed with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Only samples meeting stringent quality criteria (RNA integrity number ≥7.0 and 28S/18S ratio ≥1.5) were used for subsequent sequencing.
Total RNA was processed at CapitalBio Technology (Beijing, China). Strand-specific libraries were generated after rRNA removal (Ribo-Zero™ Kit, Epicentre, USA) employing a deoxyuridine triphosphate (dUTP)-based protocol (#E7775, NEBNext Ultra RNA Kit, NEB, USA). RNA fragments (∼300 bp) were subjected to first- and second-strand cDNA synthesis, end repair, adaptor ligation, and second-strand digestion with USER enzyme (#M5508, NEB, USA). Libraries were amplified by PCR, evaluated on an Agilent 2100 Bioanalyzer (Agilent 2100 Bioanalyzer), and quantified using the KAPA Library Quantification Kit (KK4844, KAPA Biosystems). High-throughput sequencing was conducted on the Illumina NextSeq CN500 platform (Illumina NextSeq CN500) using a paired-end sequencing strategy.
Quality control and alignment of sequencing reads
Raw paired-end reads were assessed using FastQC (v0.11.8), followed by adapter and poly(A) trimming with Cutadapt (v1.18). Reads containing >5% ambiguous bases (N) or with <70% bases above Q20 were removed using custom scripts and the FASTX Toolkit (v0.0.13). Remaining high-quality paired-end reads were synchronized with BBMap and aligned to the mouse reference genome (mm10) utilizing HISAT2 v0.7.12.
Differential miRNA expression analysis
miRNA count data were normalized and log2-transformed using the limma package (v3.48.3) in R. Differentially expressed miRNAs between SS and control groups were identified with DESeq2 (v1.32.0), applying the thresholds of p < 0.05 and |log2FoldChange| > 1 for statistical and biological significance.
Weighted gene co-expression network analysis
Genes were filtered by median absolute deviation, retaining the top 50% most variable features. Outlier genes and samples were removed using the goodSamplesGenes function in the WGCNA package. A scale-free network was constructed (minimum module size = 10; deepSplit = 6), and modules with dissimilarity <0.6 were merged. Pearson correlation analysis (p < 0.05) was used to associate modules with sample traits, and the module showing the strongest correlation with the SS phenotype was designated as the key disease-associated module for downstream investigation.
LASSO regression algorithm
To identify disease-associated hub genes, LASSO regression was implemented utilizing the glmnet package in R. A random seed was set to ensure reproducibility. The model was trained in a binary classification format, with sample group labels extracted via regular expressions to define the response variable. Model performance was evaluated through cross-validation using cv.glmnet, and the optimal penalty parameter (λ) was selected based on minimum cross-validated error. Genes with nonzero coefficients at the optimal λ were retained as candidate biomarkers for downstream analyses.
Target gene prediction
Candidate miR-23b-3p target genes were predicted using three databases: ENCOR, miRWalk, and miRTarBase. Overlapping results were integrated to increase prediction confidence. SS-related genes were retrieved from GeneCards (https://www.genecards.org/) using “Sjögren’s syndrome” as the keyword. Venn diagrams illustrating gene overlaps were generated via the Xiantao Academic online platform.
GO and KEGG pathway enrichment analysis
Functional enrichment analysis of the intersecting target genes was implemented utilizing the ClusterProfiler package in R. GO terms and KEGG pathways were analyzed, with significance thresholds set at p < 0.05. Results were visualized as bubble plots, ranked by adjusted p-values, to highlight major biological functions and signaling pathways potentially involved in SS pathogenesis.
Luciferase assay
To validate direct binding between miR-23b-3p and the 3′-UTR of IKKα, HEK293 cells (ATCC, USA) were co-transfected with a pMIR-REPORT vector containing the IKKα 3′-UTR (Promega, USA) and synthetic miR-23b-3p mimics using Lipofectamine 3000 (Invitrogen, USA). Reporter signals were assessed 48 h later, and the firefly/Renilla luciferase ratio indicated miR-23b-3p-dependent repression of IKKα expression.
