
Abstract
Aim:
Cerebral ischemia/reperfusion (I/R) injury represents a significant challenge to recanalization therapy for ischemic stroke and is critically influenced by microglial polarization. Although inhibition of Rho-associated protein kinase (ROCK) has been shown to mitigate cerebral I/R injury and associated neuroinflammation, its specific effect on the balance between M1 and M2 (anti-inflammatory) microglial polarization remains incompletely understood. This study aimed to elucidate the role and underlying mechanism of ROCK inhibition in regulating M1 and M2 microglial polarization, using the classical antidepressant fluoxetine as a positive control.
Results:
ROCK inhibitor fasudil and positive control fluoxetine effectively alleviated cerebral I/R injury and facilitated a shift in microglial polarization from the M1 to the M2 phenotype, both in vivo and in vitro. In the hippocampal tissues of cerebral I/R mice exposed to lipopolysaccharide, we observed an upregulation of thioredoxin-interacting protein (TXNIP) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2). ROCK2 knockdown promoted the M2 microglial polarization, suppressed the expression of NOX2 and TXNIP, and inhibited the activation of NF-κB P65 in mouse hippocampal tissue. Notably, pharmacological inhibition of NF-κB reduced the expression of NOX2 and TXNIP, as well as the production of reactive oxygen species (ROS), in microglia subjected to oxygen-glucose deprivation/reoxygenation. Correspondingly, inhibition of NOX2 also decreased TXNIP expression.
Conclusion and Innovation:
ROCK inhibition promotes a shift in microglial polarization from the M1 to the M2 subtype by suppressing the NF-κB/NOX2/ROS/TXNIP signaling pathway. This study provides the first evidence demonstrating the mechanism by which ROCK inhibition drives microglial polarization toward the M2 phenotype. Antioxid. Redox Signal. 45, 149–168.

Introduction
Ischemic stroke remains a leading cause of cerebrovascular morbidity and mortality worldwide (Tu et al., 2023b; Walter, 2022). The current standard clinical treatments for acute ischemic stroke, intravenous thrombolysis with tissue plasminogen activator (t-PA) and mechanical thrombectomy (Lan et al., 2024), are limited by a narrow therapeutic time window and the risk of cerebral ischemia/reperfusion (I/R) injury (Kim et al., 2017). Therefore, there is an urgent need to identify novel therapeutic targets and develop effective strategies to mitigate cerebral I/R injury.
Microglia, the resident immune cells of the central nervous system (CNS), are the first responders to cerebral ischemic stimuli and undergo rapid morphological and functional changes within minutes of stroke onset (Lv et al., 2024). Activated microglia are broadly categorized into two phenotypes: the pro-inflammatory M1 phenotype, which is associated with neurotoxicity, and the anti-inflammatory M2 phenotype, which supports neuroprotection and tissue repair (Wang et al., 2018). Promoting a shift in microglial polarization toward the M2 phenotype has emerged as a promising therapeutic strategy for alleviating cerebral I/R injury following ischemic stroke (Li et al., 2025). However, the pharmacological agents and key molecular regulators that drive the phenotypic transition of microglia remain incompletely understood.
Rho-associated protein kinase (ROCK), including the isoforms ROCK1 and ROCK2, is highly expressed in glial cells and neurons, and plays a critical role in regulating various cellular processes (Kimura et al., 2021). The therapeutic potential of ROCK inhibitors has been demonstrated in preclinical models of ischemic stroke (Sladojevic et al., 2017). Fasudil, a non-selective ROCK inhibitor, is clinically approved in East Asian countries for the treatment of cerebral vasospasm following aneurysmal subarachnoid hemorrhage (Liu et al., 2012). Our previous studies have revealed that fasudil attenuates cerebral I/R injury in mice (Ding et al., 2024) and suppresses pro-inflammatory responses in astrocytes subjected to oxygen-glucose deprivation/reoxygenation (OGD/R) (Geng et al., 2025). However, the protective mechanism and molecular targets of fasudil against cerebral I/R injury remain unclear.
Growing evidence suggests that inhibiting ROCK activity reduces neuroinflammation and microglial activation following ischemic stroke (Lu et al., 2023; Zhang et al., 2022). However, whether the suppression of ROCK activity ameliorates cerebral I/R injury by modulating microglial polarization has yet to be elucidated. In this study, we aimed to explore whether ROCK inhibition can promote a shift in microglial polarization toward the neuroprotective M2 phenotype. Given that lipopolysaccharide (LPS) is widely used to induce M1 microglial polarization both in vivo and in vitro (Su et al., 2023), we employed LPS to promote the M1 microglial polarization following cerebral I/R, thereby enabling us to assess the role of ROCK inhibition in facilitating M1-to-M2 shift of microglia.
Post-stroke depression (PSD), characterized by loss of interest and persistent low mood, is a common emotional disorder following stroke (Fan et al., 2025), affecting ∼33.5% of stroke patients (Mitchell et al., 2017). Effectively treating PSD is therefore essential for enhancing functional recovery and improving cognitive outcomes of ischemic stroke patients (Villa et al., 2018). Emerging evidence implicates the roles of ROCK pathway in the pathophysiology of depression. Pharmacological inhibition of the ROCK pathway with fasudil has also been shown to exert antidepressant effects against chronic restraint stress-induced depressive-like behaviors (Garcia-Rojo et al., 2017). Besides, ROCK inhibitors have been shown to modulate multiple signaling cascades relevant to antidepressant efficacy (Li et al., 2020).
Notably, facilitating M2 microglial polarization has been reported to alleviate depressive-like behaviors (Wang et al., 2025). However, whether ROCK inhibition exerts antidepressant effects in the context of post-stroke depression and whether such effects involve the modulation of microglial polarization remain largely unexplored. Therefore, the present study first demonstrated the effect of fasudil on depressive-like behaviors in experimental animals following cerebral I/R to further confirm the protective roles of ROCK inhibition against cerebral I/R injury.
Fluoxetine, a serotonin selective reuptake inhibitor (SSRI), remains a highly effective agent for reversing depressive-like behavior in validated animal models (He et al., 2023), acting through multiple mechanisms including anti-inflammatory effects (Qian et al., 2024). Our team and other studies have revealed that fluoxetine can inhibit M1 microglial polarization and promote polarization toward the M2 subtype (Deng et al., 2025; Su et al., 2015). Given its well-documented effects on both depressive behavior and microglial polarization, fluoxetine was employed in the present study for two purposes: (1) as a positive control to validate the sensitivity of our behavioral and immunological assays in the context of ROCK inhibition experiments, and (2) as a pharmacological intervention to investigate whether its antidepressant efficacy is associated with the regulation of microglial polarization.
Results
Fasudil reduces depressive-like behavior
To assess the affective consequences of cerebral I/R injury, depressive-like behaviors were evaluated in mice following cerebral I/R (Chang et al., 2023; Zhang et al., 2024). As shown in Figure 1, cerebral I/R induced significant depressive-like behavior in mice, as indicated by reductions in the mean moving distance, mean speed, and number of line crossings in the open-field test (OFT), along with increased immobility time in both the forced swimming test (FST) and tail suspension test (TST) (compared with the sham group, p < 0.05).

Given that LPS treatment is known to induce microglial polarization towards the M1 phenotype and trigger a robust neuroinflammatory response (Chen et al., 2022), cerebral I/R mice were administered with LPS to evaluate the role of ROCK inhibition on M1/M2 microglial polarization and cerebral I/R injury. As shown in Figure 1, LPS significantly exacerbated the cerebral I/R-induced depressive-like behaviors. Notably, treatment with the ROCK inhibitor fasudil (10 and 20 mg/kg) effectively attenuated these behavioral deficits, suggesting that ROCK inhibition ameliorates depressive-like behaviors after cerebral I/R.
Fluoxetine, a clinically used selective serotonin reuptake inhibitor (SSRI), was employed as a positive control based on its established antidepressant efficacy in rodent models (Ma et al., 2025). Consistent with its known properties and our previous findings that fluoxetine promotes microglial polarization towards the M2 subtype (Deng et al., 2025), administration of fluoxetine (20 mg/kg) similarly alleviated depressive-like behaviors in the present model.
