
Abstract
Introduction
Cystoscopy-based diagnosis and surveillance of bladder cancer (BC) remain challenging. This study presents a urine-based assay that enriches DNA-methylation signals via PCR enrichment and applies a machine-learning model to enable cost-effective early detection.
Methods
In a prospective cohort at hospital (May 2022-November 2023), 155 individuals were enrolled, BC was diagnosed and confirmed by cystoscopy-guided biopsy and histopathology. Targeted next-generation sequencing of urine DNA quantified methylation at 44 CpG sites within IRF4, PENK and PXDN. Supervised classifiers trained on these features distinguished tumor from non-tumor urine samples. Systems analyses around IRF4/PENK/PXDN mapped signaling pathways, protein interaction modules and tumor-microenvironment contexts in BC.
Results
Using 155 urine samples (68 BC, 87 non-BC) with methylation and transcript expression data in TCGA-BLCA, we found significantly increased methylation of IRF4, PENK, and PXDN in BC (P < 0.0001). Corresponding mRNA levels of IRF4 and PENK were significantly downregulated in tumor tissues, with PXDN showing a declining trend. Methylation of IRF4 and PXDN negatively correlated with their expression (P < 0.0001). A 41-CpG-site random forest classifier targeting IRF4, PENK, and PXND (BladderCando model) demonstrated excellent performance in distinguishing BC from non-BC individuals (AUC = 0.9783, F1 = 0.9773), outperforming urine cytology for low-grade BC detection. Co-expression and enrichment analyses identified DCN as a central hub gene, primarily linked to ECM functions. High expression of PXDN, IRF4, and DCN correlated with upregulated immune checkpoint genes and increased immune cell infiltration. Single-cell sequencing revealed PXDN in fibroblasts and endothelial cells, DCN in fibroblasts, and IRF4 in T cells, with expression patterns in urine mirroring tumor tissue profiles.
Conclusion
This study establishes IRF4, PENK and PXDN methylation in urine as robust molecular signatures for BC detection, with machine-learning integration markedly enhancing diagnostic precision. Beyond early surveillance, these epigenetic alterations delineate tumor-microenvironment interactions that may inform future therapeutic strategies.
Introduction
Bladder cancer (BC) is prevalent globally with nearly 614,000 newly confirmed cases and 221000 deaths announced in 2022. 1 Meanwhile in China, approximately 92,900 cases and 37,600 deaths are reported and male patients occupy about 80% of morbidity and mortality. 2 Generally, suspicious BC patients with symptoms of painless hematuria are diagnosed through cytology, followed by transurethral resection of the bladder tumor (TURBT). The wide accepted pathologically categorical scheme classifies bladder cancer into non-muscle invasive BC (NMIBC) and muscle invasive BC (MIBC), accounting for 75%-80%, 20%-25% respectively. Due to the high recurrence (31%-78%) and progression (1-45%) rates of NIMBC, continuing surveillance is inevitable and in rising demand. 3 Voided urine cytology and urethrocystoscopy (CYSTO) are currently most common strategies for early diagnosis and regular long-term follow-up. 4 Since voided urine cytology has limited sensitivity (16%) for low-grade (LG) BC and CYSTO is invasive and costly, both monitoring methods do not meet increasingly demanding clinical needs.5,6 Furthermore, the NMP22 BladderChek Test, approved by the FDA in 2002, is a clinically validated biomarker assay that plays a significant role in monitoring bladder cancer patients. But elevated NMP22 levels are not exclusively indicative of bladder cancer, as false-positive results have been documented in clinical contexts. Nonmalignant conditions, including urinary tract infections (UTIs), nephrolithiasis, or recent urological interventions involving the bladder, may contribute to increased NMP22 concentrations. 7 Thereby, a novel non-invasive, cost-effective and accurate monitoring tool is an urgent to be implemented in clinical diagnosis and routine surveillance of BC. The rapid development of molecular biology might provide feasible solutions.
