
Abstract
Background:
Dengue fever is one of the most widely distributed vector-borne infectious diseases globally, prevalent in tropical and subtropical regions. An estimated 3.9 billion people in 128 countries are at risk of infection globally, and the 2024 outbreak was the most severe. As a tropical region, Hainan Province in China serves as a significant endemic area for dengue fever, where epidemic prevention and control remain critical.
Objective:
To analyze the transmission dynamics and pathogen variation characteristics of the 2024 dengue fever outbreak in Hainan Province.
Methods:
Acute-phase serum samples from 18 confirmed dengue cases during the 2024 outbreak in Hainan Province were collected and serotyped using TaqMan real-time PCR. The viral envelope (E) gene and representative whole genomes were sequenced and compared with domestic reference strains to assess epidemic trends. Phylogenetic and molecular clock analyses were performed based on E gene and full-genome sequences to infer spatiotemporal transmission dynamics. Sequence alignment and homology modeling were used to identify E gene mutations and evaluate their potential effects on E protein functional domains and viral pathogenicity.
Results:
Among the 18 samples, 17 were dengue virus (DENV)-1, and 1 was DENV-3. Phylogenetic analysis revealed that the primary strains in this outbreak were highly homologous to those circulating in Guangdong Province; however, two strains exhibited significant genetic differences, suggesting possible independent introduction. Additionally, two unique DENV-1 E gene mutations (D147N and S338L) were identified, which we hypothesize may be associated with viral characteristics, subject to further functional verification.
Conclusion:
This study systematically reveals the epidemiological characteristics of the 2024 dengue fever outbreak and provides a scientific basis for formulating local control strategies and a research foundation for further exploring the molecular-level pathogenicity and transmissibility of DENV.
Introduction
Dengue fever is an acute, vector-borne infectious disease caused by the dengue virus (DENV), primarily transmitted by Aedes aegypti and Aedes albopictus mosquitoes. It is endemic in tropical and subtropical regions, affecting approximately 3.9 billion people in 128 countries (Cattarino et al., 2020). Common symptoms include fever, headache, rash, muscle and joint pain, nausea, and vomiting (Katzelnick et al., 2017). Severe or secondary infections with different serotypes can result in dengue hemorrhagic fever or shock syndrome (Zompi and Harris, 2013). Currently, no antiviral drugs are approved for treatment, making prevention the primary approach to reducing disease incidence (Akshatha et al., 2021).
DENV is a spherical, single-stranded positive-sense RNA virus belonging to the family Flaviviridae and genus Flavivirus (Diamond and Pierson, 2015). Its ∼10.7 kb genome contains a single open reading frame encoding three structural proteins—capsid, envelope (E), and precursor membrane—and seven nonstructural proteins (Iglesias and Gamarnik, 2011). The E protein, comprising approximately 495 amino acids, plays a central role in the viral life cycle by mediating receptor recognition and binding (Kuhn et al., 2002) and facilitating fusion between the viral E and host cell membrane (Anasir et al., 2020; Zhang et al., 2004). As the primary target of neutralizing antibodies, mutations in the E protein can influence viral infectivity, immune escape, and transmission efficiency (Sessions et al., 2015). Notably, amino acid substitutions at positions 324, 351, and 380 in domain III of the DENV-1 E protein have been shown to enhance viral infectivity, stability, and immune evasion in mammals, thereby promoting the global spread of this genotype (Jiang et al., 2024). Consequently, in-depth investigation of E protein mutations is critical for elucidating viral evolution, assessing epidemic risk, and guiding vaccine and antibody therapeutic development (Ali et al., 2025).
Based on antigenic differences in the E gene, DENV can be classified into four serotypes (DENV-1 to DENV-4), with amino acid sequence differences ranging from 25% to 40% (World Health Organization, 2009). Each serotype can be further subdivided into multiple genotypes based on genetic variations. For example, DENV-1 includes genotypes I–V, with genotype I further subdivided into lineages I–IV (Du et al., 2021). However, long-lasting cross-immune protection between different serotypes is lacking, resulting in individuals acquiring long-term immunity to one serotype after infection but remaining susceptible to others, which may even lead to more severe disease (Katzelnick et al., 2017). Therefore, accurately distinguishing DENV serotypes and genotypes is critically important for epidemic surveillance, viral tracing, vaccine development, and clinical warnings for severe cases (Katzelnick et al., 2015).