Fasudil ameliorates mouse cerebral I/R injury
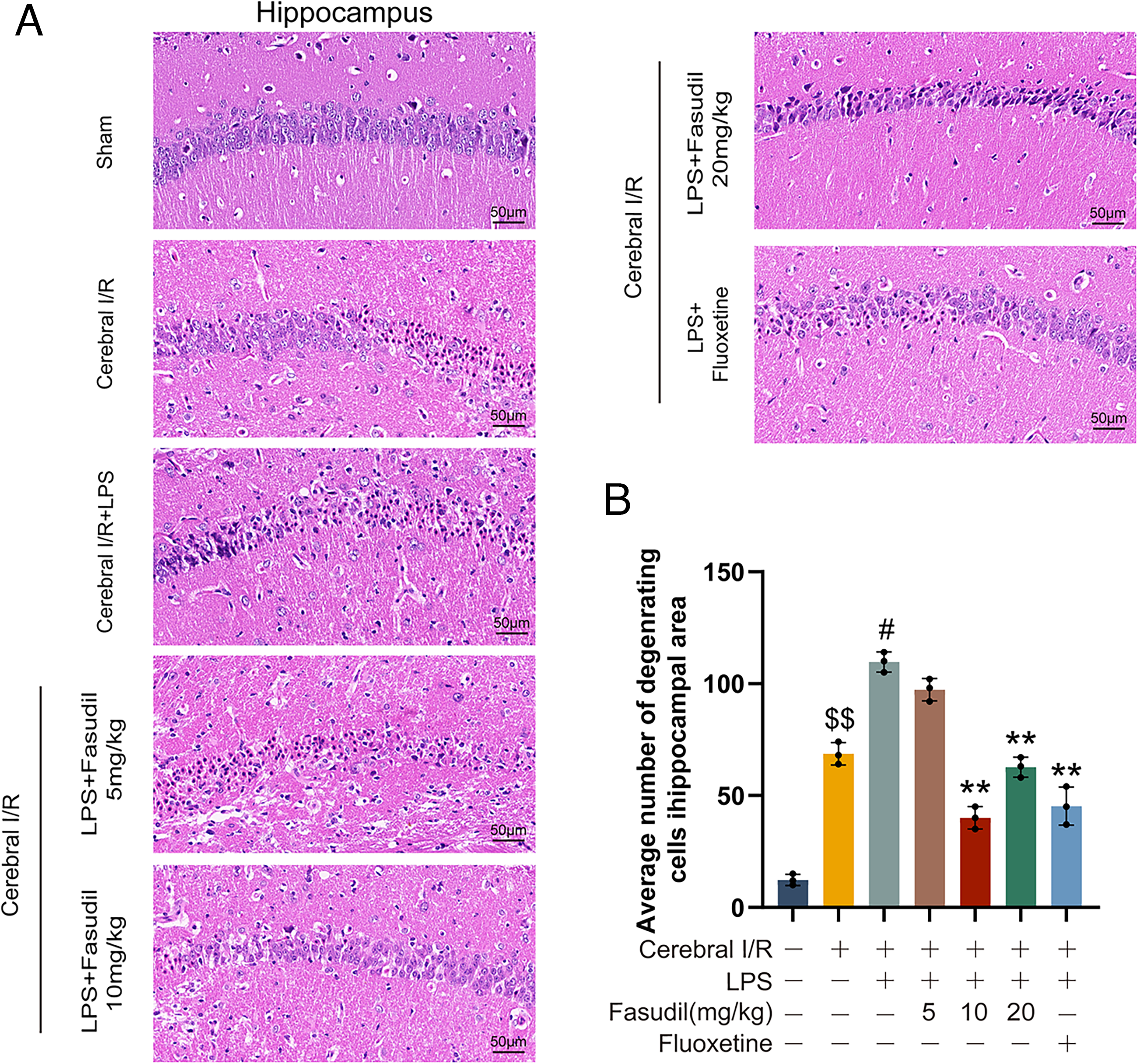
Neuronal injury following cerebral I/R was evaluated by hematoxylin and eosin (H&E) staining, and degenerating neurons in the hippocampal CA1 region were quantified according to established criteria (Yin et al., 2025). As shown in Figure 2, cerebral I/R resulted in a significant increase in neuronal degeneration compared to the sham group (p < 0.01), confirming successful model establishment. LPS administration further exacerbated this injury. In contrast, treatment with either fasudil (10 or 20 mg/kg) or fluoxetine (20 mg/kg) markedly reduced neuronal degeneration, demonstrating their neuroprotective efficacy.
Fasudil promotes M2 microglial polarization
Fasudil was administered to mice to investigate the effects of ROCK inhibition in M1 and M2 microglial polarization following cerebral I/R. As shown in Figure 3, cerebral I/R injury upregulated the expression of the microglial marker ionized calcium-binding adapter molecule-1 (Iba-1), the M1 markers CD16 and inducible nitric oxide synthase (iNOS), and the M2 marker CD206 and arginase 1 (Arg-1) in the mouse hippocampus (compared to the sham group, p < 0.05), indicating concurrent activation of both phenotypes. Administration of LPS further enhanced the expression of Iba-1, iNOS, and CD16. These results confirm the role of LPS in promoting M1 polarization.
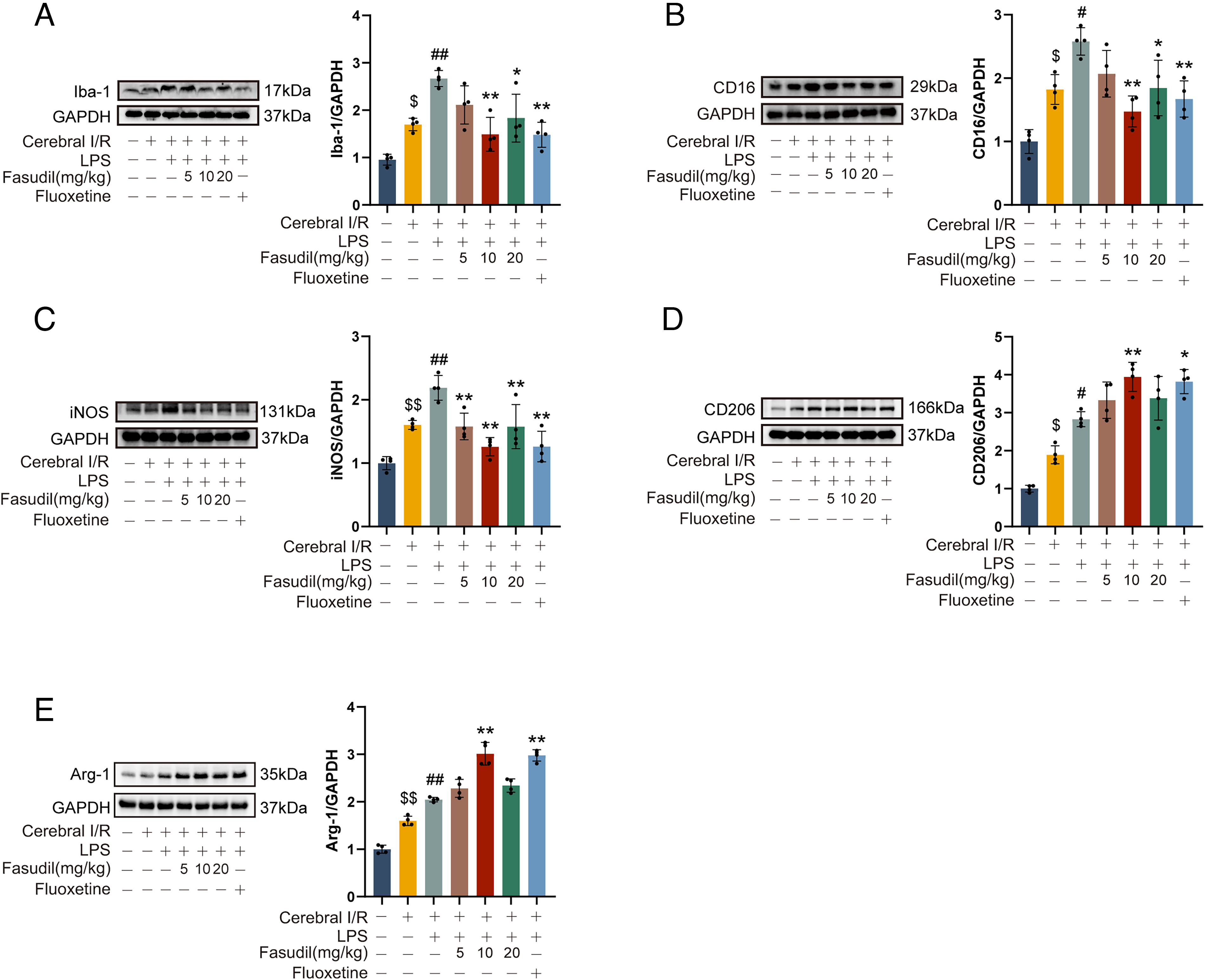
Not surprisingly, treatment with fasudil (10 and 20 mg/kg) significantly suppressed the LPS-enhanced M1 markers (Iba-1, iNOS, CD16) and increased the expression of CD206 and Arg-1. These results indicate that ROCK inhibition effectively shifts microglial polarization from the pro-inflammatory M1 toward the anti-inflammatory M2 phenotype. Similarly, fluoxetine treatment also enhanced CD206 and Arg-1 expression and reduced M1 markers, supporting its role in promoting M2 polarization.