DNA methylation has showing promising prospect in the field of the early diagnosis of various types of cancers. Multi-cancer detection and localization using methylation signatures in cell-free DNA has been widely analyzed and recognized as a relatively valid methodology to distinguish cancer and non-cancer, even could localize the tissue of origin in some extent. 8 In the promoter region of tumor suppressor genes, DNA hypermethylation is thought to be one of the earliest detectable events in bladder cancer development. 9 This methylation alteration pattern has emerged as a prospective urinary biomarker strategy, given its crucial role in downregulating tumor suppressor genes during early BC stages through hypermethylation.10,11 Compared with blood samples, the rate of methylation is relatively high and more stable in urine samples. 12 Voided urine is also known as an easy and repeatably accessible liquid biopsy source to obtain cfDNA from different sites of the urinary system. 13 Several preliminary research projects reveal inspiring preclinical performance, Hentschel et al. 4 identified hypermethylation of GHSR/MAL as urinary biomarkers appealing potential diagnostic value. The preclinical results achieved 80% sensitivity and 93% specificity with an area of under curve (AUC) of 0.89. Costa et al. 14 designed a three-gene (GDF15, TMEFF2, and VIM) panel to detect the methylation alterations in the promoter region via both tissue and urine samples, obtaining terrific sensitivity (94%) and specificity (100%). However, most ongoing tests have not proven their robustness and accuracy in clinical practice,3,5 some of them are unaffordable and require stringent storage environments. The other reason causes big concern in clinical practice is that there is lack of strong evidence to validate these biomarkers in real world. 15 Thereby cytology and cystoscopy are still the prior methods in clinical setting.
DNA methylation serves as both a cellular identifier and a key epigenomic regulator, with its stability within the genome playing a critical role in maintaining DNA structure. Methylation of cytosine residues not only enhances DNA helix stability but also increases its thermal denaturation temperature, improving the preservation of clinical samples. These characteristics make DNA methylation a reliable target for detection using methylation-specific PCR. In this study, we focused on three bladder cancer (BC)-associated genes, examining their methylation changes through targeted PCR enrichment. By integrating these epigenetic markers with machine learning algorithms, we aimed to develop an efficient, cost-effective diagnostic model for BC. This approach could potentially reduce invasive procedures and provide an early detection system for tumor recurrence, enhancing both diagnosis and disease monitoring.
Methods
Statement
The reporting of this study conforms to STARD guidelines. 16 All patients’ details were de-identified that the identity of any individual cannot be ascertained by any means.
Patient Population and Diagnosis
We conducted this retrospective clinical study at hospital. The objective was to establish a DNA methylation pattern-based machine learning model to distinguish bladder cancer and determine its sensitivity and specificity. Between May 2022 and November 2023, patients with suspected bladder tumors, identified via cystoscopy-guided biopsy, were enrolled at hospital. Participants meeting predefined inclusion/exclusion criteria provided urine specimens following standard protocols. Diagnoses integrated clinical symptoms with histopathological confirmation. Biopsies adhered to National Comprehensive Cancer Network (NCCN) or Chinese Society of Clinical Oncology (CSCO) guidelines for bladder malignancies. Clinical data were systematically recorded (see Supplemental Table 1).
Eligibility criteria for study participation were defined as follows. Inclusion criteria comprised: (1) Adult patients (aged >18 years) newly diagnosed with bladder cancer (BC) through endoscopic, imaging, histopathological, or biopsy confirmation; (2) Individuals diagnosed with benign urological conditions (e.g., urinary calculi, benign prostatic hyperplasia, renal cysts) or non-bladder malignancies (e.g., colorectal carcinoma, renal cell carcinoma, prostate adenocarcinoma); (3) Asymptomatic healthy adults with no clinically significant abnormalities detected during routine physical evaluations.
Exclusion criteria were applied to: (1) BC patients presenting with concurrent primary malignancies; (2) BC patients who had undergone prior antitumor therapies (including chemotherapy, radiotherapy, or surgical resection) preceding enrollment; or (3) Participants with non-compliant urine sample collection, storage, or processing protocols, or specimens failing to meet predefined quality control metrics.