In 2024, the global dengue fever epidemic exhibited unprecedented growth, with over 12 million cumulative reported cases and more than 8000 deaths, representing a 230% increase compared to the same period in 2023 (Venkatesan, 2024), surpassing all historical records. Against this backdrop, Hainan Province, due to its frequent international exchanges, has become a typical high-incidence area for dengue fever in China’s tropical regions and a key focus for control strategies. In 2024, local dengue fever cases emerged successively in various hospitals in the region, yet understanding of the viral genomic information remains highly limited. Analyzing the viral genomic information from local cases in Hainan Province will enable the precise determination of the sources, types, and variations of circulating strains, providing a scientific basis for formulating regional control strategies and effectively addressing the dual challenges of international importation and local transmission. This study conducted viral amplification and sequence analysis on 18 serum samples from the Haikou City Center for Disease Control and Prevention, constructing phylogenetic trees and performing molecular clock analyses to examine the epidemiological characteristics and transmission dynamics of dengue fever in Hainan Province, thereby offering scientific evidence and recommendations for subsequent control strategies.
Materials and Methods
Sample collection and processing
Between October 11 and November 13, 2024, 18 serum samples were collected from patients with dengue-like symptoms at sentinel hospitals and community health service centers in 4 administrative districts of Haikou City (Longhua, Xiuying, Meilan, and Qiongshan). Patients were included if they tested positive for DENV RNA by RT-PCR with a cycle threshold (Ct) value < 25. The cohort comprised 12 males and 6 females aged 21–55 years. Of the sequenced strains, 17 were DENV-1 and 1 was DENV-3, with detailed demographic and epidemiological information provided in Supplementary Table S1. Whole blood samples were collected in tubes without anticoagulants, centrifuged, and the separated serum was transferred to sterile 2 mL tubes. Initial screening was performed using immunochromatography and double-antibody sandwich assays, with final confirmation by molecular detection. Serum samples were transported under cold-chain conditions, delivered to the laboratory within 24 h, and stored at −80°C until further analysis.
Detection and serotyping of DENV
Viral RNA was extracted from serum samples using the Viral RNA Mini Kit (Qiagen). DENV diagnosis and serotyping were performed using an RT-Quantitative Polymerase Chain Reaction (qPCR) kit (Takara), with primers and probes designed according to the national health standard (WS 216–2018). RT-qPCR was conducted in a total reaction volume of 20 µL, containing premix, primers, probes, and RNase-free water. Primers specific for the four DENV serotypes were combined with probes labeled with different fluorophores (Supplementary Table S2). The PCR cycling conditions were as follows: reverse transcription at 52°C for 5 min, 95°C for 10 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s.
Isolation, confirmation, and amplification of the DENV E gene
A total of 500 µL of C6/36 cells at a density of 1 × 105/mL were seeded into a 6-well plate and cultured in a 28°C, 5% CO2 incubator for 1 day until a monolayer was formed. The medium was aspirated, and the cells were washed twice with phosphate-buffered saline (Gibco). Subsequently, a 20-fold diluted patient serum was added, with continued incubation at 28°C, 5% CO2, and daily observation of cytopathic effects (CPE). After 7–10 days of culture, significant CPEs were observed, and the supernatant from the affected cells was harvested and stored in a −80°C freezer for subsequent experiments. The viral supernatant was then diluted 10-fold to infect C6/36 cells, and after 24 h, immunofluorescence detection was performed. Viral nucleic acids were extracted, and one-step reverse transcription-PCR was used to specifically amplify the E genes of DENV-1 and DENV-3, with primers as shown in Supplementary Table S3. Nucleic acid electrophoresis was conducted using 1% agarose gel to confirm the correct amplification bands (DENV-1: 1740 bp, DENV-3: 2335 bp) (Fraenkel et al., 2024).
Sequencing of the E gene and full-length virus
After amplifying the E gene from the isolated viral samples (derived from the first passage [P1] following serum inoculation onto C6/36 cells), sequencing of the full-length E gene sequence was commissioned to Sangon Biotech (Shanghai) Co., Ltd., using the Sanger sequencing method. The resulting sequences were uploaded to the National Center for Biotechnology Information (NCBI) database. For whole-genome sequencing we preselected samples with RT-qPCR Ct <25 and adequate RNA quality; from this high-quality subset we prioritized serotype representation, temporal and geographic coverage, and available clinical severity (see Supplementary Table S1), employing the Illumina method for full-length viral sequencing. Genome assembly was performed using MEGAHIT v1.2.9 (completed by ProbeGene Medical Technology [Zhejiang] Co., Ltd.) with default parameters (minimum k-mer size of 21). Detailed sequencing metrics, including genome coverage and average sequencing depth for the five whole-genome samples, are provided in Supplementary Table S5. The assembled sequences were submitted to NCBI.
Phylogenetic and molecular characteristic analysis
The isolated E gene sequences were combined with representative serotype sequences from GenBank, and Basic Local Alignment Search Tool for Nucleotides (BLASTn) (v2.16.0+) was used to verify the correctness of the protein-coding sequences (CDS) individually. Subsequently, multiple sequence alignment of the reference sequences and the E gene sequences obtained in this study was performed using Multiple Alignment using Fast Fourier (MAFFT) (v7.526). Phylogenetic trees were inferred by the Maximum Likelihood method using IQ-TREE 2 (v2.1.3). The optimal substitution model was automatically selected based on the Bayesian information criterion. Branch support was assessed via ultrafast bootstrap analysis (UFBoot2) with 1000 replicates.