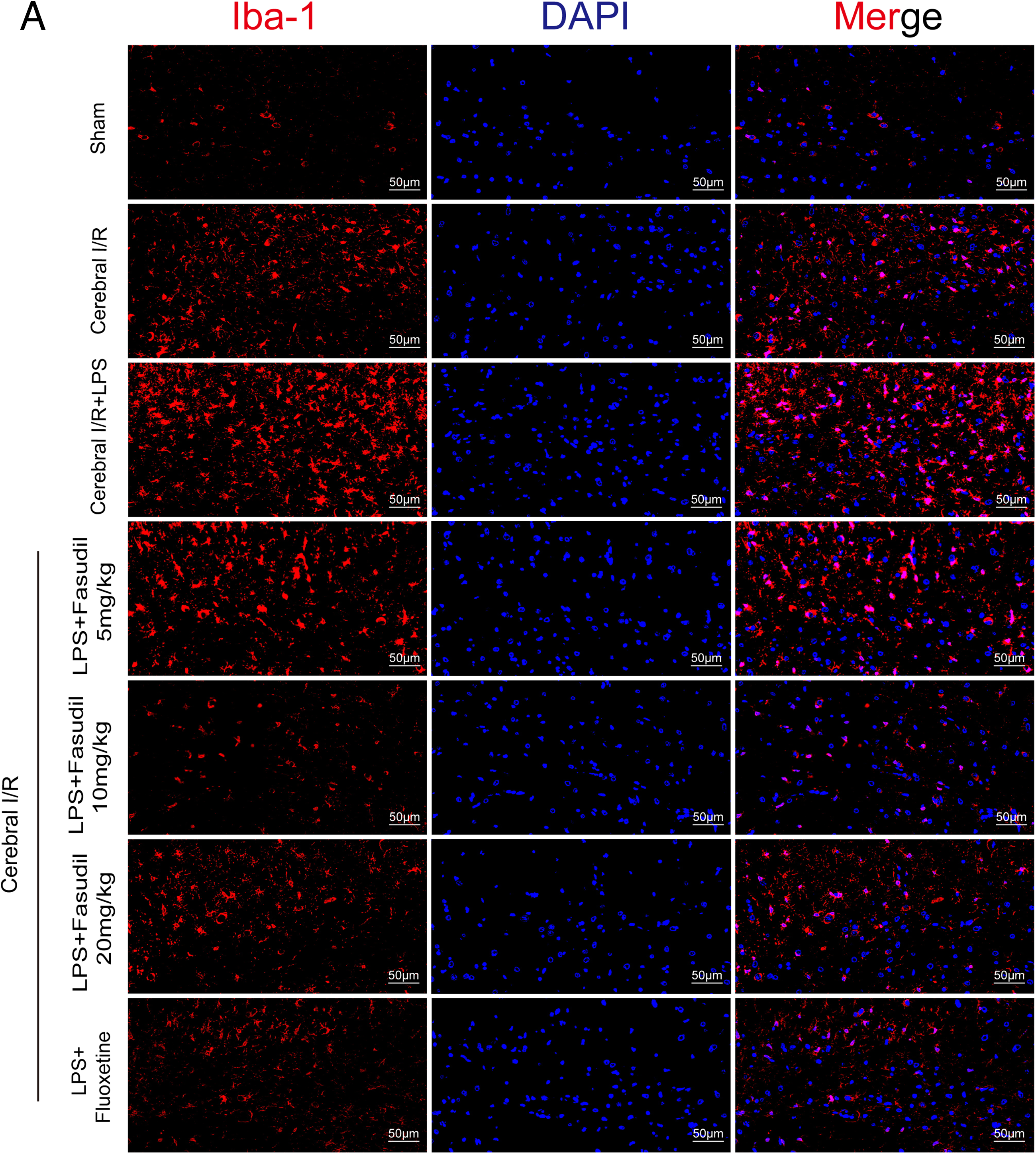
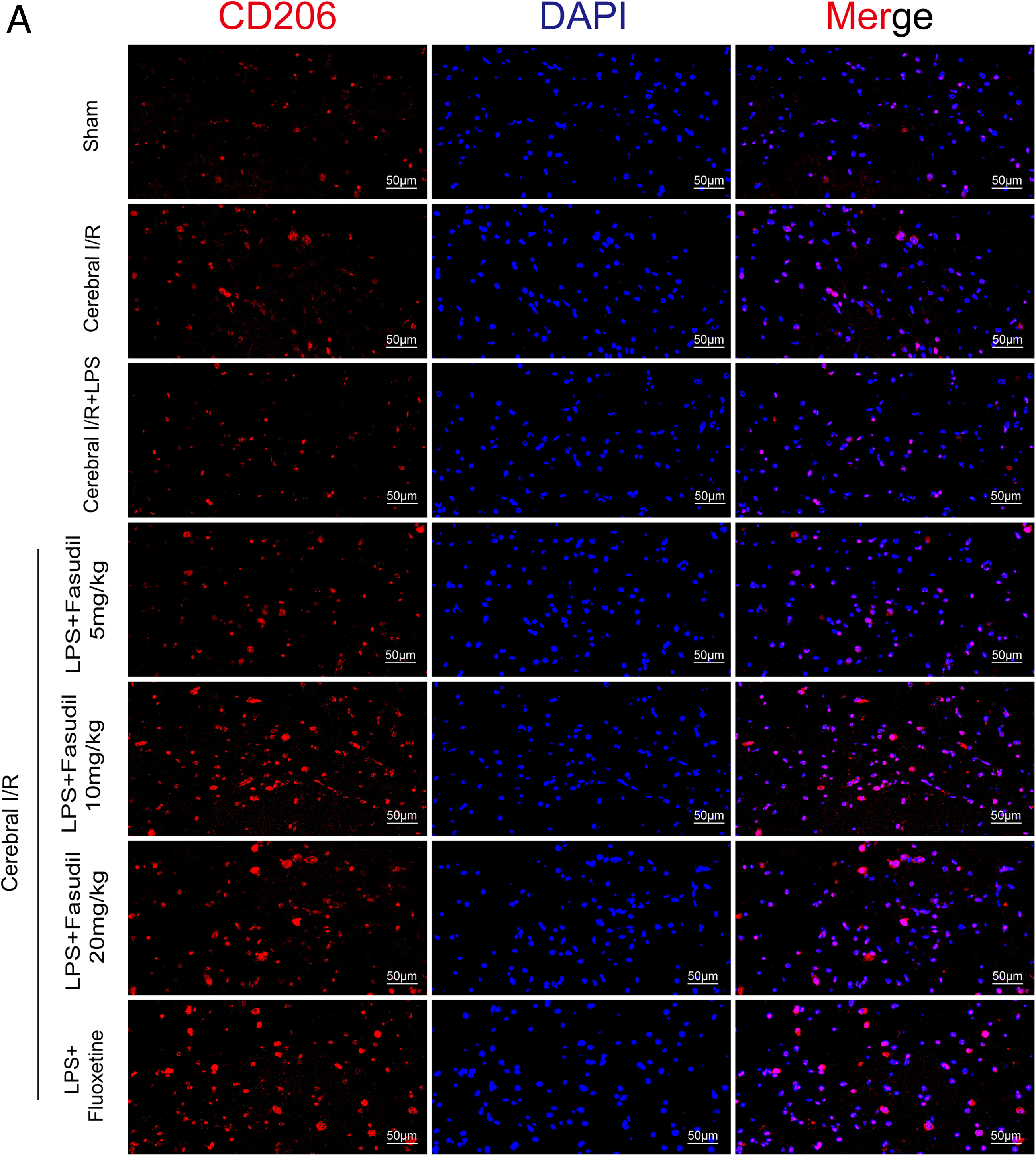
Immunofluorescence staining (Figs. 4, 5, 6, and 7A–C) provided validation of these molecular changes. Cerebral I/R injury led to a significant increase in the number of Iba-1-positive, CD16-positive, and CD206-positive cells in the hippocampus of mice. LPS administration intensified M1 polarization, as evidenced by a further increase in CD16-positive cells. Conversely, treatment with either fasudil or fluoxetine increased the number of CD206-positive cells while decreasing CD16-positive and Iba-1-positive cells in LPS-treated cerebral I/R mice. This evidence confirms that ROCK inhibition promotes M1-to-M2 phenotype shift. Additionally, we observed in the present study that the 20 mg/kg dosage of fasudil exhibited reduced efficacy compared to the 10 mg/kg dose, suggesting an inverted U-shaped dose-response relationship between fasudil and M2 microglial polarization.