RNA immunoprecipitation
To verify the interaction between miR-23b and IKKα mRNA, SGECs were seeded to 80% confluence and transfected with either miR-23b mimic or negative control mimic using Lipofectamine™ 3000 (Invitrogen, USA). After 48 h, cells were lysed in ice-cold RIP lysis buffer and clarified by centrifugation (12,000 g, 15 min) at 4°C. The supernatants were subjected to immunoprecipitation with an anti-AGO2 antibody (4 μg/mL, ab186733, Abcam) and 40 μL protein A/G magnetic beads (Sigma, USA) at 4°C for 4 h (or overnight) under gentle rotation. Beads were rinsed thrice with cold RIP wash buffer to remove nonspecific binding and then incubated with RIP elution buffer at 65°C for 10 min to release bound RNA. RNA was isolated using TRIzol reagent (Invitrogen), reverse-transcribed into cDNA, and quantified by RT-qPCR to detect IKKα mRNA enrichment.
Reverse transcription quantitative PCR
RT-qPCR was performed to quantify miR-23b-3p and IKKα expression. Total RNA was isolated from salivary gland tissues of control and SS mice (n = 6), SGECs from each experimental group, and miR-23b@SGEC-Exos using TRIzol reagent (Invitrogen, USA). RNA concentration and purity were assessed with a NanoDrop 2000 spectrophotometer. cDNA synthesis for mRNA analysis was carried out using the PrimeScript RT Reagent Kit (Takara, Japan).
For miRNA quantification, RT was performed using a poly(A) tailing kit (B532451, Sangon Biotech, China). qPCR was conducted using the 7500 Real-Time PCR System (Applied Biosystems, USA) with SYBR Premix Ex Taq™ II (DRR081, Takara, Japan). U6 snRNA and β-actin were served as normalizers for miRNA and mRNA, respectively. Relative gene expression was calculated utilizing the 2−ΔΔCt method. Primer sequences are summarized in Supplementary Table S1.
Western blot
Following total protein extraction on ice using radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific, USA) supplemented with protease and phosphatase inhibitors and protein concentration determination via a BCA assay (Thermo Fisher Scientific, USA), equal amounts of protein (30 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA). Membranes were blocked and incubated overnight at 4°C with primary antibodies targeting IKKα (ab32041, 1:1000), NF-κB p65 (phospho S536) (ab28856, 1:500), NF-κB p65 (ab32536, 1:1000), IKB alpha (phospho S36) (ab133462, 1:2000), IKB alpha (ab32518, 1:1000), AQP5 (ab305303, 1:1000), GPER (ab260033, 1:1000), CREB (ab32515, 1:1000), phospho-CREB (ab32096, 1:5000), VDAC1 (ab154856, 1:1000), Cytochrome-c (ab133504, 1:1000), Bcl-2 (ab32124, 1:1000), Bax (ab182733, 1:1000), cleaved caspase-3 (ab13847, 1:1000), RORγt (ab207082, 1:1000), FOXP3 (ab20034, 1:1000), IL-6 (ab290735, 1:1000), TNF-α (ab215188, 1:1000), beta Actin (ab8226, 1:5000), all from Abcam (UK), as well as PKA (1:1000, #4782, Cell Signaling Technology, USA), phospho-PKA (1:1000, #4781, Cell Signaling Technology, USA). Following primary incubation, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5000, Abcam) for 1 h at room temperature (RT). Protein bands were visualized using enhanced chemiluminescence (Thermo Fisher Scientific). A prestained protein marker (10–180 kDa; Thermo Fisher Scientific) was used as a molecular weight reference. Band intensities were quantified in ImageJ after background subtraction. For phosphorylated proteins, signal intensities were first normalized to their corresponding total protein levels (e.g., p-p65/p65 and p-IκBα/IκBα), and the resulting ratios were then normalized to the control group. Total protein levels, including IKKα, AQP5, Bax, Bcl-2, and cleaved caspase-3, were normalized to β-actin.