Four generated DENV-1 whole-genome sequences from this study were combined with 30 representative sequences retrieved from the NCBI database. These reference sequences were predominantly sampled from Southeast Asia across various years. The earliest available sequence, EU848545.1_USA_Hawaii_1944, was included to root the tree. CDS were aligned using the ClustalW algorithm implemented in MEGA 12.
The temporal signal was evaluated via root-to-tip regression in TempEst, demonstrating a strong temporal correlation (
Markov chain Monte Carlo chains were run for a total length of 50,000,000 generations. Two independent runs were conducted to assess reproducibility, and convergence was evaluated in Tracer, confirming highly effective sample size values for all parameters. The runs were combined using LogCombiner, and the maximum clade credibility (MCC) tree was generated with TreeAnnotator for visualization.
Structural modeling and mutation analysis of the E protein
Homology modeling was performed on the E protein variations identified in this study using the SWISS-MODEL platform and the DENV-1 amino acid sequences. For the prefusion dimer, the template Protein Data Bank Identifier (PDB ID) 7a3r was used, achieving a sequence identity of 95.95%, Global Model Quality Estimate Transform (GMQE) of 0.68, and QMEANDisCo of 0.74 ± 0.05. For the postfusion trimer, PDB ID 4gsx was selected, with a sequence identity of 96.35%, GMQE of 0.67, and QMEANDisCo of 0.72 ± 0.05. These values confirm the models’ reliability. The mutation sites (S338L and D147N) were annotated, and the structures were visualized using Python-based Molecular visualization system (PyMOL) (Delano, 2002; Nayak et al., 2009; Renner et al., 2021; Waterhouse et al., 2018).
Results
Virus isolation and amplification
During the 2024 dengue outbreak in Hainan Province, 18 acute-phase serum samples (HK-001–HK-018) confirmed by the Haikou Centers for Disease Control and Prevention (CDC) were collected. Sixteen samples were successfully isolated, with RT-qPCR serotyping identifying 15 DENV-1 and 1 DENV-3 infections. All isolates were propagated in A. albopictus C6/36 cells, where CPEs were observed from day 5 postinoculation, intensified by day 7, and became extensive by day 10 (Fig. 1). Viral supernatants were subsequently harvested and confirmed by immunofluorescence assay (Fig. 2).

Cytopathic effects of virus isolation in C6/36 cells. Serum samples were inoculated onto monolayer C6/36 cells. On day 5 postinoculation, cytopathic effects (CPE) were initially observed, manifesting as cell fusion, aggregation, and detachment (10× objective). This figure displays microscopic images from day 5 (Panel
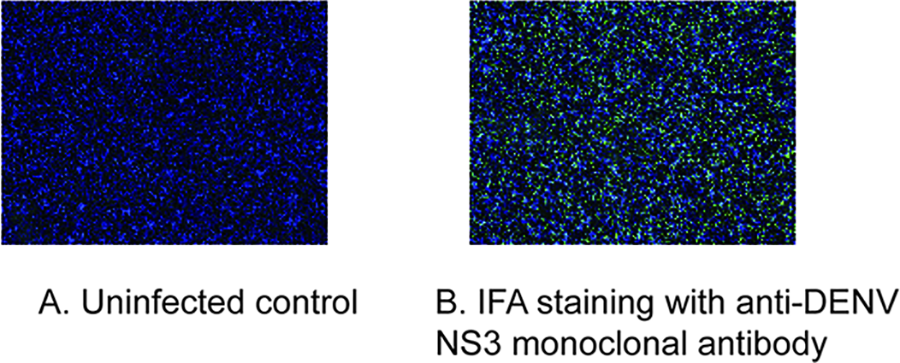
Immunofluorescence assay of dengue virus infection. C6/36 cells postvirus inoculation (using a 100-fold dilution of the virus supernatant cultured for 10 days, 24 h postinfection) were fixed and subjected to immunofluorescence analysis. Panel
Phylogenetic analysis and molecular clock model
To elucidate the molecular epidemiology of DENV in Hainan Province, we utilized the E gene sequences of 16 DENV strains determined in this study (GenBank accession numbers: PV478122–PV478135, PX317703, and PX317704; Supplementary Table S4). A comprehensive phylogenetic tree including all identified serotypes is presented in Supplementary Figure S1. Given the predominance of DENV-1 in our samples, we further constructed a serotype-specific Maximum Likelihood tree for DENV-1 to resolve the topological relationships with higher resolution (Fig. 3). The genetic evolutionary analysis of the E gene elucidated information on DENV isolates from the Haikou region in Hainan Province. Based on the E gene sequences, 15 strains (HK-001, HK-002 to HK-010, and HK-013 to HK-018) were classified as DENV-1 genotype I, with 14 local Hainan isolates located in the DENV-1 genotype I (genotype I) evolutionary branch; this suggests that the primary circulating strain in this outbreak was DENV-1. This branch formed a tight cluster with the 2023 circulating strain from Guangdong Province (PP563845.1), indicating that this outbreak may have been transmitted from Guangdong to the local area, subsequently establishing cryptic circulation locally. Although HK-010 also belongs to the DENV-1 genotype I, it did not cluster with the other DENV-1 strains in this study and shares 99.23% homology with sequences from Southeast Asian countries such as Thailand and India. HK-002, located in the same branch, is a DENV-3 strain, closely related to recent strains from India and the Philippines.
