
Abstract
Background:
Bartonella spp. are Gram-negative bacteria that cause diseases including endocarditis, lymphadenopathy, and neuroretinitis. Hantavirus (HV), belonging to the family Hantaviridae, induces illnesses such as hemorrhagic fever with renal syndrome and hantavirus pulmonary syndrome. Both pathogens exhibit host specificity—defined as a preference or restriction to specific host species or ranges. Rodents and shrews are primary hosts for these pathogens, and their high coinfection rates often indicate elevated risk of human exposure. To our knowledge, however, data on Bartonella spp.–HV coinfection in rodents and shrews from Eastern China remain limited.
Materials and Methods:
Between 2020 and 2023, rodents (n = 311) and shrews (n = 16) were investigated for coinfection with Bartonella spp. and HV in Qingdao, eastern China. Nested Polymerase Chain Reaction (PCR) was used for the detection of RNA-dependent RNA polymerase (RdRp) gene of HV and the Internal Transcribed Spacer, citrate synthase (gltA) and RNA polymerase beta subunit (rpoB) genes of Bartonella spp.
Results:
The overall infection rates of Bartonella spp., HV, and coinfection were 21.4%, 6.7%, and 4.0%, respectively. The highest rates were observed in Apodemus agrarius (53.8%, 21.3%, and 15.0%). Coinfection rates differed significantly by species (p < 0.05), with A. agrarius exhibiting the highest rate (15.0%). Notably, the coinfection rate was significantly higher in male (28.9%) than female A. agrarius (7.1%) (p < 0.05).
Conclusions:
This study confirms the coinfection of Bartonella spp. and HV in rodents in the eastern region of China. Enhanced monitoring of rodent and shrew densities, as well as their carried pathogens, is essential. Additionally, timely screening, diagnosis, and treatment should be conducted for high-risk populations in the region to reduce the incidence of related zoonoses.
Keywords
Introduction
The genus Bartonella consists of Gram-negative bacteria that are primarily transmitted by blood-sucking arthropods. Over 40 species of Bartonella have been described, which can infect various animals, with rodents and shrews being significant hosts (Huang et al., 2019). Humans may become infected with bartonellosis either through arthropod vectors or through direct contact with infected hosts (Morick et al., 2010). After human infection with Bartonella, a range of mild and nonspecific clinical symptoms and signs may manifest, but it can also lead to severe, potentially life-threatening diseases (Breitschwerdt et al., 2010; Garcia-Quintanilla et al., 2019), such as trench fever (B. quintana), cat scratch fever (B. henselae), and Carrion’s disease (Jacomo et al., 2002). In the past decade, an increasing number of cases of rodent-borne Bartonella species infections and related febrile illnesses or endocarditis have been reported, indicating that human exposure to these pathogens is more common than previously suspected (Krügel et al., 2022; Okaro et al., 2017; Vayssier-Taussat et al., 2016).
Hantavirus (HV) is an emerging global public health threat, among which Hantaan virus (HTNV) and Seoul virus (SEOV) are the two main pathogens. Both belong to the genus Orthohantavirus, family Hantaviridae, order Bunyavirales, and are classified as Old World HVs. Their primary hosts are Apodemus agrarius and Rattus norvegicus, respectively (Hansen et al., 2015). HTNV can cause a severe form of hemorrhagic fever with renal syndrome (HFRS), which is characterized by renal failure and an estimated mortality rate of 5–15%; SEOV induces moderate HFRS with a case fatality rate of <1% (Fang et al., 2015). Humans primarily become infected with HV through contact with infected rodents and shrews or their excreta (Zou et al., 2016), which can lead to severe diseases such as hantavirus pulmonary syndrome and HFRS following infection (Tarig and Kim, 2022; Watson et al., 2014). In recent years, the incidence of HFRS in China has shown an upward trend, with new cases reported in all 31 provinces (autonomous regions) nationwide (Wang et al., 2021).
Coinfection, defined as the simultaneous infection of a host with multiple pathogens, can result in various symptoms and significantly influence epidemiological characteristics as well as the severity of diseases (Rothenburger et al., 2019). Research indicates that coinfections are more likely to have adverse effects on host health compared with single infections. For instance, mice coinfected with Borrelia burgdorferi and human granulocytic ehrlichiosis agent can modulate the host immune response, suppressing the activation of immune cells, which leads to increased bacterial load, Lyme arthritis, and the transmission of pathogens to vectors (Thomas et al., 2001). Meanwhile, a study on HV and Leptospira spp. indicates that infection with either of the two pathogens can impair the immune capacity of the infected individuals, thereby facilitating the infection by the other pathogen (Jeske et al., 2021). This suggests that when host populations are infected with multiple pathogens, it may expand the transmission range of the pathogens and exacerbate the associated clinical symptoms.
Additionally, the high coinfection rate observed in rodents suggests that humans also face the risk of simultaneous infection with these pathogens (Md-Lasim et al., 2021). In recent years, an increasing number of coinfection phenomena have been detected in arthropods and rodents worldwide. For instance, fleas can harbor pathogens from various genera simultaneously, including Bartonella spp., Rickettsia, and Wolbachia (Manvell et al., 2022). Previous studies in China have reported the detection of coinfections with Bartonella spp., Leptospira spp., HV, and Orientia tsutsugamushi in R. norvegicus (Zhao et al., 2024).
China is recognized as a country rich in biodiversity, housing approximately 200 species of rodents across 12 families (Wu et al., 2012). These rodents are capable of transmitting various pathogens, including those from the families Arenaviridae, Hantaviridae, Reoviridae, Borrelia spp., Bartonella spp., Leptospira spp., and Coxiella burnetii, which may exhibit multiple coinfection scenarios among different hosts. However, such coinfections remain poorly studied in Qingdao, a coastal city in eastern China’s Shandong Peninsula with unique ecological and demographic characteristics that justify its selection as the study area.