Sample Collection and DNA Extraction
The entire DNA methylation analysis processes were divided into three steps. The first step was to extract DNA from urine samples. Biopsy specimens were submitted to the pathology department for formalin fixation, paraffin embedding, hematoxylin-eosin staining, and blinded histopathological evaluation by two independent board-certified pathologists. Urine samples were collected from the middle part of morning urine and participants were required to remain NPO the day before urine collection. To preserve nucleic acid integrity during clinical handling and preprocessing phases, urine samples were immediately transferred to specialized preservation tubes (Yunying Biotechnology, Zhejiang, China) post-collection. Each tube was gently inverted 3–5 times to ensure homogeneous interaction between the specimen and stabilizing solution, followed by refrigeration at 4°C for short-term storage prior to downstream processing.
Prior to nucleic acid isolation, preserved specimens were homogenized via 5-second vortex agitation. A 50 mL aliquot of urine was systematically transferred using triple-phase pipetting to eliminate potential stratification artifacts. Automated DNA extraction and purification were performed using the Human DNAplus isolation kit (Yunying Biotechnology, Zhejiang, China) integrated with the Auto-Pure20 workstation (Allsheng Instruments, Zhejiang, China). Bisulfite modification of purified DNA was subsequently executed with the MetPro bisulfite conversion system (Yunying Biotechnology, Zhejiang, China), following standardized manufacturer specifications. Processed DNA eluates were cryopreserved at -20°C in temperature-regulated environments pending analytical procedures.
Library Preparation and Targeted Methylation DNA Sequencing
After DNA extraction, the obtained DNA was amplified by PCR, followed by bisulfite sequencing of the amplification products. The BC-uDMR NGS panel, developed by Jiaxing Yunying Medical Testing Co., Ltd., is a specialized assay for detecting DNA methylation in urine samples for bladder cancer diagnosis. The panel targets 44 specific CpG sites across three genes—IRF4, PENK, and PXDN. Detailed information on these methylation sites and the corresponding primers is provided in Supplemental Table 2.
Bisulfite-treated DNA mentioned was utilized in a multiplex PCR assay to generate barcoded sequencing libraries. Each 30 μL first-round reaction contained 19.5 μL of mixed reaction buffer (Vazyme, Nanjing, Jiangsu, China), 5 μL of template DNA, 5 μL of a pre-designed primer mix (2 μM each primer; synthesized by Sangon, Shanghai, China; see Table S2), and 0.5 μL of AceTaq DNA polymerase (Vazyme). PCR was performed in a CFX96 PCR machine (Eastwin, Beijing, China) under the following conditions: 95°C for 10 min, 35 cycles with an increment of 0.2°C per cycle (95°C for 30 s, 46–53°C for 30 s, 72°C for 30 s), followed by 72°C for 5 min, and a final hold at 4°C.
The second-round multiplex PCR was performed in a 30 μL reaction containing 20.5 μL of mixed reaction buffer (Vazyme), 2 μL of adapter primer (Sangon), 2 μL of index primer (Sangon), 0.5 μL of AceTaq DNA polymerase (Vazyme), and 5 μL of the first-round PCR product. Cycling conditions were as follows: 95 °C for 5 min; 20 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; with a final extension at 72 °C for 5 min and a hold at 4 °C.
PCR products were purified using a DNA magnetic bead purification kit (Yunying, Jiaxing, Zhejiang, China). Library concentrations were quantified with the EqualBit 1× dsDNA HS Assay Kit (Vazyme). Paired-end sequencing (150 bp) was performed on a MiniSeq platform (Illumina, San Diego, CA, USA) using the MiniSeq Mid Output Reagent Cartridge (Illumina), following the manufacturer’s instructions (Illumina, 2023).