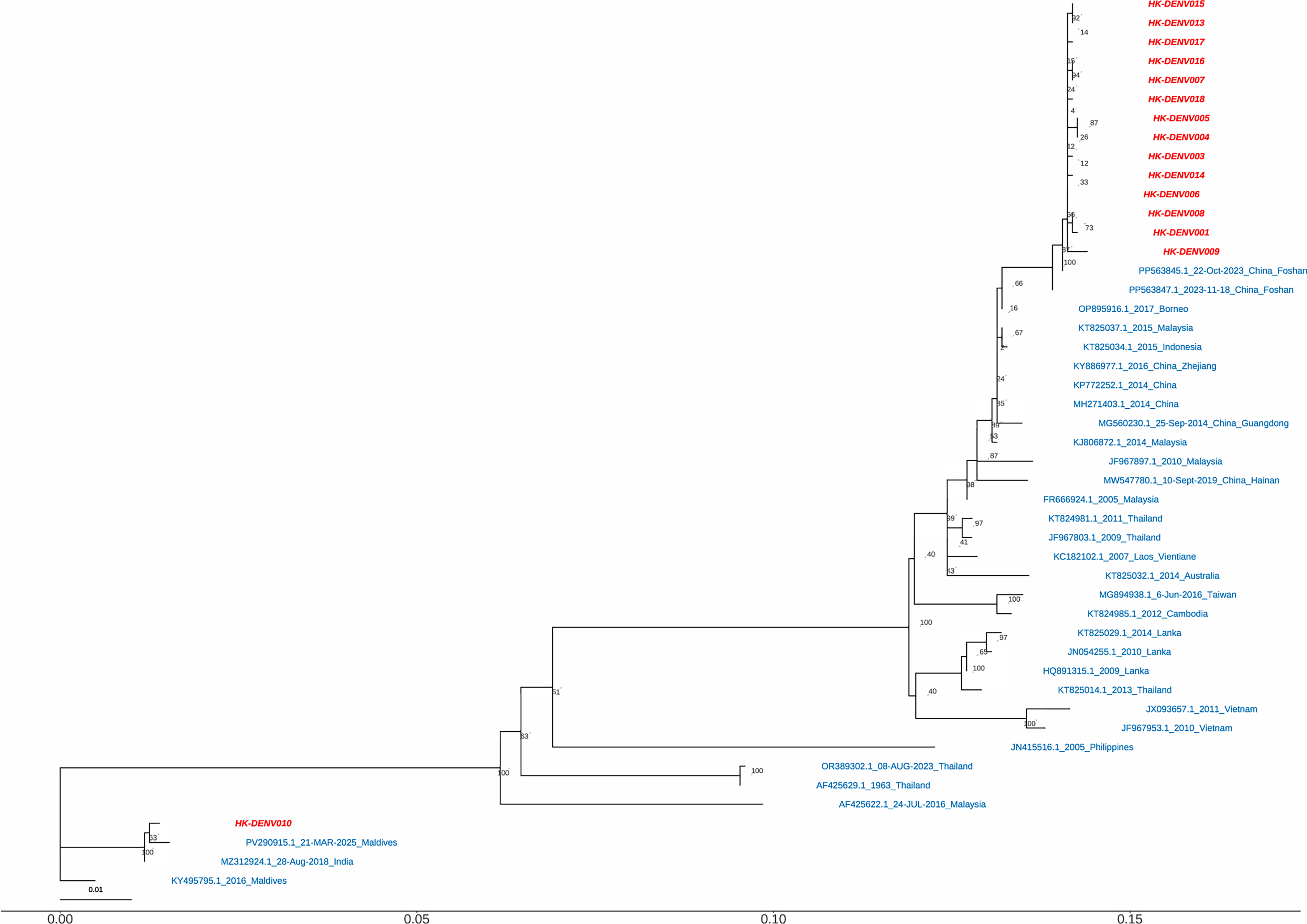
Phylogenetic analysis of DENV-1 strains based on E gene sequences. The Maximum Likelihood (ML) tree was inferred using IQ-TREE 2 (v2.1.3) based on E gene sequences of 16 DENV isolates from the 2024 Hainan outbreak (GenBank accession nos. PV478122–PV478135, PX317703, and PX317704) and representative reference strains. Multiple sequence alignment was performed using MAFFT (v7.526). The optimal substitution model was selected based on the Bayesian information criterion (BIC). Ultrafast bootstrap support values (UFBoot2) calculated from 1000 replicates are shown at nodes (values ≥70% are displayed). The scale bar represents nucleotide substitutions per site. The strains from the Hainan outbreak are highlighted in red, while all other reference strains are shown in blue.