Qingdao (35°35′–37°09′ N, 119°30′–121°00′ E) has a temperate monsoon climate, diverse habitats, and serves as a major coastal transportation/tourist hub, facilitating human–rodent interactions and pathogen transmission (Romanello et al., 2023). The rapid urbanization and intensive agricultural development in Qingdao in recent years have altered rodent community structure (Chen, 2021), facilitated pathogen exchange, and increased human exposure to infected rodents. Its distinct rodent community, high human mobility, and ongoing environmental changes make Qingdao uniquely significant for coinfection research. Thus, this study investigates the prevalence, regional distribution, species differences, and host-related influencing factors (age, mass, sex) of Bartonella spp. and HV coinfections in Qingdao rodents, providing a reference for understanding coinfection impacts on hosts and populations.
Materials and Methods
Sample collection
We continuously collected samples for seven nights at four sites located in Huangdao (120.18°E, 35.96°N), Jimo (120.45°E, 36.39°N), Laixi (120.53°E, 36.88°N), and Pingdu (119.95°E, 36.78°N). Among the four sampling sites, the Huangdao area features a mountain-sea interactive landscape, predominantly consisting of hills; the Laixi area is primarily flat; the Jimo area has extensive coastal plains and more inland hills; and the Pingdu area is relatively flat, traversed by the Jiaolai River. Additionally, all four regions engage in agricultural production activities and harbor a significant population of rodents and shrews, making them highly representative. All animals were trapped using the squirrel-cage method (Zhao et al., 2024). These traps were placed at the edges of farmland, with 100–200 traps set per site for one night; cages were placed before sundown and collected the following early morning. The capture date, location, habitat environment, and quantity were recorded. Cages containing captured animals were placed into cardboard boxes to prevent escape and quickly transferred to the laboratory, where euthanasia was performed via cervical dislocation, while making every effort to minimize their suffering (Shomer et al., 2020). In the process of capturing and transferring samples, we utilized the following protective equipment: medical disposable protective clothing (Dongbei, China), single-use sterilized rubber gloves (Xinaoli, China), Medical Isolation Shoe Cover (Chao Jian, China), and medical protective masks (3M, China). Given that body mass is frequently used as an indicator of rodent age (Costa et al., 2014), each specimen was weighed accordingly. The species of rodents and shrews were initially identified morphologically, with uncertain species confirmed by sequencing the mitochondrial cytochrome b gene (Zhang et al., 2018). Spleen (20 mg) and lung (20 mg) tissues were sampled using aseptic techniques, placed in 1.8 mL freezing tubes, and stored at −80°C until nucleic acid extraction.
Nested PCR detection of Bartonella and HV
For each rodent and shrew specimen, 20 mg of spleen tissue and 20 mg of lung tissue were added to a homogenization tube (Roche Diagnostics GmbH, Hilden, Germany) containing 1000 μL of Dulbecco's Phosphate-Buffered Saline (DPBS) (Gibco, Grand Island, NY, USA) and metal beads (Hilden, Germany). Tissues were homogenized at 6000 rpm for 3 min (The centrifugal force is 28174 g) using a tissue grinder (Hilden, Germany). A total of 200 μL of the resulting tissue homogenate was mixed with 250 μL of lysis buffer (Hilden, Germany), and nucleic acids (DNA and RNA) were extracted using a nucleic acid extractor (Hilden, Germany). From each specimen’s spleen and lung tissues, 100 μL of total RNA and 100 μL of total DNA were extracted separately. Total RNA extracted from lung tissues was subjected to cDNA synthesis using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Applied Biosystems, Lithuania). The reverse transcription reaction was performed in a final volume of 20 μL containing 2 μL buffer, 2 μL random primers, 1 μL Deoxynucleoside Triphosphate (dNTP) mix, 1 μL MultiScribe reverse transcriptase, 4 μL nuclease-free water, and 10 μL RNA template. The thermal cycling conditions were set as follows: one cycle of 25°C for 10 min, one cycle of 37°C for 120 min, and one cycle of 85°C for 5 min.
For HV, cDNA generated from lung tissue samples was used for nested PCR amplification of the RdRp gene fragment. For Bartonella spp. nested PCR targeting the Internal Transcribed Spacer (ITS) gene fragment was performed using DNA extracted from spleen samples. For samples identified as coinfected, additional PCR amplifications were conducted targeting the gltA and rpoB genes of Bartonella (Table 1). Due to limitations in funding, laboratory consumables, and equipment availability, positive controls were not included in this study. While this may introduce a certain risk of false-negative results, similar approaches have been adopted in previous investigations (Qin et al., 2019). To minimize the risk of false-positive PCR findings, DNA extraction, reaction mixture preparation, PCR amplification, and amplicon analysis were conducted in physically separated laboratory rooms, and sterile water was used as the PCR negative control.
Primers for Amplification of Bartonella Spp. ITS, gltA and rpoB, and HV RdRp

HV, Hantavirus; ITS, Internal Transcribed Spacer.
All PCR products were resolved by electrophoresis on a 1% agarose gel, and presumptive positive amplicons were subjected to Sanger sequencing (Tsingke Biotechnology Company Limited, Beijing, China).