Sequencing Data Processing
The acquired bisulfite sequencing data then underwent analysis with a standard NGS pipeline. The raw data BCL files were subjected to quality control using Sequencing Analysis Viewer (v1.8; Illumina, San Diego, CA, USA). Sequences with a Q30 base percentage exceeding 70% were retained as high-quality. The bcl2fastq software (v2.20.0.422; Illumina) was used to convert qualified BCL files to FASTQ files. After adapter and low-quality base removal with fastp (v0.20.1), 17 the Burrows-Wheeler Aligner (BWA; v0.7.17) 18 aligned sequences from quality-controlled FASTQ files to a custom methylated reference sequence (GRCh37). Aligned sequences were sorted by genomic coordinates using samtools (v1.2) 19 to generate BAM files and indices. Base depth analysis was performed with igvtools (v2.3.98), 20 filtering out sites with sequencing depth below 1000×. Methylation levels at CpG and non-CpG sites were calculated using the internally developed MetSeq module (Yunying, Jiaxing, Zhejiang, China). Quality control metrics Q30 percentage, sequencing depth, methylation rates were aggregated into a TSV file.
Constructing BladderCando Models for Cancer Diagnosis
Machine learning models were developed to distinguish cancer and non-cancer participants using per-locus DNA methylation rates as features. A total of 44 CpG sites across three candidate genes (IRF4, PENK, and PXDN) were quantified in 155 samples. The per-locus DNA methylation rates were further examined by the Benjamini-Hochberg method to control the false discovery rate under 1% and 3 loci were removed. Missing values were observed exclusively in the non-cancer group. To avoid data leakage, missing values were imputed using the mean methylation value calculated from the corresponding training fold only, and the imputation parameters were subsequently applied to the testing fold.
To systematically evaluate the classification contribution of each gene and their combinations, seven datasets were constructed: three single-gene datasets (IRF4, PENK, PXDN), three dual-gene datasets (IRF4+PENK, IRF4+PXDN, PENK+PXDN), and one three-gene dataset (IRF4+PENK+PXDN). Each dataset contained the CpG sites corresponding only to the specified gene set, enabling assessment of incremental predictive value when additional genes were incorporated. For each dataset, three supervised machine learning classifiers were implemented: Support Vector Machine (SVM), Random Forest (RF), and Extreme Gradient Boosting (XGBoost). The parameters of each model were obtained from the GridSearchCV algorithm (5-fold, ‘scoring’ method ‘f1’). ‘C’, ‘gamma’ and ‘epsilon’ were selected as the parameters of SVM models need to be optimized; ‘n_estimators’, ‘max_depth’, ‘min_impurity_decrease’ were the parameters under optimizing of random forest models, while ‘criterion’ was set to ‘entropy’ and ‘random_state’ was 42; in XGBOOST, the underlying parameters included ‘max_depth’, ‘min_child_weight’, ‘gamma’, ‘subsample’, ‘colsample_bytree’ and ‘reg_alpha'.
Model performance was evaluated using the area under the receiver operating characteristic curve (AUC) and F1 score. AUC was selected to quantify overall discriminative ability independent of threshold selection, whereas F1 score was used to assess the balance between sensitivity and precision, particularly under potential class imbalance. To account for sampling variability and ensure that results were not dependent on a single train-test split, a bootstrapping strategy with 120 iterations was applied. In each iteration, samples were resampled with replacement from the training set, models were retrained, and performance metrics were recorded in the testing set. Mean performance and 95% confidence intervals were calculated across all iterations.
Across all 21 candidate models (7 gene-combination datasets × 3 classifiers), the final BladderCando model was selected based on predefined criteria: 1) High mean AUC and F1 score in the testing set; 2) Stable performance across bootstrap iterations (narrow confidence intervals). The model demonstrating both strong discriminative performance and robustness was chosen for subsequent analyses.
Furthermore, DNA methylation is influenced by environmental and demographic factors, especially smoking status and age. In order to explore the potential impact of smoking status and age on the final model, we treated smoking status and age as additional features along with the DNA methylation rates. The AUC of ROC curve and f1 scores were utilized to evaluate the influence of smoking status and age.