To further resolve the evolutionary dynamics indicated by the E gene phylogeny, an MCC tree was reconstructed based on the whole genomes of four representative isolates: PX352011 (HK-DENV017), PV478125 (HK-DENV006), PX317703 (HK-DENV001), and PX352012 (HK-DENV010). The whole-genome topology (Fig. 4) was consistent with the E gene analysis. HK-DENV017, HK-DENV006, and HK-DENV001 formed a monophyletic clade that clustered tightly with recent strains from Guangdong, including the late-2023 Foshan isolate (PP563845.1). This genomic proximity supports the epidemiological link to Guangdong. In contrast, HK-DENV010 exhibited distinct genetic divergence and segregated into a separate lineage. It clustered alongside a 2018 Indian isolate (MZ312924.1), corroborating the independent origin previously suggested by the E gene data.
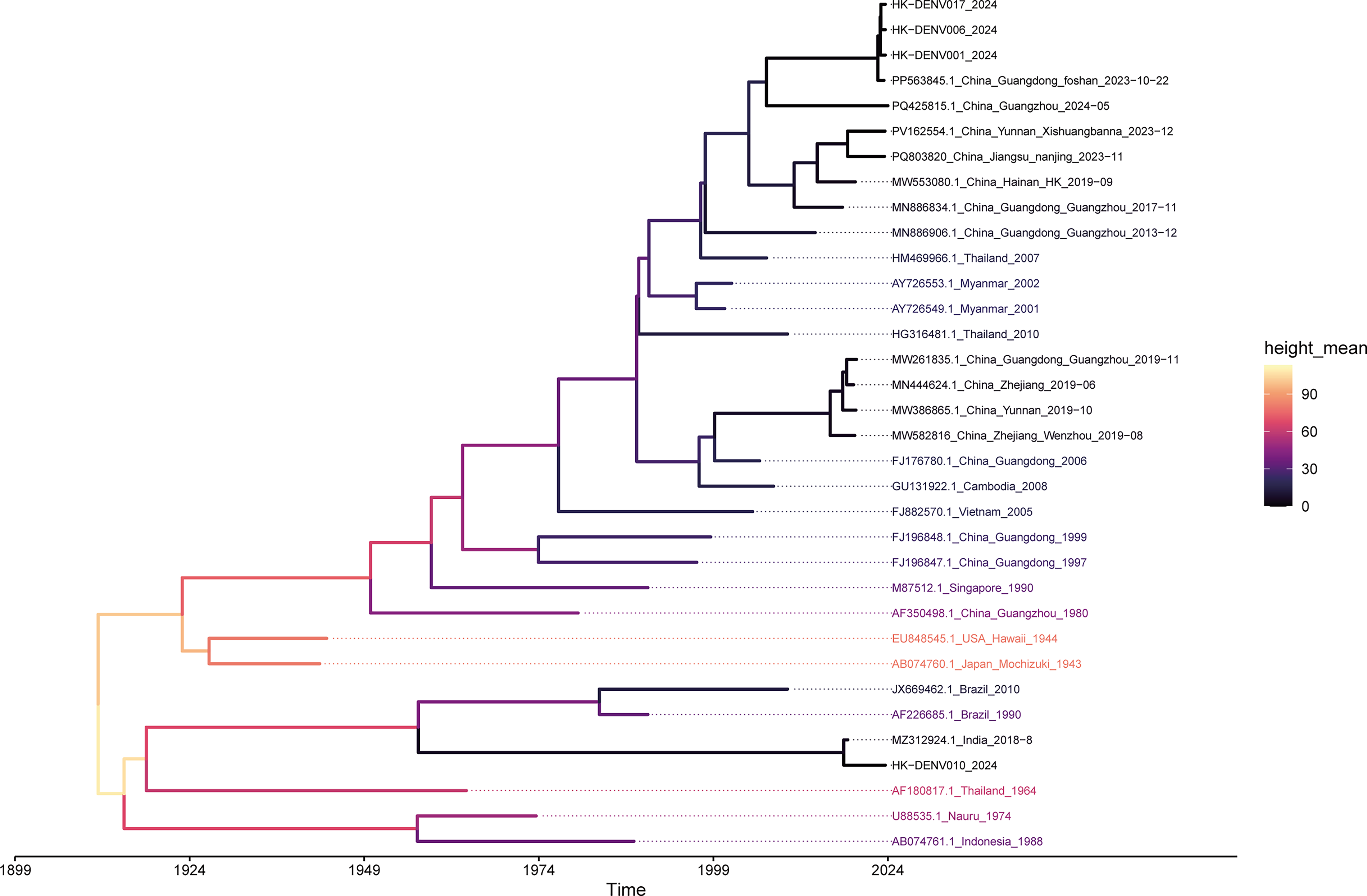
Time-scaled phylogenomic analysis of DENV-1. A maximum clade credibility (MCC) tree was reconstructed using BEAST X v10.5.0 based on the coding sequences (CDS) of four representative isolates from the current outbreak (HK-DENV001, HK-DENV006, HK-DENV010, and HK-DENV017) and 30 global reference genomes. The reference dataset primarily includes representative strains from Southeast Asia and China, along with historical sequences (such as a 1944 Hawaii isolate) to root the tree. Sequence alignment was performed using the ClustalW algorithm in MEGA 12. Following temporal signal assessment via TempEst (R2 = 0.904), the evolutionary timescale was estimated under a strict molecular clock model. The horizontal axis represents evolutionary time in years, spanning from approximately 1899 to 2024. Branch colors correspond to the estimated mean node height, reflecting the temporal progression from ancestral lineages to recent divergence events.