Cell culture
The human SGEC A253 (CL-0860, Wuhan Pricella Biotechnology Co., Ltd.) was utilized as an in vitro model for SGECs, based on its phenotypic similarity and frequent use in SS-related studies investigating metabolic and inflammatory dysfunction (Feng et al., 2025; Xu et al., 2023). Cells were cultured in RPMI-1640 medium enriched with 10% fetal bovine serum (FBS), 2 mM
Immunofluorescence
A253 cells were washed with PBS (14190144, Gibco, USA), fixed at RT with 4% paraformaldehyde (PFA, 15710, Electron Microscopy Sciences, USA) for 25 min, and permeabilized with 0.25% Triton X-100 (T8787, Sigma-Aldrich, USA) for 10 min. After three PBS washes, cells were blocked with a solution containing 1% bovine serum albumin (BSA) (A2153, Sigma-Aldrich, USA), 2% skim milk (9999, Cell Signaling Technology, USA), and 0.1% Tween-20 (P9416, Sigma-Aldrich, USA) for 1 h at RT. Cells were then incubated overnight at 4°C with anti-AQP5 antibody (AB3559, 1:200, Millipore, USA). After washing, Alexa Fluor 488-conjugated goat antirabbit IgG secondary antibody (A11008, 1:500, Invitrogen, USA, link) was applied for 1 h at RT in the dark. Nuclei were counterstained with DAPI (D1306, 1:1000, Invitrogen, USA) for 5 min, and slides were mounted with Prolong Gold Antifade Mountant (P36930, Invitrogen, USA). Images were acquired under an Olympus BX51 fluorescence microscope (Olympus Corporation, Japan) (Takakura et al., 2024).
Isolation and purification of SGEC-Exos
Exos were isolated from the culture supernatant of human SGECs. Cells were maintained in Dulbecco’s modified Eagle medium (DMEM) enriched with 10% FBS (Gibco, USA) and 1% penicillin-streptomycin (Gibco, USA). After 48 h of culture, the conditioned medium was sequentially centrifuged to remove contaminants: 300 g for 10 min to eliminate cells, 2000 g for 20 min to remove organelles, and 10,000 g for 30 min to exclude large vesicles. Crude Exos was pelleted by ultracentrifugation at 100,000 g for 2 h using a Beckman Coulter ultracentrifuge (USA). To enhance purity, the Exo fraction was subjected to immunoaffinity capture using magnetic beads coated with anti-CD63 antibodies (Santa Cruz Biotechnology, USA). The enriched SGEC-Exos were washed, resuspended in PBS, and stored at −80°C for downstream analysis.
Loading of miR-23b-3p into SGEC-Exos
Synthetic miR-23b-3p mimics were encapsulated into SGEC-derived Exos by electroporation to generate SGEC-Exos@miR-23b-3p. Electroporation was performed using the CUY21EDIT II system (BEX, Japan) in a 0.4 cm chilled cuvette with the following settings: poring voltage 110 V, poring time 6 ms, interval 10 ms, transfer voltage 25 V, and capacitance 940 μF, for 10 cycles. Following electroporation, Exos were incubated at 37°C for 30 min to promote membrane recovery. Free, unincorporated miR-23b-3p was removed via ultracentrifugation.
The morphology, particle size distribution, and zeta potential of Exos from each sample were analyzed using TEM (Talos L120C G2, Thermo Fisher Scientific, USA) and DLS (NS-90Z, OMEC, China), respectively. DLS enables rapid measurement of exosomal particle size distribution and zeta potential (Zhu et al., 2025). Owing to its low cost, minimal sample requirement, and operational simplicity, DLS was selected for Exo characterization in this study. Total protein content was quantified employing the Pierce BCA Protein Assay Kit (Aspen, China). To assess miRNA stability within Exos, SGEC-Exos@miR-23b-3p and free miR-23b-3p mimics were incubated at 37°C for five days. Residual miRNA levels were measured by RT-qPCR.