Integrated Bioinformatics Analysis of Gene Co-expression Networks
To further explore the potential biological roles and molecular mechanisms associated with IRF4, PENK, and PXDN in BC, we conducted an integrated bioinformatics analysis using TCGA BLCA transcriptomic data. Gene co-expression analysis was performed using the LinkFinder module of the LinkedOmics database (https://www.linkedomics.org) in a cohort of BLCA samples (n = 408). Pearson correlation coefficients were calculated to assess gene-gene associations and identify genes significantly correlated with the expression of IRF4, PENK, and PXDN. Common co-expressed targets were subsequently subjected to Gene Ontology (GO) functional enrichment analysis to elucidate their involvement in biological processes and molecular pathways. A protein–protein interaction (PPI) network was constructed using the STRING database (confidence score > 0.4) and visualized with Cytoscape software (v3.7.2). To further evaluate the potential immunological relevance of these genes, immune-related analyses were performed using the IOBR 2.0 platform. Based on bulk RNA-seq expression profiles, the ESTIMATE and TIMER algorithms were applied to calculate tumor immune infiltration scores.
Single-Cell RNA-Seq Analysis
To characterize the cell-type-specific expression patterns of IRF4, PENK, and PXDN in BC, we analyzed the publicly available single-cell RNA sequencing (scRNA-seq) dataset GSE267718 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE267718), generated using the 10X Genomics platform. Quality control was performed based on unique molecular identifier (UMI) counts and the proportion of mitochondrial gene expression per cell, and low-quality cells were excluded. After data normalization, highly variable genes were identified for downstream analyses.
Following data normalization, we identified highly variable genes and conducted principal component analysis (PCA) to reduce dimensionality. Based on the top 30 principal components, we applied Uniform Manifold Approximation and Projection (UMAP) to visualize the high-dimensional data in a two-dimensional space. Subsequently, cell clustering was performed using the FindClusters function in the Seurat package, with the clustering resolution set to 0.3 to achieve clear delineation of cell populations while avoiding over-segmentation. Prior to clustering, we applied the Harmony algorithm to correct for batch effects and eliminate systematic biases across samples. Cell type annotation was performed based on canonical marker genes identified through differential expression analysis, conducted using the FindAllMarkers function in Seurat. Marker genes were defined by a log-fold change of no less than 0.25 and expression in at least 25% of the cells within the corresponding cluster.
Statistical Analysis
Statistical analysis and data visualization were conducted using Python (v.3.9) and R (v4.3.2). Significance analysis of intergroup differences was conducted based on the normality of data distribution, using Wilcoxon rank-sum test. The ‘sklearn’ package was used to build the random forest model and was used to calculate ROC curves and f1 scores for evaluating the performance of the machine learning models as well. 95% confidence intervals of the confusion matrix were calculated according to the efficient-score method (corrected for continuity) described by Robert Newcombe, based on the procedure outlined by E. B. Wilson in 1927. Various plots, including ROC curves, boxplots, were created using the Python package “matplotlib” and the R package “ggplot2”.