E gene mutation analysis
To compare the genetic characteristics of the isolated DENV-1 strains, amino acid-based multiple sequence alignment was performed using reference DENV-1 sequences from the same phylogenetic branch. High sequence similarity was observed in the E protein region; however, the 14 isolates harbored 2 unique amino acid substitutions, D147N and S338L. D147 is located in domain I, which is associated with conformational changes and membrane fusion, whereas S338 resides in domain III, an immunoglobulin-like domain containing key receptor-binding sites and neutralizing antibody epitopes involved in tissue tropism and immune escape (Hu et al., 2019) (Figs. 5 and 6).

Schematic diagram of DENV-1 E protein conformation. Domain I (light blue) consists of three discontinuous segments; Domain II (pink) includes two discontinuous segments; Domain III (dark gray) is located at the N-terminus of the black stem region. The C-terminal light gray segment represents the transmembrane anchor region. Two unique mutation sites are marked in red.
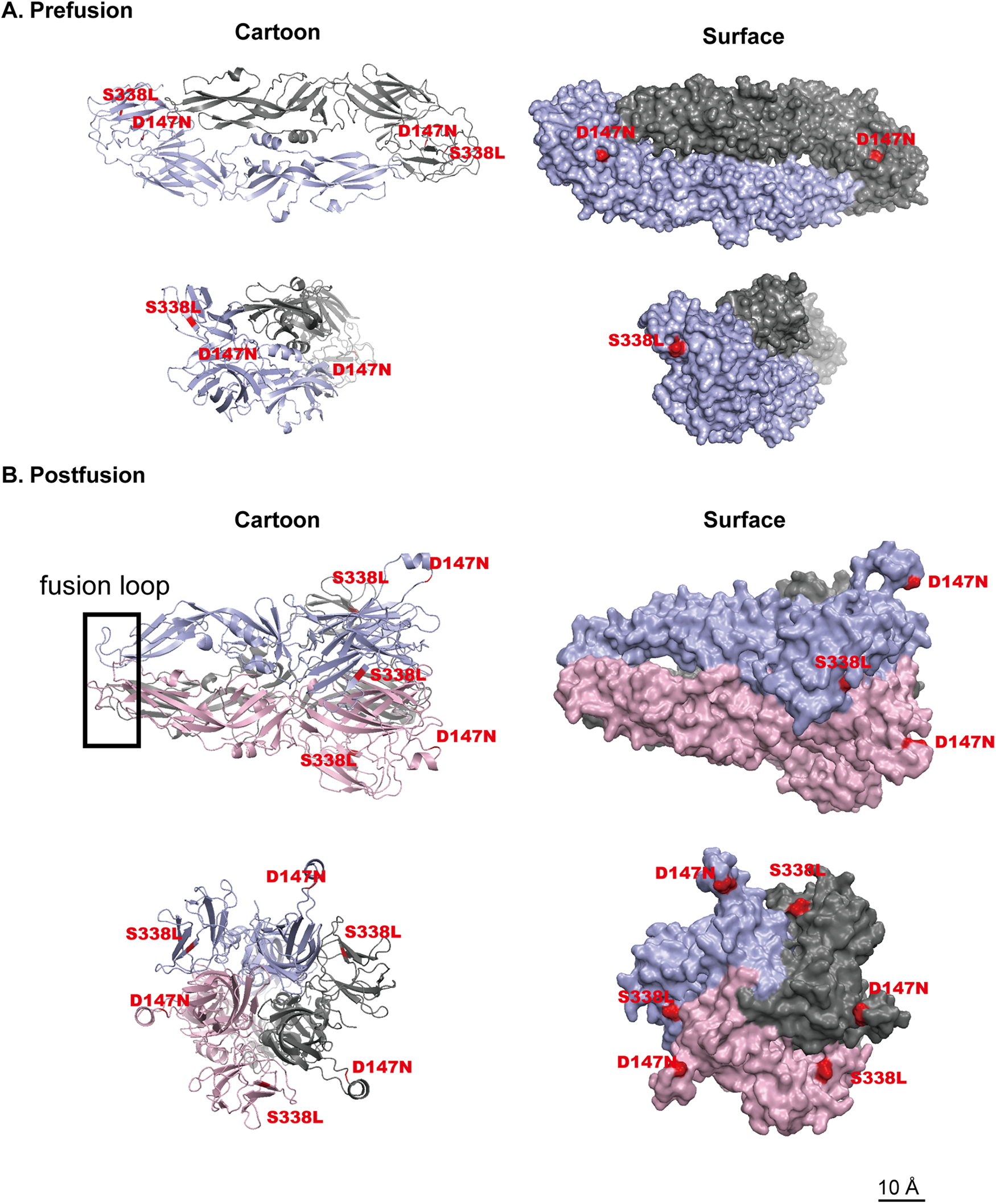
Structural prediction of DENV-1 E protein prefusion and postfusion conformations and localization of key mutation sites.