Northern blot
For Northern blot analysis, 50 μg of SGEC-Exos@miR-23b-3p was collected after ultracentrifugation and 0.22 μm filtration. RNA was extracted using TRIzol™ Reagent (15596026, Invitrogen) and denatured at 70°C for 10 min with RNA Loading Dye (AM8550, Invitrogen) and 0.1% bromophenol blue. Samples, along with a 20–80 nt RNA ladder (N0362S, NEB), were loaded onto a 15% denaturing polyacrylamide/8M urea gel (161-0144, Bio-Rad) and electrophoresed in 1× TBE buffer (15581-028, Invitrogen) at 200 V for 1.5 h. RNA was transferred to Hybond™-N+ nylon membranes (RPN303B, Cytiva) using a semidry blotting system (170–3940, Bio-Rad) at 25 V, 1.3 A for 30 min, followed by UV crosslinking (120 mJ/cm2; CL-1000, UVP). Membranes were prehybridized in DIG Easy Hyb buffer (11603558001, Roche) at 42°C for 30 min, then hybridized overnight at 42°C with a digoxigenin-labeled locked nucleic acid (LNA) probe specific for miR-23b-3p (sequence 5′-DIG-TCACATTGCCAGGGATTACC-3′, Exiqon). The next day, membranes were washed twice with 2× SSC/0.1% SDS at RT (10 min each), followed by two washes in 0.1× SSC/0.1% SDS at 50°C (20 min each). Blocking was performed for 30 min at RT, after which membranes were incubated with anti-digoxigenin-AP Fab fragments (1:10,000, 11093274910, Roche) for 30 min. After washing, CDP-Star chemiluminescent substrate (12041677001, Roche) was applied for 5 min, and signals were captured using an Amersham™ Imager 680 (29165762, Cytiva, USA). A specific band at 22 nt confirmed successful loading of miR-23b-3p into Exos and excluded free miRNA contamination (Koscianska et al., 2011).
Cellular uptake of SGEC-Exos@miR-23b-3p
To assess Exo internalization, SGEC-Exos@miR-23b-3p was labeled with DiI and diluted in serum-free medium (10 μg/mL), followed by incubation with SGECs at 37°C for 24 h. After incubation, cells were fixed with 4% PFA and stained with 1% DAPI solution (62248, Thermo Scientific, USA) for nuclear labeling. Cell membranes were counterstained using PKH67 dye (MINI67, Sigma-Aldrich, USA). Observation of intracellular uptake of labeled Exos was completed using a confocal laser scanning microscope (CLSM; Zeiss LSM710, Germany).
Cell culture and grouping
SGECs were cultured in DMEM (Gibco, USA) enriched with 10% FBS at 37°C with 5% CO2 until reaching 80% confluence. Cells were then assigned to six experimental groups. Control group: SGECs were cultured without any treatment. Model group: SGECs were treated with IFN-γ (20 ng/mL, 285-IF-100, R&D Systems, USA) for 24 h to simulate the pathological state of SS. SGEC-Exos@miR-23b-3p group: Model group cells were additionally treated with SGEC-Exos@miR-23b-3p at a final 100 μg/mL concentration for 24 h. SGEC-Exos group: SGECs in the Model group were additionally treated with SGEC-Exos at a final concentration of 100 μg/mL for 24 h. Rescue group (miR-23b@SGEC-Exos + oe-IKKα): Model group cells were first treated with SGEC-Exos@miR-23b-3p for 24 h, followed by transfection with a plasmid overexpressing IKKα, with incubation continued for an additional 48 h (Hu and Zheng, 2022; Lustig et al., 2021). Positive control group (BMS-345541 group): Based on the Model group, BMS-345541, an inhibitor of the IKK/NF-κB signaling pathway [HY-10519, MedChemExpress (MCE), USA], was added and administered at a final concentration of 5 μM for 12 h (Ping et al., 2016; Yang et al., 2006). To examine the changes in IKKα protein levels and ROS levels over time following IFN-γ induction, A253 cells were incubated with IFN-γ for 2, 4, 8, 16, and 24 h, respectively.