Finally, an in vitro OGD/R model without LPS was used to further validate the effect of fasudil on microglial polarization. As shown in Figure 7D–F, OGD/R upregulated the expression of Iba-1, iNOS, and CD206 in microglia (p < 0.01 vs. control group), suggesting concurrent M1 and M2 activation. Treatment with fasudil (5 and 10 μmol/L) selectively suppressed the OGD/R-induced expression of Iba-1 and iNOS (M1 marker) while further enhancing CD206 (M2 marker) expression (p < 0.01 vs. OGD/R). Moreover, fasudil also significantly improved microglial cell viability under OGD/R (Fig. 7G). Together, these in vitro data confirm that ROCK inhibition directly induces microglial polarization toward the M2 phenotype, independent of other CNS cell types.
ROCK inhibition induces the expression of NOX2 and TXNIP in mouse hippocampal tissues
To elucidate through which ROCK2 inhibition promotes M2 microglial polarization, we performed RNA-Sequencing (RNA-Seq) on hippocampal tissues obtained from sham, cerebral I/R + LPS, and cerebral I/R + LPS + fasudil mice at 9 days after cerebral I/R. Differentially expressed genes (DEGs) were identified using thresholds of a false discovery rate (FDR) p value < 0.05 and |log2(fold change) | >1. Comparative analysis revealed 1339 DEGs between the sham and cerebral I/R + LPS groups (Fig. 8A), 611 DEGs between the cerebral I/R + LPS and cerebral I/R + LPS + fasudil groups (Fig. 8B), with 263 DEGs overlapping DEGs across all three conditions (Fig. 8C). Hierarchical clustering revealed distinct transcriptomic profiles for each experimental group (Fig. 8D).

Additionally, Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were conducted to identify biological pathways associated with these DEGs. The top 20 most significantly enriched KEGG pathways and top 20 enriched terms from each GO category across the comparisons were selected for detailed interpretation and presented as bubble diagrams in Figure 9. Several key pathways and genes implicated in neuroinflammation and oxidative stress were modulated by fasudil. Notably, the gene encoding NOX2, termed CYBB or gp91phox, was associated with enriched pathways such as “Phagosome” and “AGE-RAGE signaling pathways in diabetic complications” (Fig. 9A). NOX2 expression was elevated in cerebral I/R + LPS mice and attenuated by fasudil treatment. Similarly, TXNIP (thioredoxin interacting protein), a regulator of oxidative stress and inflammation linked to the GO terms “Response to mechanical stimulus” and “Epithelial cell differentiation”, was upregulated by cerebral I/R + LPS and downregulated by fasudil treatment (Fig. 9B). The complete inventories of all statistically enriched KEGG pathways and GO terms are provided in Supplementary Tables S1, S2, S3, and S4. Given that NOX2-derived ROS are known to promote M1 microglial polarization (Shi et al., 2024), we further examined NOX2 expression at the protein levels in mouse hippocampal tissues. As shown in Figure 8E, cerebral I/R injury significantly upregulated NOX2 expression compared with the sham group (p < 0.01), and this effect was further enhanced by LPS administration. Notably, fasudil treatment reduced NOX2 expression in the hippocampal tissues of LPS-exposed cerebral I/R mice.
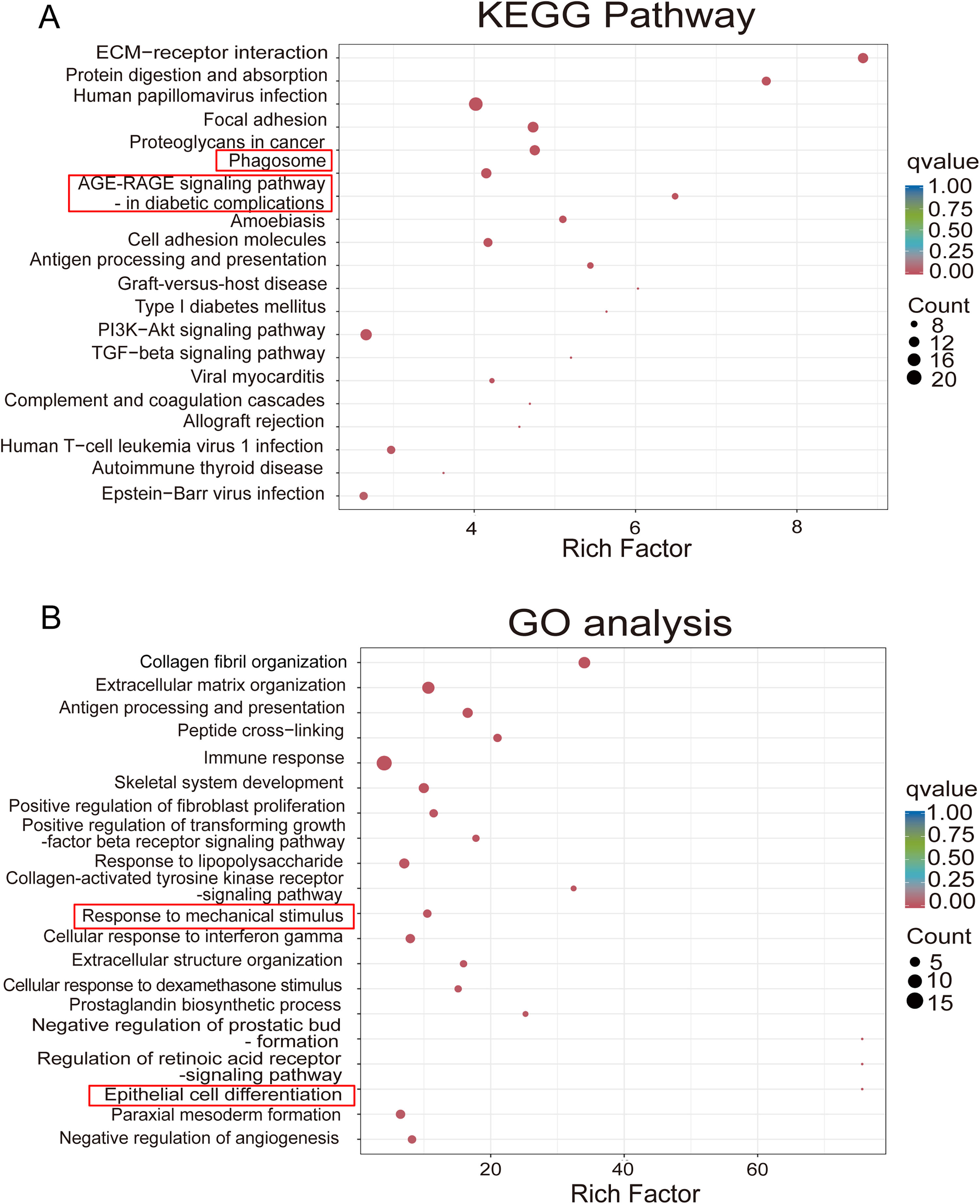
To further clarify the role of ROCK inhibition in regulating NOX2 and TXNIP expression, ROCK2 knockdown (ROCK2+/−) mice were subjected to the cerebral I/R model. Knockdown efficiency of ROCK2 was confirmed by assessing ROCK2 mRNA (Yin et al., 2025) and protein levels (Fig. 10A). As shown in Figure 10B and C, cerebral I/R induced a significant upregulation of both NOX2 and TXNIP in hippocampal tissues compared to the sham group (p < 0.01). Notably, ROCK2 deficiency markedly reduced the expression of NOX2 and TXNIP.