HV and Bartonella species shared similar thermal cycling conditions, except for the extension temperature, as detailed in Table 1. The first-round PCR was conducted in a total volume of 20 μL, which included 10 μL of 2× Taq Master Mix (Dye Plus) (Nanjing Vazyme Biotech Company Limited, China), 2 μL of forward primer, 2 μL of reverse primer, 2 μL of extracted cDNA/DNA, and 4 μL of nuclease-free water. The second round of PCR was identical to the first round, except that 2 μL of extracted DNA was replaced with 2 μL of first-round PCR products. The same thermal cycling conditions were applied for both rounds, which included 95°C for 5 min; 35 cycles of 95°C for 40 s, 48°C for 1 min, and 72°C for 1 min; and one cycle of 72°C for 10 min. Bartonella species in this study were adapted from previously validated, peer-reviewed literature (Qin et al., 2019). For HV detection, our assay protocol was based on a previously validated published method (Tian et al., 2023). However, amplification conditions were appropriately optimized to accommodate differences in the reaction mixture, reagents, and laboratory equipment used. Specifically, the number of thermal cycles was reduced, reaction temperatures were adjusted, and incubation times were extended. The specificity of the established assay was confirmed by sequence analysis of all positive amplicons.
Phylogenetic analysis of HV and Bartonella in all the coinfection samples
One pair of primers targeted the ITS region of Bartonella spp., and the other targeted the RdRp gene segment of HV. For samples identified as coinfected, additional PCR amplification was performed for the Bartonella spp. gltA gene and rpoB gene. Bidirectional sequencing was performed for all positive amplicons. For inconsistent bases identified during sequencing, further comparison and confirmation were conducted via peak maps to ensure sequencing accuracy. DNA sequence analysis was carried out using BLAST on the GenBank nucleotide database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Phylogenetic trees of HV and Bartonella spp. were constructed using the maximum likelihood method in MEGA 7.0, with Rift Valley fever virus and Brucella abortus as outgroups; bootstrap values were calculated based on 1000 replicates. To ensure consistency of the Bartonella genus phylogenetic tree, the gltA and rpoB sequences of each Bartonella species were concatenated. To maximize information content and meet the requirements for standardization and comparability, only results with bootstrap values >60% are presented (Yu et al., 2022a; Zhang et al., 2022). Isolated Bartonella species were considered homologous to validated Bartonella species if the sequence similarity of gltA and rpoB fragments was ≥96% (Scola et al., 2003). Isolates with ≤90% nucleotide identity in the HV RdRp gene sequence compared with representative reference strains of known HVs were not regarded as novel genotypes or species (Laenen et al., 2019).
Statistical analysis
The population diversity of rodents and shrews in the study area was quantified using the Shannon–Wiener diversity index (H’), Simpson diversity index (D), and Pielou evenness index (J’). Infection positivity rates among rodent and shrew species, as well as positivity rates of Bartonella spp., HV, and coinfections across rodent sexes, were analyzed using the chi-squared test or Fisher’s exact test. The Pearson chi-squared test was used if sample size ≥40 and all expected frequencies ≥5; the continuity-corrected chi-squared test was applied if sample size ≥40 but at least one expected frequency = 1–5; and Fisher’s exact test was utilized if sample size <40 or any expected frequency <1. To assess associations between body mass/age (by species) and infection status, we first tested data for normality (Kolmogorov–Smirnov test) and homogeneity of variance (Levene test). Concurrently, 95% confidence intervals (CIs) for body mass were calculated based on the t-distribution. Due to heteroscedasticity in R. norvegicus body mass data, Welch’s t-test was used for this subset. In contrast, body weight data of A. agrarius and M. musculus were analyzed using the independent samples t-test. All analyses were two-tailed and performed using SPSS 19.0 (IBM Corp., Armonk, NY, USA). Statistical significance was defined as p < 0.05.
Ethical standard
As the capture of rodents and shrews was conducted as part of regular pest control measures, including environmental governance, disinfection, and killing, in accordance with the “Healthy China 2030” Planning Framework for Vector Prevention and Control Strategy, no ethical approval was required (Zhao et al., 2024).
Results
Prevalences and distribution of Bartonella spp. and HV
A total of 327 wild small mammals were captured in Qingdao City, Shandong, China. We collected 98, 38, 161, and 30 samples in Huangdao, Jimo, Laixi, and Pingdu, respectively. The collected rodents and shrews were identified into seven distinct species: R. norvegicus (119/327), Mus musculus (102/327), A. agrarius (80/327), Crocidura lasiura (12/327), Crocidura shantungensis (4/327), Tscherskia triton (5/327), and Cricetulus griseus (5/327). R. norvegicus and M. musculus were identified as the dominant species. Population diversity analysis yielded a Shannon–Wiener index (H’) of 1.355, a Simpson index (D) of 0.7087, and a Pielou evenness index (J’) of 0.696, indicating moderate diversity and good evenness in the region. From 2020 to 2023, the number of samples collected was 17, 86, 142, and 82, respectively. The total number of rodents and shrews included 224 males (68.5%) and 103 females (31.5%) (Table 2).