Discussion
China has experienced multiple dengue outbreaks since the disease reemerged in Foshan, Guangdong, in 1978 (Xiong and Chen, 2014). Following its inclusion in the list of notifiable diseases in the 1990s (Lai et al., 2015), outbreaks were mainly concentrated in southern provinces, though the geographical range and frequency of outbreaks have gradually increased in recent years (Ni et al., 2024). Hainan Province has faced three major dengue outbreaks: 1978 (type 3), 1985–1988 (type 2), and 1991 (type 1) (Fan et al., 1989; Liu et al., 2021; Qiu et al., 1991). In July 2024, the WHO classified DENV as a high-risk pathogen, urging global prevention and control efforts (World Health Organization, 2024). Despite repeated cases in Hainan, there is still limited understanding of the genetic characteristics and epidemic patterns of local strains.
Phylogenetic analysis indicated that the 2024 dengue outbreak in Hainan was dominated by DENV-1 genotype I, forming a tight cluster with the Guangdong strain (PP563845.1). While this genetic similarity suggests a potential introduction from Guangdong, the exact transmission route remains unclear without detailed mobility and vector surveillance data. Among the 18 specimens, one was a DENV-3 infection (HK-002), and another (HK-010) did not match the local circulating strains, suggesting independent transmission chains possibly introduced from Southeast Asia. These strains may be in the early stages of transmission and require close monitoring.
The E protein is the primary antigenic component of DENV and the main target of host immune pressure (Katzelnick et al., 2015; Sessions et al., 2015), comprising three domains (I–III; Table 1). Comparative analysis with reference DENV-1 E proteins identified two mutations: D147N in domain I and S338L in domain III. Structural analysis of the prefusion E protein (HK-008) showed that residues D147 and S338 are surface-exposed, suggesting potential involvement in host receptor interactions and altered binding affinity.
Structural Domain Architecture of Dengue Virus-1 Envelope Glycoprotein
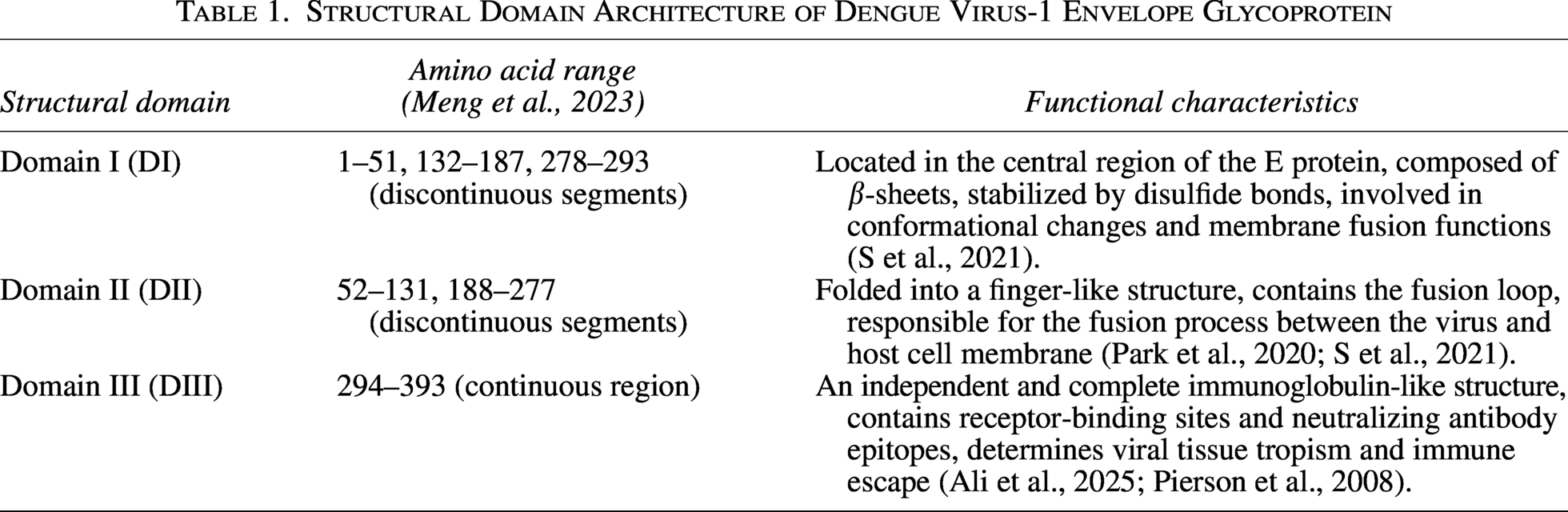
Domain I consists of discontinuous segments in the E protein and participates in viral conformational changes and membrane fusion processes. Studies have shown that T226K/G228E enhances viral infectivity in mosquito or mammalian hosts (Chen et al., 2022), while mutations at position 160 in the E protein increase antibody neutralization activity, leading to replacement of circulating genotypes (Wang et al., 2016). These findings indicate that changes in domain I can affect viral infectivity; therefore, we speculate that the change at D147 might influence the virus’s infectivity (Table 2). Domain III contains type- and subtype-specific structural antigenic epitopes that can induce neutralizing antibodies (Park et al., 2020). Additionally, domain III features an independent and complete immunoglobulin-like structure, containing receptor-binding regions and neutralizing antibody epitopes, playing important roles in the viral life cycle.
Predicted Functional Impact of Key Mutations in Dengue Virus-1 E Protein Domains

Thus, the S338 mutation may contribute to immune escape by enhancing receptor-binding capability; however, this inference is based on structural modeling and requires experimental validation. This hypothesis is supported by recent studies on the 2023 autochthonous outbreak in Italy (Carletti et al., 2024) and broader analyses of host–virus interactions (Ghosh et al., 2025), which highlight the critical role of E protein surface mutations in viral adaptation and antibody evasion. The co-occurrence of D147N and S338L is reported here for the first time, and their effects on viral transmission and pathogenicity, including potential synergistic interactions, warrant further investigation using reverse genetics and in vivo models.