Results
Patient Enrollment and Clinical Characteristics
As depicted in Figure 1, a total of 201 participants were initially enrolled in this study. Of these, 46 participants were excluded due to the exclusion criteria, while 25 candidates refused to undergo cystoscopy, indicating a relatively low acceptance rate for this invasive diagnostic procedure. Consequently, 155 participants were included in the final analysis. Pathological examination confirmed bladder cancer in 68 patients (43.9%), comprising 52 high-grade, 15 low-grade, and 1 case of unspecified grade. The remaining 87 participants were assigned to the non-bladder cancer group, consisting of 57 patients with non-malignant lesions and 30 healthy volunteers. The overall cohort had a mean age of 60.8 years (SD 18.8; median 65, range 22-92) and was predominantly female (85.2%). Most participants were non-smokers (78.7%) and reported minimal or no alcohol consumption (91.0%); hematuria was present in 29.7% of cases. Among the confirmed cancer cases (n=68), 33.5% were high-grade, and the tumor stages were as follows: Tis/Ta 4.5%, T1 14.8%, and ≥T2 24.5% (Table 1). The flowchart of total enrollment participants and the detailed number of participants excluded in each step Clinical Characteristics of Participants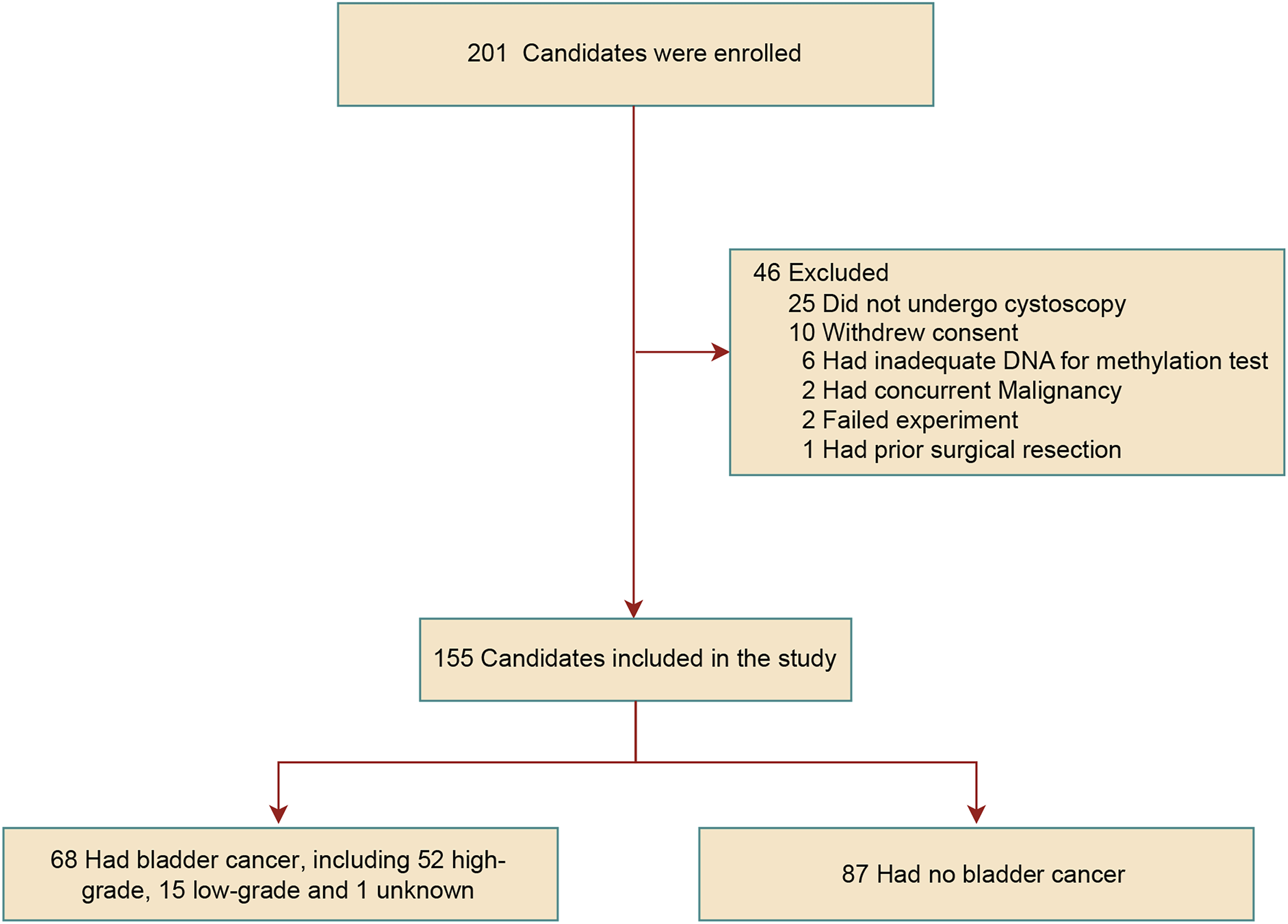
Methylation Characteristics of Bladder Malignant Tumors
The targeted methylation sequencing data for all 155 urine samples passed the quality control standards. To further explore the methylation differences between the BC group and the non-BC control group, we calculated the average methylation level of all CpG sites for the three genes in the BC-uDMR NGS panel, defining this as the average methylation rate of the gene. Statistical analysis revealed that the methylation