Infection and Coinfection Rates of Bartonella Spp. and HV in Different Host Species and Sexes
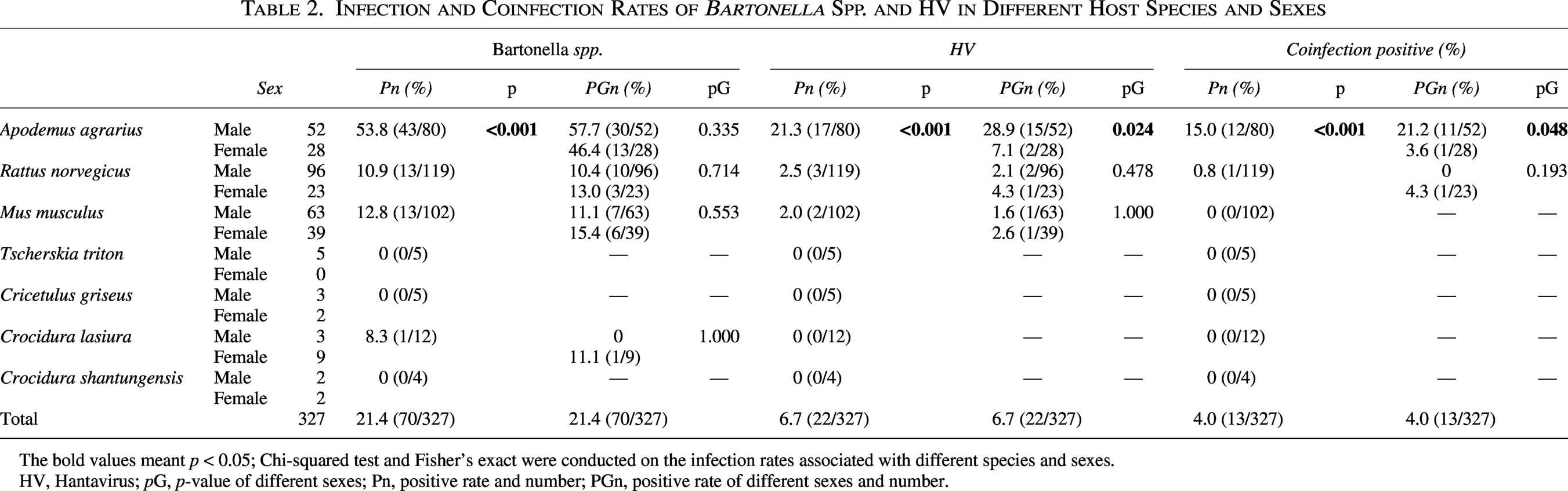
The bold values meant p < 0.05; Chi-squared test and Fisher’s exact were conducted on the infection rates associated with different species and sexes.
HV, Hantavirus; pG, p-value of different sexes; Pn, positive rate and number; PGn, positive rate of different sexes and number.
All seven species were tested for the ITS gene of Bartonella spp. and the RdRp gene of HV. Bartonella spp. infection rates varied by species: A. agrarius (53.8%), R. norvegicus (10.9%), M. musculus (12.8%), and C. lasiura (8.3%); no infection was detected in the remaining three species. Fisher’s exact test revealed a significant difference in Bartonella spp. infection rates across all species (p < 0.001). HV infection was detected in three rodent species: A. agrarius (21.3%), R. norvegicus (2.5%), and M. musculus (2.0%) (Table 2). Fisher’s exact test confirmed a significant difference in HV infection rates across all species (p < 0.05).
Bartonella spp. and HV infection rates by sex are presented in Table 2. Chi-squared and Fisher’s exact tests showed a significant difference in HV infection rates only between male and female A. agrarius.
Coinfection with Bartonella spp. and HV was detected only in 12 A. agrarius and one R. norvegicus in Huangdao, with an overall coinfection rate of 4.0%. The coinfection rate was 15.0% in A. agrarius and 0.8% in R. norvegicus. Statistical analysis using chi-squared test and Fisher’s exact test demonstrated a significant difference in coinfection positivity rates among rodent and shrew species (p < 0.05). Using Fisher’s exact test, the 95% CI for the difference in coinfection rates between A. agrarius and R. norvegicus was (6.18%, 22.14%). Among coinfected A. agrarius, 11 were male and 1 was female. The coinfection rate was significantly higher in males (28.9%) than in females (7.1%) (p < 0.05) (Table 2).
The relationship between mass/age and infection rate
For A. agrarius, the likelihood of infection with Bartonella spp., HV, and coinfection all increased with increasing body mass, with statistically significant differences (all p < 0.001; Table 3). Specifically, the mean body mass of A. agrarius infected with Bartonella spp. was 34.7 (32.5–36.8) g, with HV was 38.8 (34.9–42.6) g, and with coinfection was 38.9 (36.0–41.8) g. In contrast, the mean body mass of noninfected A. agrarius was 28.8 (25.9–31.8) g for Bartonella spp., 30.1 (28.2–32.0) g for HV, and 30.7 (28.7–32.7) g for coinfection.
Infection of Bartonella Spp., HV, and Coinfection in Different Species Based on Mass
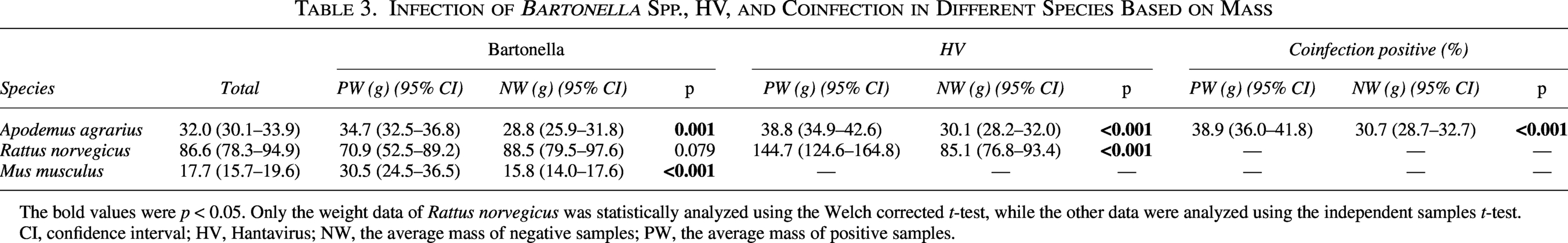
The bold values were p < 0.05. Only the weight data of Rattus norvegicus was statistically analyzed using the Welch corrected t-test, while the other data were analyzed using the independent samples t-test.
CI, confidence interval; HV, Hantavirus; NW, the average mass of negative samples; PW, the average mass of positive samples.