From a public health perspective, these findings underscore the need to strengthen local control strategies in Hainan by integrating sustained mosquito surveillance with real-time genomic monitoring for early detection of viral introductions. In addition to conventional vector control, targeted vaccination of high-risk populations may represent an important complementary approach to reducing outbreak risk in this tropical region.
This study has several limitations. First, the limited sample size and the lack of detailed transmission dynamics restrict attribution analyses and may not fully capture the genetic diversity circulating in the population, although the samples were sufficient for initial pathogen identification and phylogenetic placement. Second, although amino acid substitutions (D147N and S338L) were identified in functionally important regions, their biological effects were inferred solely from structural modeling, without experimental validation, such as neutralization or pseudovirus infectivity assays. Therefore, any inferred effects on viral transmissibility or immune escape should be considered hypothesis-generating and require further in vitro and in vivo investigation.
The 2024 epidemic underscores the growing challenge of dengue in China. As the southernmost province, Hainan’s persistently high temperatures, abundant rainfall, and frequent trade exchanges increase its susceptibility to viral introduction and establishment. Surveillance data indicate that A. albopictus has largely replaced A. aegypti as the predominant vector in the region, a situation further complicated by documented insecticide resistance in local mosquito populations (Chen et al., 2020; Zhao et al., 2023). In addition, the tropical climate accelerates transmission by shortening the viral extrinsic incubation period and sustaining high vector densities, resulting in a substantially prolonged transmission season compared with temperate regions of China (Yue et al., 2021).
Against this high-risk ecological background, our study characterizes the molecular and epidemiological features of the 2024 Haikou outbreak. By clarifying viral origins and transmission chains, these findings provide a scientific basis for optimizing local control strategies, including targeting insecticide-resistant vectors, and offer a framework for future research into DENV pathogenicity and transmissibility.
Ethics Review
The experimental protocol was approved by the Ethics Review Committee of the Naval Medical University, having passed all legal and ethical reviews. All experiments were conducted in accordance with relevant guidelines and regulations.
Authors’ Contributions
Z.-M.C.: Investigation (lead), data curation (lead), and writing—original draft (lead). B.-H.X.: Methodology (lead) and data curation (lead). P.L.: Resources (lead) and data validation (lead). X.-H.N.: Conceptualization (lead). Z.-H.Z.: Formal analysis (lead). Q.Z.: Investigation (supporting). H.R. and Y.-Z.Z.: Supervision (lead) and writing—review and editing (equal for both). All authors contributed critically to the drafts and gave final approval for publication.
Footnotes
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.