ROCK2 knockdown promotes the microglial polarization towards the M2 subtype
To further demonstrate the role of ROCK2 knockdown in modulating microglial phenotypic polarization, the expression levels of Iba-1, CD16, and CD206 were evaluated in hippocampal tissues of both wild-type (WT) and ROCK2+/− mice. As shown in Figure 10D–F, ROCK2 knockdown significantly suppressed the expression of Iba-1 and the M1 marker CD16, while enhancing CD206 expression, compared to the WT cerebral I/R group (p < 0.05). These results indicate that ROCK2 knockdown promotes a shift in microglial polarization from the pro-inflammatory M1 phenotype toward the anti-inflammatory M2 phenotype.
ROCK2 knockdown inhibits expression and phosphorylation of NF-κB P65 at ser 276
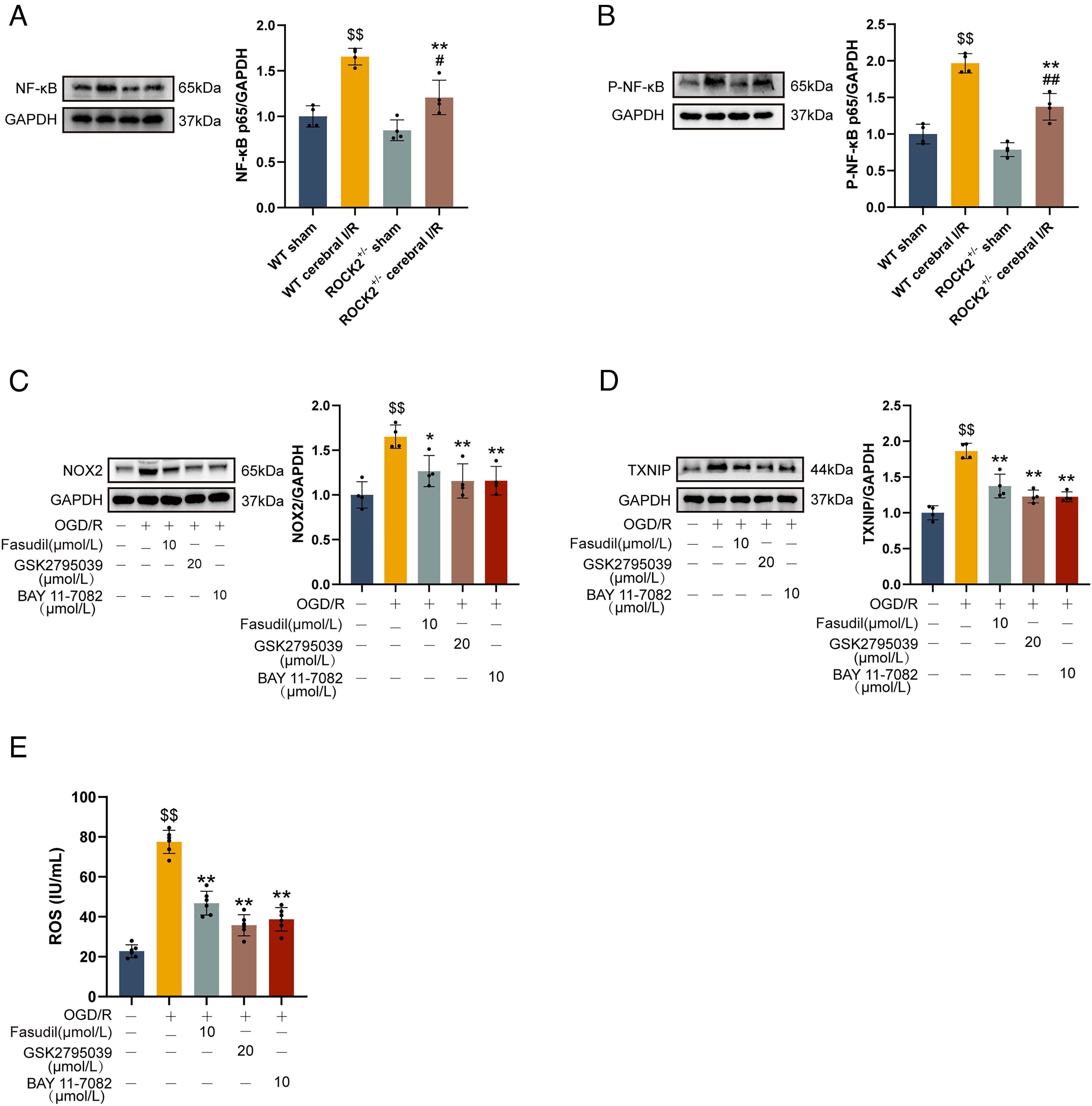
Previous studies have demonstrated that the activation of NF-κB pathway upregulates the expression of NOX2 and TXNIP (Liu et al., 2023b; Ye et al., 2024). Therefore, we examined the effect of ROCK knockdown on the expression and phosphorylation status of NF-κB P65 using ROCK2+/−mice. As shown in Figure 11A and B, cerebral I/R significantly increased both the total protein level and the phosphorylation of NF-κB P65 at Ser 276 compared to the WT (ROCK2+/+) sham group (p < 0.05). Notably, ROCK2 knockdown suppressed the expression of NF-κB p65 and attenuated its phosphorylation at Ser276 compared with WT mice subjected to cerebral I/R (p < 0.01).
NF-κB inhibitor suppresses the NOX2/ROS axis and TXNIP expression in OGD/R-treated microglia
The specific NF-κB inhibitor BAY11-7082 was used to explore whether the NF-κB pathway mediates the regulation of the NOX2/ROS axis and TXNIP expression in microglial cells under OGD/R conditions. As illustrated in Figure 11C and D, treatment with BAY11-7082 (10 μmol/L) significantly suppressed the expression of both NOX2 and TXNIP in OGD/R-exposed microglial cells (p < 0.01 vs. OGD/R group). Consistent with the suppression of NOX2, BAY11-7082 also inhibited ROS levels in the culture supernatant of OGD/R-treated microglial cells (Fig. 11E, p < 0.01 vs. the OGD/R group). Similarly, ROCK inhibitor fasudil likewise reduced the expression of NOX2 and TXNIP, as well as ROS levels. These findings suggest that the downregulation of NOX2 and TXNIP expression mediated by ROCK inhibition is associated with suppression of the NF-κB pathway.
NOX2/ROS inhibition suppresses TXNIP expression in microglial cells
To determine whether the inhibitory effect of ROCK inhibition on TXNIP expression is mediated through the NOX2/ROS axis, BV-2 mouse microglial cells were treated with the specific NOX2 inhibitor GSK2795039 (Wang et al., 2023a). Not surprisingly, OGD/R induced a significant increase in TXNIP expression in microglia compared to the normal control group (p < 0.01; Fig. 11D). Both GSK2795039 (20 μmol/l) and fasudil (10 μmol/l) effectively reduced NOX2 expression in OGD/R-treated microglia and decreased ROS levels in the culture supernatant (compared with OGD/R group, p < 0.01; Fig. 11C and E). Importantly, the increased TXNIP expression observed in OGD/R-treated microglia was also inhibited by both GSK2795039 and fasudil (compared with the OGD/R group, p < 0.01; Fig. 11D). These results demonstrate that TXNIP is a downstream effector of the NOX2/ROS axis, and that the suppression of TXNIP by ROCK inhibition is, at least in part, mediated through the inhibition of this axis.