For R. norvegicus, higher body mass was associated with a significantly increased likelihood of HV infection (p < 0.001). The mean body mass of HV-infected R. norvegicus was 144.7 (124.6–164.8) g, compared with 85.1 (76.8–93.4) g for noninfected individuals. However, no statistically significant correlation was observed between body mass and Bartonella spp. infection likelihood in R. norvegicus (p = 0.079 > 0.05), with mean body masses of 70.9 (52.5–89.2) g for infected and 88.5 (79.5–97.6) g for noninfected individuals.
For M. musculus, increasing body mass was significantly correlated with a higher likelihood of Bartonella spp. infection (p < 0.001). The mean body mass of Bartonella spp.-infected M. musculus was 30.5 (24.5–36.5) g, compared with 15.8 (14.0–17.6) g for noninfected individuals.
Phylogenetic analysis of sequences in the samples coinfected with HV and Bartonella spp
The phylogenetic tree of the RdRp gene of HV from 13 coinfected samples showed that HV strains from rodents clustered into two clades: 1 strain grouped with SEOV, and 12 strains grouped with HTNV (Fig. 1).
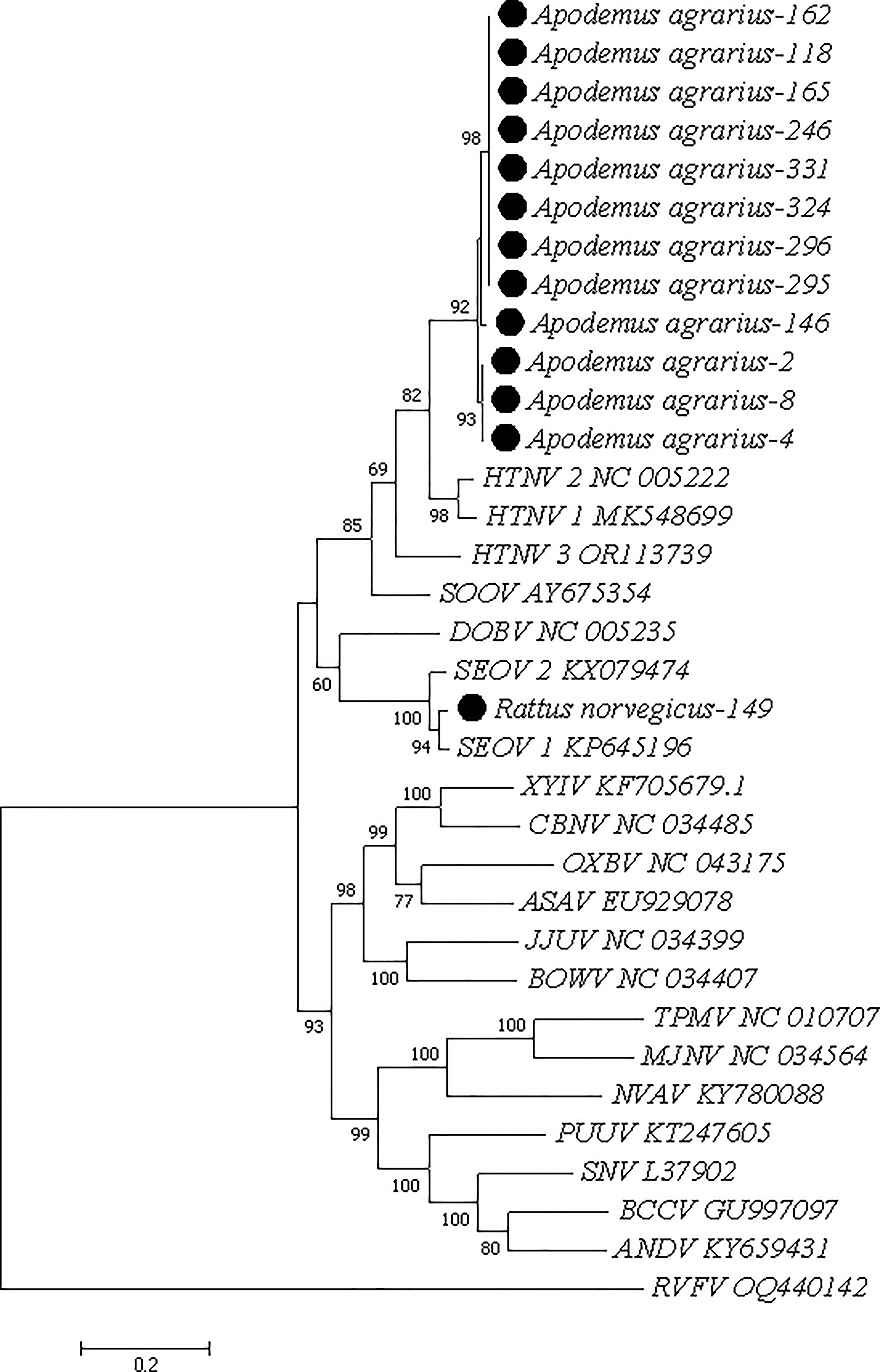
Phylogenetic tree of the RdRp gene of HV. HTNV, Hantaan virus; HV, Hantavirus; SOOV, Soochong virus; DOBV, Dobrava virus; SEOV, Seoul virus; XYIV, Xinyi virus; CBNV, Cao Bang virus; OXBV, Oxbow virus; ASAV, Asama virus; JJUV, Jeju virus; BOWV, Bowe virus; TPMV, Thottapalayam virus; MJNV, Imjin virus; NVAV, Nova virus; PUUV, Puumala virus; SNV, Sin Nombre virus; BCCV, Black Creek Canal virus; ANDV, Andes virus; RVFV, Rift Valley fever virus. The phylogenetic trees based on HV L segment. The tree was generated using the Maximum Likelihood method, and 1000 replicates for bootstrap testing in MEGA 7.0 software. Only bootstrap values >60% were shown. The scale bar indicates nucleotide substitutions per site. The HV species’ name and complete genome of GenBank accession numbers of reference sequences were shown in each line.