To ensure the reproducibility of cell samples, freeze–thaw cycles were minimized to maintain a highly consistent cellular state upon each recovery. The key principles included slow freezing, rapid thawing, batch management, and standardized documentation. Cells selected for cryopreservation were healthy and at 80%–90% confluence. Prior to freezing, the culture medium was replaced with fresh medium to ensure that cells were in an optimal physiological state. The cryopreservation medium consisted of complete culture medium supplemented with 10% dimethyl sulfoxide (DMSO) and 20% serum. Serum from the same batch was used and heat-inactivated in advance, while fresh, high-purity DMSO was selected to avoid repeated freeze–thaw cycles. The slow-freezing principle was strictly followed, with a cooling rate of 1°C/min until −80°C. A Mr. Frosty™ controlled-rate freezing container (5100-0001, Thermo Scientific) was used to achieve gradual cooling, after which the cryovials were transferred to liquid nitrogen for long-term storage. The rapid-thawing principle was also rigorously applied. After removal from liquid nitrogen, cryovials were immediately placed in a 37°C water bath and gently agitated, allowing complete thawing within ∼1 min. Following thawing, cells were immediately diluted with prewarmed fresh culture medium, and the suspension was centrifuged at low speed to remove the DMSO-containing supernatant, thereby minimizing the cytotoxic effects of DMSO. Batch management was implemented by establishing a seed cell banking system, which is critical for maintaining long-term experimental reproducibility. First, a master cell bank was generated by cryopreserving a large batch of early-passage cells upon acquisition. Subsequently, a working cell bank was established: For each experiment, one vial from the master bank was thawed and expanded, and then a batch of cells was re-cryopreserved as working stocks. Routine experiments used cells directly from the working bank, and once depleted, a new vial was retrieved from the master bank. This strategy effectively minimized cellular drift caused by prolonged passaging. Finally, comprehensive labeling and documentation were maintained. All cryovials were clearly labeled with cell line name, passage number, freezing date, and operator. In addition, detailed laboratory records were maintained to document key variables, including cell source, culture medium batch number, serum batch number, freezing date, and operator, thereby ensuring traceability of experimental conditions.
To ensure experimental rigor and reproducibility, biological replicates (n = 3) were defined as independently cultured and independently treated cell samples, rather than technical replicates derived from the same sample. For cell-based assays, each biological replicate originated from a separate well or independently prepared culture under the same standardized conditions. To minimize variability associated with long-term passaging, all experiments were performed using low-passage cells derived from the working cell bank.
Measurement of ROS
Intracellular ROS levels were assessed using the fluorescent probe dihydroethidium (DHE; 309800, Sigma-Aldrich, USA). Treated SGECs were incubated with 10 μM DHE at 37°C for 30 min in the dark. After washing three times with PBS, cells were analyzed immediately by flow cytometry (BD Biosciences, USA) without fixation or storage to prevent signal degradation. A fixed gating strategy was applied using FlowJo software (Tree Star, USA) to identify the ROS-positive population, and ROS levels were quantified as the percentage of positive cells. At least 10,000 events were acquired per sample under identical instrument settings.
Mitochondrial membrane potential (Δψm) assay
To evaluate changes in Δψm, SGECs were incubated with 5 μg/mL JC-1 dye (C2003S, Beyotime, China) at 37°C for 20 min (dark condition). Cells were subsequently washed twice with PBS, and fluorescence was visualized utilizing a fluorescence microscope (Leica Microsystems, Germany). Red fluorescence indicates JC-1 aggregation in polarized mitochondria, whereas green fluorescence reflects mitochondrial depolarization. The red/green fluorescence ratio was quantified using ImageJ (NIH, USA).