Discussion
Microglia, the resident immune cells of the CNS, play a crucial role in maintaining brain homeostasis through continuous surveillance of the neural environment (Madore et al., 2020). Shifting microglial polarization from the M1 to the M2 phenotype represents a promising therapeutic strategy for cerebral I/R injury. ROCK is a well-established regulator of various cellular processes in neurons and blood-brain barrier (BBB) components within the CNS (Chen et al., 2025). Its expression is upregulated in brain tissues during cerebral ischemia, particularly in early stages (Shi et al., 2026). Pharmacological inhibition of ROCK has demonstrated neuroprotective effects in animal models of cerebral I/R injury, including reduced infarct volume and improved functional outcomes (Liu et al., 2025; McKerracher et al., 2020). Our present study elucidates a novel mechanism by which ROCK inhibition confers neuroprotection following cerebral I/R injury. We demonstrate that pharmacological inhibition of ROCK or genetic knockdown of ROCK2 shifts microglial polarization from a detrimental M1 state toward a protective M2 phenotype.
Crucially, we revealed that the modulatory effect of ROCK inhibition on microglial phenotypes correlates with improved outcomes of cerebral I/R, including reduced neuronal damage and alleviation of post-stroke depressive-like behaviors. PSD is a common and frequent sequela of ischemic stroke, and is positively correlated with impaired cognitive function, slow functional rehabilitation, high mortality, and poor quality of life after stroke (Partoazar et al., 2021). Accumulating studies have revealed an association between depressive-like behaviors and M1 microglial polarization, whereas facilitation of the M2 phenotype has been shown to ameliorate such behaviors (Yang et al., 2024). Our findings support the concept that ROCK inhibition-mediated M2 microglial polarization alleviates depressive-like behaviors following cerebral I/R.
Notably, we observed that LPS challenge potentiated M1 microglial polarization in mouse hippocampus after cerebral I/R. Treatment with the ROCK inhibitor fasudil, however, not only countered this M1 polarization but also concomitantly enhanced M2 polarization. These results suggest that inhibiting ROCK activity can redirect microglial polarization from a detrimental M1 toward a protective M2 phenotype. These data extend previous findings from our group and others on the general anti-neuroinflammatory effects of ROCK inhibition (Yin et al., 2025; Zhang et al., 2022), by pinpointing the reprogramming of microglial phenotype as a critical cellular mechanism. This aligns with the understanding that microglia possess dynamic plasticity in the pathological context of stroke (Tian et al., 2022).
While intraperitoneal LPS administration is a well-established model for studying neuroinflammation (Verma et al., 2024), M1 microglial polarization (Si et al., 2024), and depressive-like behaviors (Liu et al., 2024), it is important to acknowledge a potential confound in interpreting our data. Systemic LPS challenges not only induce microglial M1 polarization but also provoke a robust peripheral acute-phase response and sickness behavior (Konsman et al., 2008). Consequently, the observed behavioral deficits and microglial polarization may be influenced by peripheral-derived cytokines infiltrating the brain parenchyma through a compromised BBB (Li et al., 2023b).
To determine whether the effects of fasudil on microglial polarization are cell-autonomous and independent of peripheral inflammatory signals, we examined the response of microglia to fasudil under OGD/R conditions in the absence of LPS. Strikingly, fasudil treatment alone significantly shifted microglial polarization toward the M2 phenotype, as evidenced by increased expression of M2 marker CD206 and decreased expression of M1 marker iNOS. These findings indicate that fasudil can directly modulate microglial function without the confounding influence of systemic LPS-induced inflammation.
In addition to the considerations regarding LPS-induced systemic inflammation, another important aspect in interpreting in vivo findings is the cellular specificity of ROCK inhibition. ROCK is expressed not only in microglia but also in neurons and astrocytes, raising the possibility that the observed microglial M2 shift could be an indirect consequence of fasudil’s effects on other cell types. Our in vitro data using a microglial OGD/R model address this concern by demonstrating that fasudil exerts direct, cell-autonomous effects on microglial polarization, independent of neuronal or astrocytic signaling. Nevertheless, we acknowledge that in the complex in vivo environment, indirect effects may also contribute. Future studies employing cell-type-specific conditional knockout mice (e.g., microglial-specific ROCK2 KO) will be essential to fully dissect the relative contributions of direct versus indirect mechanisms.
After CNS injury, microglia at the lesion site initially undergo M2 polarization, a state associated with neuroprotective and reparative functions. However, approximately one week after cerebral ischemic injury, a phenotypic shift towards the M1 state occurs. M1 microglial polarization exacerbates neural damage through sustained release of pro-inflammatory mediators (Tozihi et al., 2023). In the present study, we found that ROCK inhibition promotes M2 microglial polarization both in vivo and in vitro. Notably, ROCK inhibition not only exerted this effect in mouse hippocampus at 9 days post-cerebral I/R, a subacute time point typically dominated by a shift towards the detrimental M1 phenotype (Huang et al., 2017; Petrovic-Djergovic et al., 2016). This interference with the phenotypic shift highlights the potential of targeting ROCK to extend the therapeutic window for stroke recovery.
To decipher the molecular pathway underlying ROCK2-mediated microglial phenotypic shift, we used RNA-seq, a high-throughput next-generation sequencing (NGS) technology (Ozsolak and Milos, 2011; Wang et al., 2024), to identify the DEGs in hippocampal tissues of mice and revealed the upregulation of TXNIP and NOX2 genes enriched in several pathways in hippocampal tissue of cerebral I/R mice exposed to LPS. Fasudil treatment decreased the expression of both TXNIP and NOX2 in mice exposed to LPS. As reported in previous studies, both TXNIP and NOX2 are established contributors to neuroinflammation, oxidative stress, and microglial activation (Tu et al., 2023a; Zhuo et al., 2023). Increased oxygen supply after cerebral I/R induces a surge in inflammatory factors and oxidative stress, which promotes the M1 microglial polarization (Wang et al., 2023b) and exacerbates cerebral I/R injury (Yang et al., 2023).
NOX2, the predominant isoform of NADPH oxidase, is highly expressed in microglia (Bedard and Krause, 2007) and catalyzes the production of ROS, a process known as the oxidative burst (Sareila et al., 2011). During early inflammatory responses, activation of NOX2 and subsequent ROS generation promote microglial polarization toward the M1 phenotype (Griffiths et al., 2017). Conversely, NOX2 deficiency has been shown to facilitate microglial polarization toward the M2 phenotype following traumatic brain injury in mice (Kumar et al., 2016). TXNIP, initially identified as an endogenous inhibitor of the thioredoxin system (Panda et al., 2025), is upregulated following ischemic stroke and exacerbates neuroinflammation by promoting microglial activation (Zhuo et al., 2023). Elevated TXNIP expression is likewise associated with oxidative stress and M1-like microglial transformation (Chen et al., 2024).
In order to further confirm the relationship between ROCK inhibition and NOX2 and TXNIP in the microglial polarization, we next examined the effect of ROCK inhibition on the expression of NOX2 and TXNIP at protein levels using ROCK2 knockdown (ROCK2+/−) mice, as the ROCK2 isoform is predominantly expressed in brain tissues (Yuan et al., 2023) and complete deletion of ROCK2 leads to placental dysfunction and intrauterine growth retardation, resulting in ∼90% embryonic lethality in C57BL/6N and 129/Sv mouse strains (Julian and Olson, 2014). Our results showed that expression of both NOX2 and TXNIP was upregulated in hippocampal tissues of cerebral I/R mice, whereas ROCK2 knockdown significantly inhibited their expression. These findings suggest that ROCK inhibition-mediated M2 microglial polarization is related to inhibition of NOX2 and TXNIP. Strikingly, the direct effect of ROCK inhibition on microglial polarization was further corroborated in vivo that ROCK2 knockdown similarly promoted M2 polarization without LPS challenge. Collectively, these findings demonstrate that inhibiting the ROCK pathway intrinsically drives microglial M2 programming, independent of the confounding influences of systemic LPS-induced inflammation.