rates of IRF4, PENK, and PXDN were all significantly higher in BC patients compared to non-bladder cancer groups (Mann-Whitney U test, P < 0.0001, Figure 2C). To further validate this result, we also analyzed the methylation differences of these three genes between cancerous and adjacent tissues in the TCGA-BLCA cohort using 450K methylation data from the UALCAN database. The results were highly consistent with our findings (P < 0.0001, Figure 2A). Fundamental characteristics of IRF4, PENK and PXDN. (A) Methylation levels of the IRF4, PENK and PXDN in UALCAN databases between primary bladder urothelial cancer and normal cases (t test, **** p<0.0001). (B) Expression levels of the IRF4, PENK and PXDN in UALCAN databases (t test, * p<0.05, ns not significant). (C) Average methylation levels of IRF4, PENK and PXDN in bladder cancer and non-bladder cancer subgroups (Mann-Whitney U Test, **** p<0.0001)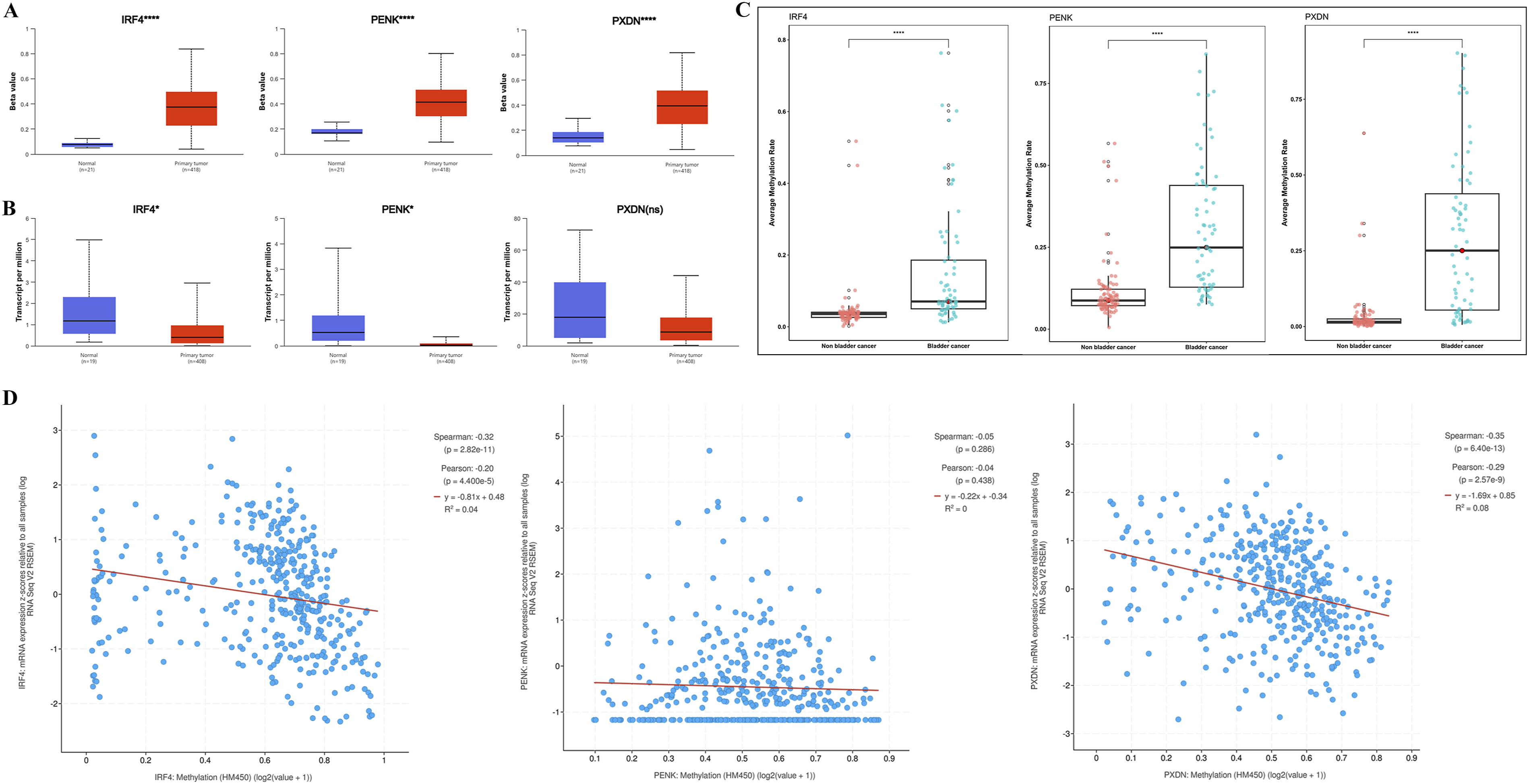
To evaluate the impact of methylation on gene expression, we concurrently analyzed mRNA expression levels of IRF4, PENK, and PXDN in the TCGA-BLCA dataset. The results demonstrated significantly reduced mRNA expression of these genes in tumor tissues compared to adjacent normal tissues (Figure 2B). Although PXDN did not reach statistical significance, its median expression level exhibited a marked decline. Further correlation analysis revealed strong inverse relationships between methylation rates and mRNA expression for IRF4 and PXDN, with Spearman ρ = -0.32 and -0.35, and Pearson r = -0.20 and -0.29, respectively (all P<0.0001, Figure 2D). In contrast, PENK showed no significant correlation between methylation and expression.
Establishment and Application Evaluation of Machine Learning Models