Enzyme-linked immunosorbent assay
Cytokine concentrations in cell culture supernatants were measured using ELISA kits from R&D Systems (USA), including TNF-α (DTA00D), IL-1β (DLB50), IL-6 (D6050), TGF-β (DB100B), IL-10 (D1000B), IL-17 (D1700), and IL-23 (D2300B). All assays were performed according to the manufacturers’ protocols, with samples analyzed in triplicate. Absorbance was recorded at 450 nm using a microplate reader (Bio-Rad, USA), and cytokine concentrations were calculated based on standard curves.
Cell coculture experiment
Jurkat T cells (ATCC, USA) were maintained in RPMI-1640 medium (Gibco, USA) enriched with 10% FBS and 1% penicillin-streptomycin. SGECs were grouped into Control, Model, SGEC-Exos@miR-23b-3p, and Rescue conditions as described previously. For coculture experiments, SGECs and Jurkat T cells were plated in a Transwell system (0.4 μm pore size, Corning, USA) at a 1:5 ratio, with SGECs seeded in the lower chamber and T cells in the upper chamber to avoid direct cell–cell contact. After 48 h of coculture, downstream analyses were conducted.
Flow cytometry
Flow cytometry was employed to evaluate the proportions of Treg (CD4+CD25+Foxp3+) and Th17 (CD4+IL-17A+) cells, as well as to assess apoptosis within the coculture system. Following 48-h coculture, T cells were harvested, PBS-washed, and gated on FSC-A versus SSC-A plots to isolate lymphocyte populations while excluding debris and dead cells (DAPI-). For Treg analysis, cells were stained with anti-CD4-FITC (555346, BD Biosciences), anti-CD25-PE (555432, BD), and anti-Foxp3-APC (17-4777-42, eBioscience). For Th17 detection, anti-CD4-FITC and anti-IL-17A-PE (560436, BD) were used. To minimize interference from Th1/Th2 cells, additional staining for IFN-γ (558771, BD) and IL-4 (560699, BD) was performed, and only IFN-γ-/IL-4- double-negative cells were included in Th17 analysis (Hoseinzadeh et al., 2023).
For apoptosis assessment, Annexin V-FITC/PI staining (C1062M, Beyotime) was conducted. Samples were analyzed using a BD FACSVerse flow cytometer (BD Biosciences), and data were processed with FlowJo (Tree Star). At least 10,000 events were acquired per sample.
CCK-8 assay
Cell proliferation was examined utilizing CCK-8 (CK04, Dojindo, Japan). After 24-h incubation of cells (5 × 103/well) in 96-well plates, 10 μL of reagent was added and incubated for 2 h. Absorbance at 450 nm was tested utilizing a microplate reader to quantify viability against control levels.
Caspase-3 activity assay
Caspase-3 activity was tested to assess apoptosis using a commercial assay kit (ab39401, Abcam, UK). Following lysis, samples were incubated with the caspase-3 substrate. The absorbance was recorded at 405 nm using a microplate reader, and activity levels were normalized and expressed as fold change.
In vivo functional validation
To assess whether SGEC-Exos@miR ameliorates salivary gland dysfunction in SS, in vivo studies were conducted using female NOD/Ltj mice. Animals were randomly assigned to three groups (n = 6 per group): SGEC-Exos@miR treatment, positive control receiving HCQ, and disease control receiving PBS. HCQ (catalog no. HY-W031727) was purchased from MCE (USA). Mice in the treatment group received SGEC-Exos@miR via tail vein injection once weekly at a dose of 25 mg/kg for 10 consecutive weeks. Mice in the positive control group were administered HCQ (60 mg/kg) by oral gavage, while mice in the disease control group received an equal volume of PBS. Salivary flow rate was monitored weekly beginning after the first treatment. After 10 weeks, mice were euthanized, and submandibular glands and venous blood were collected for further analyses.