Furthermore, we likewise focused on NF-κB, a master regulator of ischemic stroke-induced brain injury (Kong et al., 2022), and found that ROCK2 knockdown inhibited NF-κB expression and reduced phosphorylation of NF-κB p65 at Ser276 in the hippocampus of cerebral I/R mice. NF-κB is an inducible transcription factor involved in the microglial response to cerebral ischemia stimuli, thereby promoting microglia-mediated neuroinflammation (Kong et al., 2022). The transcriptional activity of NF-κB is positively modulated by phosphorylation of p65 at Ser 276, and decreased phosphorylation of p65 at this site reduces the activity of NF-κB p65 (Lin et al., 2021). Accumulating studies have revealed that upregulation of NOX2 and TXNIP is strongly correlated with activation of the NF-κB pathway (Liu et al., 2023b; Ye et al., 2024). These observations indicate that the downregulation of TXNIP and NOX2 mediated by ROCK inhibition decreased expression may occur by targeting NF-κB pathway.
To further investigate the interplay among NF-κB, NOX2/ROS axis and TXNIP, BV-2 microglial cells exposed to OGD/R were treated with the NF-κB inhibitor BAY11-7082 (Geng et al., 2025) or the NOX2 inhibitor GSK2795039 (Singh et al., 2025). Our results revealed that BAY11-7082 markedly suppressed the expression of NOX2 and TXNIP in OGD/R-treated microglia and reduced ROS production. Furthermore, GSK2795039 treatment also decreased ROS production and reduced TXNIP expression in microglial cells under OGD/R conditions. These findings suggest that the mechanisms by which ROCK inhibition promotes microglial polarization towards the M2 phenotype may involve suppression of NF-κB, thereby suppressing the NOX2/TXNIP pathway.
Furthermore, we found that fluoxetine, used as positive control, promoted the polarization state of microglia toward the M2 subtype in vivo. Previous reports support the plausibility of our observations. We previously demonstrated that fluoxetine effectively shifted microglial polarization toward the M2 phenotype following cerebral I/R (Deng et al., 2025). Consistently, Wang et al. reported that fluoxetine and S-citalopram suppress M1 and promote M2 microglial activation at both transcriptional and translational levels (Su et al., 2015). Similarly, fluvoxamine, another SSRI, has been found to promote M2 macrophage/microglial polarization in both in vivo and in vitro inflammatory models (Shi et al., 2022).
Taken together, these previous findings, combined with our current data, suggest that the modulation of microglial polarization may represent a previously underappreciated mechanism underlying the broader neuroprotective effects of fluoxetine. Nevertheless, several important questions remain. For example, whether fluoxetine regulates microglial polarization via direct serotonergic signaling on glial cells, or through indirect neuronal modulation, requires further investigation.
Moreover, an unexpected finding of the present study is that fasudil at 20 mg/kg appeared less effective than the 10 mg/kg regimen in ameliorating cerebral I/R-induced depressive-like behavior and regulating microglial polarization, challenging the typical expectation of a monotonic dose-response relationship. This inverted U-shaped or biphasic pattern, while counterintuitive, is not unprecedented in CNS pharmacology. For instance, biphasic dose-response effects are well-documented for many centrally acting drugs. For instance, 17-beta-estradiol (E2) enhances hippocampal plasticity in animals in a monotonically increasing (dose-dependent) manner, but this relationship can also exhibit an inverted U-shaped function, depending on the hippocampal subregion examined (Bayer et al., 2018). Similarly, ketamine exerts an inverted U-shaped dose-response in antidepressant-sensitive behavioral tests (Zanos et al., 2023).
One potential mechanism underlying the reduced efficacy of fasudil observed at 20 mg/kg compared to 10 mg/kg is that the lower dose may already achieve saturation of fasudil’s primary therapeutic targets. Administration of a higher dose could lead to receptor overactivation, potentially triggering rapid desensitization. Alternatively, higher doses of fasudil may engage off-target effects that counteract its therapeutic benefits. Fasudil has been reported to affect other kinases, such as inhibiting p38 mitogen-activated protein kinase (p38 MAPK) (Kondo et al., 2014) and to activate ion channels, including the voltage-gated potassium channel subunit Kv7.4 (Liu et al., 2023a). These potential confounding effects from non-ROCK targets may disrupt the finely balanced signaling networks required for optimal behavioral recovery and microglial polarization. To validate this hypothesis, future studies should systematically investigate the molecular, pharmacological, and metabolic alterations induced by high-dose fasudil administration.
Despite these limitations, the present study provides the first evidence that fasudil reduces depressive-like behavior of mice following cerebral I/R and that these effects are associated with modulation of microglial polarization. The unexpected dose-response pattern observed here highlights the complexity of translating preclinical dosing to clinical regimens and underscores the need for rigorous dose-finding studies in future translational research. Addressing the limitations outlined above will be essential to fully elucidate the mechanisms underlying the antidepressant effects of fasudil and to optimize its therapeutic potential.
Conclusion
We demonstrate that ROCK inhibition promotes microglial polarization from the M1 to the M2 phenotype, representing a promising therapeutic strategy for mitigating cerebral I/R injury. The protective effects of ROCK inhibition are mediated through suppression of the NOX2/ROS/TXNIP signaling axis via inhibition of the NF-κB pathway.
Innovation
Although our previous studies demonstrated that fasudil attenuates cerebral I/R injury in mice and suppresses pro-inflammatory responses in astrocytes subjected to OGD/R, the role of ROCK inhibition in microglial phenotype switching remained unclear. This study provides the first evidence that ROCK inhibition induces a shift in microglial polarization from the M1 to the M2 phenotype by suppressing the NF-κB/NOX2/ROS/TXNIP signaling pathway.
Materials and Methods
Animal model
WT C57BL/6J mice (6–8 weeks old, weighing 18–22 g) were obtained from the Animal Center of Anhui Medical University. Heterozygous ROCK2 knockout (ROCK2+/−) mice (6–8 weeks old, weighing 18–22 g) were provided by Shanghai Southern Model Biological Technology Co., Ltd. WT C57BL/6J and ROCK2+/− mice were housed under standard conditions with a 12-h light/dark cycle, at 22 ± 1°C and 55 ± 5% humidity, with free access to food and water. All experimental procedures were approved by the Ethics Committee of Anhui Medical University and conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 2011).
Mouse cerebral I/R model was established via transient occlusion of the bilateral common carotid artery (2-VO) under anesthesia. Briefly, mice were anesthetized by intraperitoneal injection of 1.2% tribromoethanol (0.2 mL per 10 g body weight). Ischemia was induced by 2-VO for 1 h, followed by removal of the occlusion to allow reperfusion for nine days (Zhang et al., 2021) (Fig. 12). At the end of the reperfusion period, mice were deeply anesthetized with 1.2% tribromoethanol (0.2 mL/10 g) and euthanized by cervical dislocation. Brains and serum samples were collected for subsequent biochemical and histological analyses. Reagents and drugs were shown in Supplementary Data.

Experimental protocol of mouse experiment. Fasudil or fluoxetine were administered once daily for 8 consecutive days, starting from the second day after acute cerebral I/R. Mice in the LPS treatment group were administered LPS for three consecutive days, starting from the third day after cerebral I/R. On the eighth day, mice were subjected to the open field test, tail suspension test, and forced swimming test.
Cell culture
BV-2 microglial cells were cultured in high-glucose DMEM supplemented with L-glutamine, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin, and maintained at 37°C in a 5% CO2 incubator. Cells were assigned to the following groups: Control, OGD/R, OGD/R + Fasudil (2.5 μmol/L), OGD/R + Fasudil (5 μmol/L), and OGD/R + Fasudil (10 μmol/L). Concentrations of fasudil were selected based on our previous work (Li et al., 2023a). In a parallel experiment, cells were randomly divided into: Control, OGD/R, OGD/R + Fasudil (10 μmol/L), OGD/R + GSK2795039 (25 μmol/L), and OGD/R + BAY 11–7082 (10 μmol/L). Concentrations of GSK2795039 and BAY 11–7082 were determined according to our previous studies (Deng et al., 2025; Geng et al., 2025) and preliminary experimental data.
Cells in the control group were maintained under normal culture conditions. Cells in OGD/R groups were exposed to OGD for 2 h in glucose-free medium within a tri-gas incubator (94% N2, 5% CO2, 1% O2) (Xu et al., 2021). Subsequently, the glucose-free medium was replaced with high-glucose culture medium, and cells were returned to normoxic conditions for 22 h of reoxygenation. Pharmacological agents, including fasudil, GSK2795039, and BAY 11-7082, were administered at the start of the reoxygenation phase.