DNA sequence analysis revealed 99.7% identity between three HV strains and Hantaan orthohantavirus detected in Myospalax psilurus (OQ129474); 99.4% identity between eight strains and Hantaan orthohantavirus detected in A. agrarius (KX775459); 98.8% identity between one strain and orthohantavirus hantanense detected in A. agrarius (KX775458); and 99.1% identity between one strain and SEOV detected in R. norvegicus (HM748794).
Among coinfected rodents, the HV infection rate was 92.3% (12/13) for HTNV and 7.7% (1/13) for SEOV.
For the Bartonella gltA gene: Three sequences (2, 4, 295) from A. agrarius showed 100% identity with uncultured Bartonella spp. sequences isolated from A. agrarius (DQ155392) and 95.56% identity with B. krasnovii sequences isolated from Synosternus cleopatrae (MH618795); Six sequences (8, 162, 165, 246, 296, 324) from A. agrarius showed 100% identity with B. fuyuanensis sequences isolated from A. agrarius (MH748113); One sequence (146) from A. agrarius showed 100% identity with uncultured Bartonella spp. sequences isolated from rodents (KJ175030) and 99.40% identity with B. grahamii sequences isolated from A. agrarius (AY584855); One sequence (149) from R. norvegicus showed 100% identity with B. tribocorum sequences isolated from R. tanezumi (PQ279879).
For the Bartonella rpoB gene: 11 sequences (2, 4, 8, 118, 146, 162, 165, 246, 295, 296, 331) from A. agrarius showed 100% identity with B. fuyuanensis sequences isolated from A. agrarius (MH748129); One sequence (324) from A. agrarius showed 100% identity with B. fuyuanensis sequences isolated from A. agrarius (MH748124); One sequence (149) from R. norvegicus showed 100% identity with B. tribocorum sequences isolated from R. norvegicus (MH748138).
Due to inconsistent gltA/rpoB matching results for some sequences and 100% identity of certain sequences with uncultured Bartonella spp., we concatenated the gltA and rpoB genes to ensure result accuracy and uniqueness. Phylogenetic analysis of concatenated sequences showed all A. agrarius-derived strains clustered with B. fuyuanensis, while one R. norvegicus-derived strain clustered with B. tribocorum (Fig. 2). Although gltA sequences were not detected in two samples (118, 331), their rpoB sequences confirmed clustering with B. fuyuanensis.
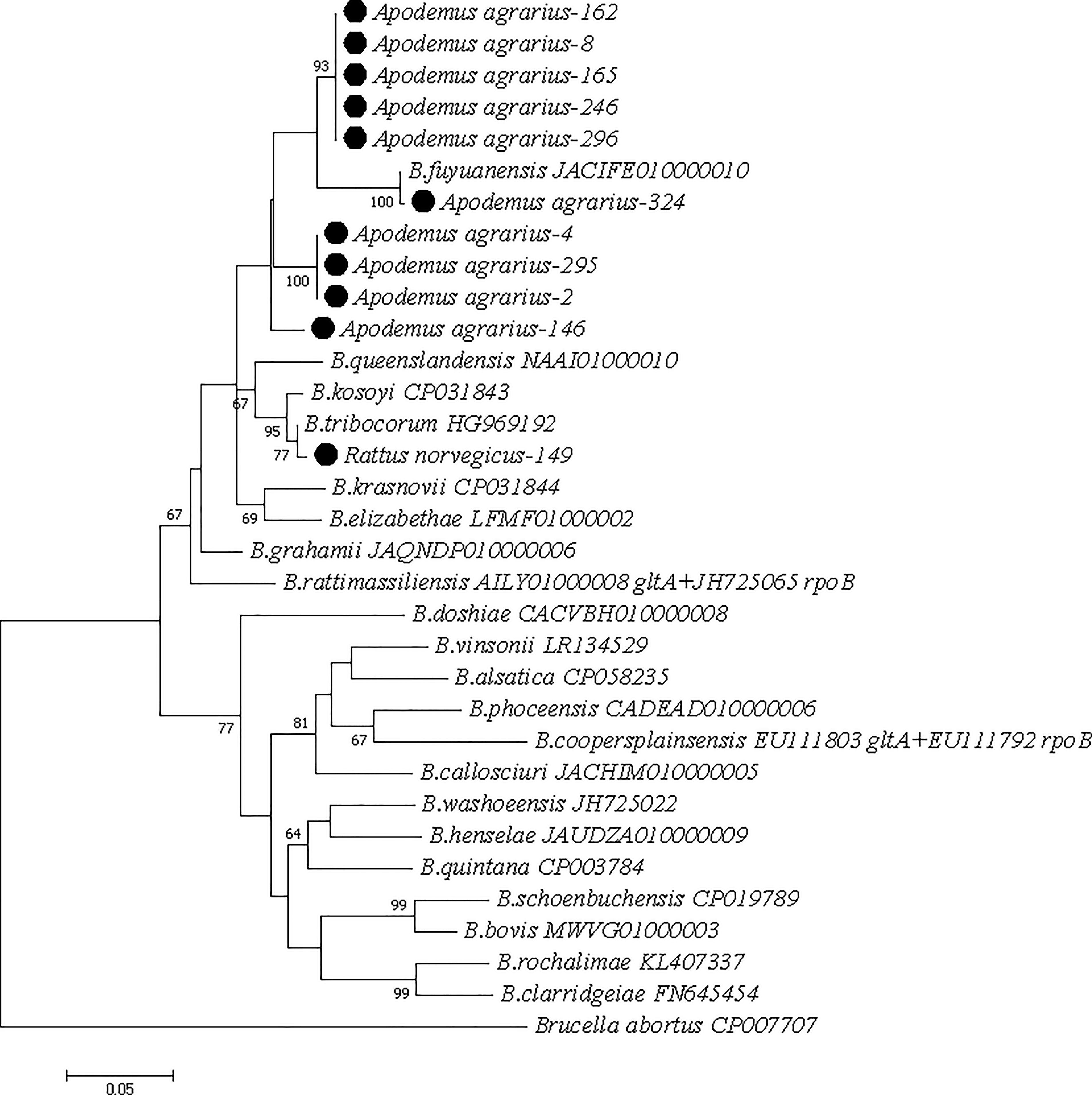
Phylogenetic tree of the concatenation of Bartonella spp. gltA and rpoB gene. Only the Bartonella sequence portions comparable to our sequence were utilized. Since the gltA fragment was not detected in Apodemus agrarius-118 and Apodemus agrarius-331, and only the rpoB fragment was detected, their genes were not concatenated; they were excluded from the alignment. The phylogenetic trees were constructed using the concatenated sequences of the gltA gene and rpoB gene. The tree was generated using the Maximum Likelihood method and 1000 replicates for bootstrap testing in MEGA 7.0 software. Only bootstrap values >60% were shown. The scale bar indicates nucleotide substitutions per site. The Bartonella species’ name and complete genome GenBank accession numbers of reference sequences were shown in each line.