Measurement of salivary flow rate
Following body weight recording, mice were anesthetized and injected subcutaneously with pilocarpine hydrochloride (0.1 mg/kg) to stimulate salivary secretion. Ten minutes later, saliva was collected for 10 min using a glass capillary tube positioned along the sublingual lateral wall and transferred into preweighed tubes. Salivary flow rate was calculated based on the difference in tube weight before and after collection.
Histology and IHC
Following euthanasia, submandibular glands were excised and fixed in 4% PFA, paraffin-embedded, and sectioned at 4 μm. Sections were stained with H&E for histopathological evaluation. For IHC, sections were deparaffinized, rehydrated, subjected to antigen retrieval in sodium citrate buffer, and treated with 1% hydrogen peroxide to quench endogenous peroxidase activity. After blocking with goat serum, sections were incubated overnight at 4°C with primary antibodies against AQP5 (1:200) and GPER (1:200). Immunoreactivity was detected using appropriate secondary antibodies and 3,3′-diaminobenzidine. Images were captured using a light microscope (Olympus, Tokyo, Japan).
Measurement of serum anti-SSA/SSB antibodies
Venous blood was collected by cardiac puncture, and serum was isolated by centrifugation. Serum anti-SSA/Ro and anti-SSB/La antibody levels were quantified using mouse ELISA kits (mL-1117538 and mL-1117539, Mlbio, Shanghai, China). Absorbance was recorded at 450 nm with a microplate reader, and antibody concentrations were calculated from standard curves.
Experimental standardization and quality control
To justify the low inter-replicate variability observed in this study, additional measures were implemented for sample handling and quantification. Samples from each experimental group and its corresponding control group were handled in parallel whenever possible by the same operator using the same batch of reagents. For ROS analysis, all samples were stained and analyzed immediately after harvest without fixation or storage. For phosphoprotein analysis, cell lysates were prepared on ice in phosphatase inhibitor-supplemented buffer and processed under matched conditions, with all lysates handled using the same extraction protocol and immunoblot workflow to minimize signal loss and batch-related variation. Raw data from individual biological replicates were retained for statistical analysis and verification, and uncropped WBs together with individual flow cytometry data points are provided in the Supplementary Material.
Statistical analysis
All data were analyzed utilizing GraphPad Prism 9.0 (GraphPad Software, USA) and are detailed as mean ± standard deviation. For comparisons between two groups, unpaired Student’s t-tests were used. Multiple group comparisons were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s or Dunnett’s post hoc tests. For data not conforming to a normal distribution, the Mann–Whitney U test or Kruskal–Wallis test was applied. Statistical significance was defined as p < 0.05.
Authors’ Contributions
Y.C. and J.F. designed and performed the experiments, analyzed the data, and drafted the article. X.W. contributed to the study conception, bioinformatics and machine learning analyses, and critical revision of the article. C.E. supervised the overall project, provided clinical and experimental guidance, and finalized the article. All authors reviewed and approved the final version of the article and agreed to be accountable for all aspects of the work.
Data Availability Statement
The bulk RNA sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA1403644. The dataset includes control samples (SRA accession numbers: SRR36862469, SRR36862468, SRR36862465, SRR36862464, SRR36862463, SRR36862462) and case samples (SRA accession numbers: SRR36862461, SRR36862460, SRR36862459, SRR36862458, SRR36862467, SRR36862466). The data are publicly available at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1403644.
Ethical Statement
All animal experiments were approved by the Animal Ethics Committee of HFK Bioscience Co., Ltd. All animal experiments were designed, conducted, and reported in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Footnotes
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This work was supported by the Department of Finance of Jilin Province (Grant No. JCSZ2025678-13).
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.