Drug treatments
WT C57BL/6J mice were randomly assigned to seven experimental groups (n = 10, with equal numbers of male and female): sham, cerebral I/R, cerebral I/R + LPS (2 mg/kg), cerebral I/R + LPS + Fasudil (5 mg/kg), cerebral I/R + LPS + Fasudil (10 mg/kg), cerebral I/R + LPS + Fasudil (20 mg/kg), and cerebral I/R + LPS + Fluoxetine (20 mg/kg). Fluoxetine, a clinically established antidepressant, served as the positive control (Capitao et al., 2023). Doses of LPS and fluoxetine were chosen based on previous literature (Li et al., 2021) and preliminary experimental data. Fasudil doses were determined according to our earlier research (Ding et al., 2024).
Fasudil (5, 10, or 20 mg/kg) was administered intraperitoneally once daily, while fluoxetine (20 mg/kg) was given orally once daily. Both fasudil and fluoxetine were administered on the second day after cerebral I/R injury and continued for eight consecutive days. Mice in the LPS-treated groups received intraperitoneal injections of LPS for three consecutive days starting on day 3 post-injury. In another in vivo experiment, ROCK2+/− mice and WT mice were randomly divided into the following groups: WT Sham, WT Cerebral I/R, Het Sham, and Het Cerebral I/R.
Western blot assay
Total proteins were extracted from mouse hippocampal tissues or BV-2 microglial cells and quantified using commercial protein assay kits (Beyotime Biotechnology, Shanghai, China). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked for 1 h at room temperature with 5% skim milk in TBST buffer (pH 8.0; 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.1% Tween-20). Subsequently, membranes were incubated overnight at 4°C with primary antibodies against Iba-1, iNOS, CD16, CD206, and NADPH oxidase 2 (NOX2). After washing three times with TBST, membranes were incubated for 1 h with horseradish peroxidase (HRP)-conjugated secondary antibodies in TBST containing 5% skim milk. Protein bands were visualized using an enhanced chemiluminescence kit (Biosharp, Beijing, China). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control.
Open-field test
The OF was performed to evaluate locomotor activity and exploratory behavior in mice according to established protocols (Chang et al., 2024) with minor modifications. Briefly, each mouse (n = 7 per group) was individually placed in the open-field apparatus and allowed to acclimate for 3 min. During the subsequent 3 min test period, parameters including moving speed, total moving distance, and the time that experimental animals spent in the center square were recorded to assess locomotor activity and exploratory behavior of mice.
Forced swimming test
The FST was undertaken to evaluate depression-like behaviors in mice (Chen et al., 2023). A transparent cylindrical bucket (20 cm in height and 15 cm in diameter) was filled with water to a depth of 14 cm, maintained at 33–35°C. Each mouse (n = 7 per group) was gently placed into the bucket and allowed to swim freely for 6 min. Immobility time, defined as floating motionlessly or making movements necessary to keep the head above water, was recorded as an index of behavioral despair.
Tail suspension test
The TST was carried out according to previously described with minor modifications (Cheng et al., 2018). Each mouse (n = 7 per group) was suspended by the tail from an iron hook positioned 45 cm above the bench surface using adhesive tape. Animals typically display initial escape-oriented behaviors followed by periods of immobility. The total duration of immobility during a 6-minute test period was recorded to evaluate the depressive-like behavior of mice.
Hematoxylin and eosin staining
Hematoxylin and eosin (H&E) staining was performed to assess neuronal injury in the hippocampus of mice following cerebral I/R (Yin et al., 2023). Mouse brains were fixed in 4% paraformaldehyde (Biosharp, Hefei, China) for 48 h, dehydrated through a graded ethanol series, cleared in xylene, and embedded in paraffin. Subsequently, brains were sectioned coronally into transverse sections (4-μm thickness), which were dewaxed, rehydrated, and stained with H&E. Histopathological changes in the hippocampus, including nuclear condensation, vacuolar degeneration, and necrotic neurons, were examined and recorded as degenerating cells.
Immunofluorescence labeling
Mouse brains were randomly selected from each group (n = 3) and fixed in 10% formalin. Subsequently, brain samples were dehydrated, embedded in paraffin, and sectioned coronally at a thickness of 7 μm. Brain sections were dewaxed, rehydrated, and subjected to antigen retrieval using heated citrate buffer (pH 6.0). After blocking with a protein-blocking solution for 1 h at room temperature, brain sections were incubated overnight at 4°C with primary antibodies against CD206, CD16, or Iba-1. Nuclei were counterstained with 4’,6-diamino-2-phenylindole (DAPI). After three washes, sections were incubated with secondary antibodies: Cy3-conjugated goat anti-rabbit IgG and fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (GB21303) (Servicebio Hubei, China). Iba-1-positive cells, CD206-positive cells, and CD16-positive cells were counted and analyzed using Image J software (v1.8.0, National Institutes of Health, Bethesda, Maryland, USA).
Biochemical measurements and assessment of BV-2 microglial viability
The level of ROS in the supernatant of microglial cell cultures (n = 6) was quantified using commercial ELISA kits according to the manufacturer’s protocol. Briefly, samples were added to a pre-coated plate, which was sealed and incubated at 37°C for 30 min. After washing, 50 µL of enzyme-conjugated detection antibody was added to each well (except blanks) and incubated for another 30 min at 37°C. After a second wash, color was developed by sequentially adding 50 µL each of substrate solution A and B, followed by a 10-min incubation at 37°C in the dark. The reaction was terminated by adding 50 µL of stop solution, and the absorbance was immediately measured at 450 nm using a microplate reader, with the blank well as reference. Results from six independent replicates (n = 6) are expressed as IU/mL.
Microglial viability was assessed using the Cell Counting Kit-8 (CCK-8) assay. Cells were seeded in 96-well plates and subjected to the respective treatments. Following treatment, 10 µL of CCK-8 solution was added to each well, and plates were incubated at 37°C for 1 h. The absorbance at 450 nm was measured using a microplate reader. Cell viability was calculated as a percentage relative to the control group.
RNA sequencing and bioinformatic analysis
Total RNA was extracted from mouse hippocampal tissues using TRIzol Reagent (Life Technologies, California, USA). RNA concentration and purity were measured with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA integrity was assessed using the RNA Nano 6000 Assay Kit on an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Poly(A) + mRNA was enriched from total RNA using oligo(dT)-coated magnetic beads. Sequencing libraries were constructed using the Hieff NGS Ultima Dual-mode mRNA Library Prep Kit for Illumina [Yeasen Biotechnology (Shanghai) Co., Ltd.], and library quality was validated on the Agilent Bioanalyzer 2100 system. Whole-transcriptome RNA sequencing was performed on an Illumina platform by Beijing Biomarker Biotechnology Co., Ltd.
Raw reads were subjected to quality control, and clean reads were aligned to the GRCm38 (release 95) reference genome using HISAT2 software. Differential expression analysis was conducted with the DESeq2 package, with genes meeting the criteria of |log2(fold change) | > 1 and p value <0.05 considered differentially expressed genes (DEGs). Functional enrichment analysis of these DEGs was performed based on Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using the clusterProfiler package in R.
Statistical analysis
All data were analyzed using GraphPad Prism 8.0 software (GraphPad Software, RRID: SCR_002798) and are presented as mean ± standard deviation. Electronic laboratory notebook was not used in this study. Homogeneity of variance was assessed using Levene’s test, and normality of distribution was evaluated using the Shapiro–Wilk test. Comparisons between groups were performed by one-way ANOVA followed by Duncan’s post hoc test. A p value < 0.05 was considered statistically significant.
Authors’ Contributions
X.X. and Z.W.: Conceptualization, methodology, writing original draft, data analysis. F.H., L.W., B.G., and Z.L.: Investigation, methodology, formal analysis. J.W.: Supervision, project administration, funding acquisition, writing—review and editing.
Footnotes
Availability of Data and Materials
Data will be made available on request.
Ethics Approval and Consent to Participate
All experimental protocols in our study complied with the rules, which were set by the animal care and use committee of Anhui Medical University (Certification No. LLSC 20231096; Certification date: 1/3/2023).
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This study was supported by Natural Science Foundation of Colleges and Universities in Anhui Province (No. 2023AH050672) and the Natural Science Foundation of Anhui Province (NO. 2308085MH302).
Supplemental Material
Supplemental Material
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.