Among rodents coinfected with HV and Bartonella spp., the Bartonella species infection rates were 92.3% (12/13) for B. fuyuanensis and 7.7% (1/13) for B. tribocorum.
The sequences of pathogens obtained in this study were deposited in GenBank with accession numbers: HV RdRp: PV394103–PV394107. Bartonella ITS: PV455977–PV455982. Bartonella gltA: PV394108–PV394111. Bartonella rpoB: PV421268–PV421270.
Discussion
This study investigated the prevalence and genetic diversity of Bartonella spp., HV, and their coinfections in rodents and shrews from Qingdao, China. To our knowledge, this is the first study focusing on such coinfections in this region. The coinfection rate ranged from 0.8% to 15.0%, with the highest rate observed in A. agrarius. A. agrarius is native to Asia, with a wide geographic distribution, and exhibits strong adaptability to diverse habitats (Han et al., 2015). Furthermore, A. agrarius is a known reservoir and vector for multiple zoonotic pathogens, including HV, Norovirus, Leptospira spp., Bartonella spp., and various parasites (Farkas et al., 2012; Hansen et al., 2015; Krügel et al., 2022; Zhang et al., 2022), posing a multifaceted risk for zoonotic disease transmission. Collectively, our findings suggest that the local human population may face a heightened risk of zoonotic disease exposure associated with A. agrarius and other rodent/shrew hosts.
Previous studies have reported Bartonella spp. infection rates in rodent populations across China, including 30.1% in Qinghai (Yu et al., 2022), 38.6% in the Qaidam Basin (Rao et al., 2021), 57.7% on Heixiazi Island (Li et al., 2015), 49.5% in the Zhongtiao Mountains (Latrofa et al., 2022), and 6.4% in Guangzhou (Yao et al., 2022). Meanwhile, HV infection rates in rodents from other regions have been documented as 5.0% in Shaanxi (Yu et al., 2015) and 4.1% in Jiangxi (Du et al., 2023). These discrepancies may be attributed to differences in rodent sample size, species composition, habitat characteristics, and arthropod vector abundance (Meheretu et al., 2013). Notably, the Bartonella spp. infection rate in A. agrarius in our study was significantly higher than the 11.1% (8/72) reported in Jiaonan (p < 0.001) (Qin et al., 2019). This difference may stem from urbanization and rising temperatures (Chen, 2021), which can alter rodent density and modulate vector–host interactions (Jonsson et al., 2024; Su et al., 2020; Xiang et al., 2018). Rapid urbanization drives the fragmentation of natural habitats and increases the population density of highly adaptable species (Galbraith et al., 2015; Hassell et al., 2017). Elevated temperatures prolong the reproductive period and expand the activity range of rodents, while also expanding the distribution of vectors and extending their transmission seasons (Matthee et al., 2020; Reisen, 2010). Collectively, these findings allow for the inference that A. agrarius may be emerging as an increasingly significant potential reservoir for bartonellosis.
Rodents and shrews in this study inhabited areas near human settlements and farmlands—habitats that increase their proximity to humans and frequency of human contact, thereby elevating the risk of human infection with the pathogens (Kerins et al., 2018; Su et al., 2020). Given the shared transmission routes between Bartonella spp. and HV (Morick et al., 2010; Wesley et al., 2010), coinfected rodents and shrews pose a heightened public health risk. Coinfections with multiple pathogens can significantly alter disease severity: they may induce more severe clinical manifestations, modulate host immune responses, and change host behavior—all of which can further enhance pathogen shedding and transmission (Bordes and Morand, 2011; Ezenwa, 2016; Garza-Cuartero et al., 2014; Vaumourin et al., 2015; Webster, 2001). Consequently, it can be inferred that humans coinfected with HV and Bartonella spp. may face a potential increase in the risk of severe health outcomes.
Coinfections involving zoonotic pathogens have been documented globally, such as dual infections of Anaplasma phagocytophilum and Bartonella spp., and triple infections of A. phagocytophilum, Bartonella spp., and Babesia spp. in the Netherlands (Wijburg et al., 2022). In our study, all coinfected A. agrarius harbored only B. fuyuanensis and HTNV, while one R. norvegicus was coinfected with B. tribocorum and SEOV, this finding confirms the host specificity of Bartonella spp. and HV, consistent with previous reports (Qin et al., 2019; Wang et al., 2020), and explains the significant variation in coinfection rates across rodent species.
Notably, A. agrarius exhibited the highest coinfection rate. To our knowledge, this is the first documentation of B. fuyuanensis–HTNV coinfection in A. agrarius. Previously, only Bartonella spp.–DOBV coinfection had been reported in A. agrarius in Croatia, with a much lower rate of 1.89% (1/53) (Tadin et al., 2016). Additionally, the coinfection rate in R. norvegicus (0.84%, 1/119) was significantly lower than the 11.8% reported in Guangzhou, China (Zhao et al., 2024). These differences may be attributed to variations in pathogen detection methods, rodent density, and environmental conditions. Guangzhou has a warm and humid subtropical climate that facilitates the reproduction of R. norvegicus populations and pathogen transmission. In addition, nucleic acid detection kits were employed for pathogen identification in the study conducted in Guangzhou.
The relatively high Bartonella spp.–HV coinfection rate in our study may be attributed to overlapping transmission routes between the two pathogens. Both can be transmitted via arthropod vectors (Bown et al., 2004; Chomel et al., 1996; Jiang et al., 2019; Kabeya et al., 2010; Shao et al., 2015). Additionally, susceptible rodents can spread HV and Bartonella spp. through the fecal–oral route; HV can also be vertically transmitted via the placenta (Kosoy et al., 1998; Swanink et al., 2018; Tołkacz et al., 2018), and mating or fighting among rodents further facilitates transmission of both pathogens (Forbes et al., 2018; Kosoy et al., 1998; Le Duc, 2023). More importantly, both pathogens can establish chronic infections in hosts, enabling long-term pathogen carriage and shedding (Bai et al., 2011; Ma et al., 2021). Collectively, these factors may increase the persistence and complexity of transmission risks, potentially altering rodent population size and structure, disrupting interspecific competition, and complicating epidemiological monitoring and traceability.
Interestingly, the infection rate of HV and coinfection in male A. agrarius was significantly higher than that in female. Given that both the Qingdao region and the Mecsek Mountain are located in the midlatitude regions of the Northern Hemisphere and share similar climatic and geographical characteristics, we speculate that this phenomenon may be attributed to the significantly higher survival rate of male A. agrarius during winter (Madai et al., 2020). Additionally, due to sex-specific behaviors such as aggression and biting, males may have advantages over females in terms of food competition and health status (Escutenaire et al., 2002). Therefore, we speculate that the underlying causes of this phenomenon are twofold. On the one hand, the behavioral characteristics and larger activity ranges of male A. agrarius may expose them to a higher risk of pathogen exposure. On the other hand, due to the immunomodulatory effect of testosterone (Klein and Calisher, 2007), male A. agrarius infected with HV tend to develop chronic infections rather than succumbing to the infection, thereby exhibiting higher survival rates.
In addition to sex, body mass/age was positively correlated with Bartonella spp. and HV infection in rodents. Previous studies had indicated that as body mass/age increases, pathogen infection rates also rise—likely due to cumulative exposure over time (Costa et al., 2014; Faria et al., 2008). A study on coinfection of HV and Leptospira also confirmed that older individuals have a higher likelihood of coinfection (Jeske et al., 2021). Given the opportunistic pathogenicity of Bartonella spp. (Krügel et al., 2022) and their ability to persist in hosts without causing severe harm (Schülein et al., 2001), older/heavier rodents are more likely to encounter and harbor Bartonella spp. This aligns with a study on R. norvegicus in Guangzhou, which found higher Bartonella detection rates in larger individuals and a positive correlation between body mass and HV prevalence (Zhao et al., 2024). Notably, in our study, Bartonella spp. infection rates in R. norvegicus did not correlate with increasing body mass (p > 0.05). This discrepancy may be explained by the narrower body mass range of our R. norvegicus specimens (78.3–94.9 g) compared with the broader range (105–201 g) reported in Guangzhou. Collectively, it can be inferred that larger, more mobile rodent species harboring multiple zoonotic pathogens may potentially expand pathogen transmission ranges, increase the likelihood of severe human infections, complicate clinical diagnosis and treatment processes, and elevate potential public health risks.
Our study had several limitations. First, the total sample size was insufficient, with small sample sizes for certain rodent and shrew species and a limited number of coinfection-positive specimens. This precluded multivariable analysis of the data, restricting us to univariate analyses of key factors (e.g., species, sex, body mass). Second, we did not investigate the effects of Bartonella spp.–HV coinfection on host immunology. This gap prevented us from exploring the biological consequences of coinfection at the molecular and immunological mechanistic levels. Third, body mass in rodents is influenced by factors beyond age: for example, rodents in warmer regions or areas with abundant food resources tend to have higher body mass than those in cooler or food-scarce environments, and individuals in better overall health typically weigh more than those in poor health. Finally, although positive controls play a more critical role in quantitative real-time PCR assays, the lack of such a control in the present study does carry a certain risk of false-negative results, which may lead to the missed detection of some pathogens. Accordingly, the negative findings of this study should be interpreted with caution. In conclusion, this study reports the first detection of B. fuyuanensis and HTNV coinfection in A. agrarius, identifying host age/mass and sex as influential factors. While direct evidence of human disease impact remains to be established, these results highlight the complexity of zoonotic pathogen dynamics in reservoir hosts. This underscores the necessity for integrated, long-term ecological and virological surveillance to better assess the potential risks associated with multipathogen infections in endemic regions.
Authors’ Contributions
S.-H.S., L.-Z.F., and H.-P.D. designed the study, and S.-H.S. performed the experiments. R.-Z.L., D.Z., and L.-Y.D. participated in collecting and processing the sample. S.-H.S., J.J., and K.-Z.L. wrote the article. K.-Z.L., H.-Q.Z., and H.-P.D. proofread the article. Z.-G.W. provided the financial support. All the authors read and approved the final article.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This research was supported by the Science and Technology Benefiting Policy of Qingdao (No.