In this study, 155 participants (68 BC cases, 57 non-BC individuals, and 30 healthy controls) were stratified and randomly allocated into a training set (n=103) and a testing set (n=52) at a 2:1 ratio. Clinical baseline characteristics and disease status proportions remained balanced between the two cohorts (Table 1). Using SVM, RF, and XGBoost, we constructed seven classifiers from single genes (IRF4, PENK, PXDN) and their combinations to distinguish BC from non-cancerous individuals, with hyperparameters optimized by GridSearch.
Among the various alternative models, those based on XGBoost and RF classifiers demonstrated superior overall performance compared to SVM (Supplemental Figure S2). Notably, the 41-CpG-site random forest classifier targeting IRF4, PENK, and PXND exhibited a relatively stable performance and achieved the optimal results in the testing set, with an AUC of 0.976 and an F1 score of 0.9545 (Figure 3A). Importantly, the bootstrapping results illustrated the 41-CpG-site random forest classifier targeting IRF4, PENK, and PXND had reliable performance with the most accurate and robust performance, where AUC was 0.9732 (95%CI, 0.9497-0.9925) and f1 score was 0.9237 (95%CI, 0.8443-0.9778) in the testing set (Figure S2, Table S5-6). As a result, the 41-CpG-site random forest classifier targeting IRF4, PENK, and PXND was selected as BladderCando model for subsequent analyses. Characteristic analysis of model BladderCando. (A) ROC curves and f1 scores of the random forest models based on the combinations of IRF4, PENK and PXDN. (B) Boxplots overlaid with dot plots of displaying the distribution of BladderCando scores in bladder cancer and non-bladder subgroups and the distribution of BladderCando scores stratified by various clinical information, containing age, gender, tumor stage, tumor grade and smoking status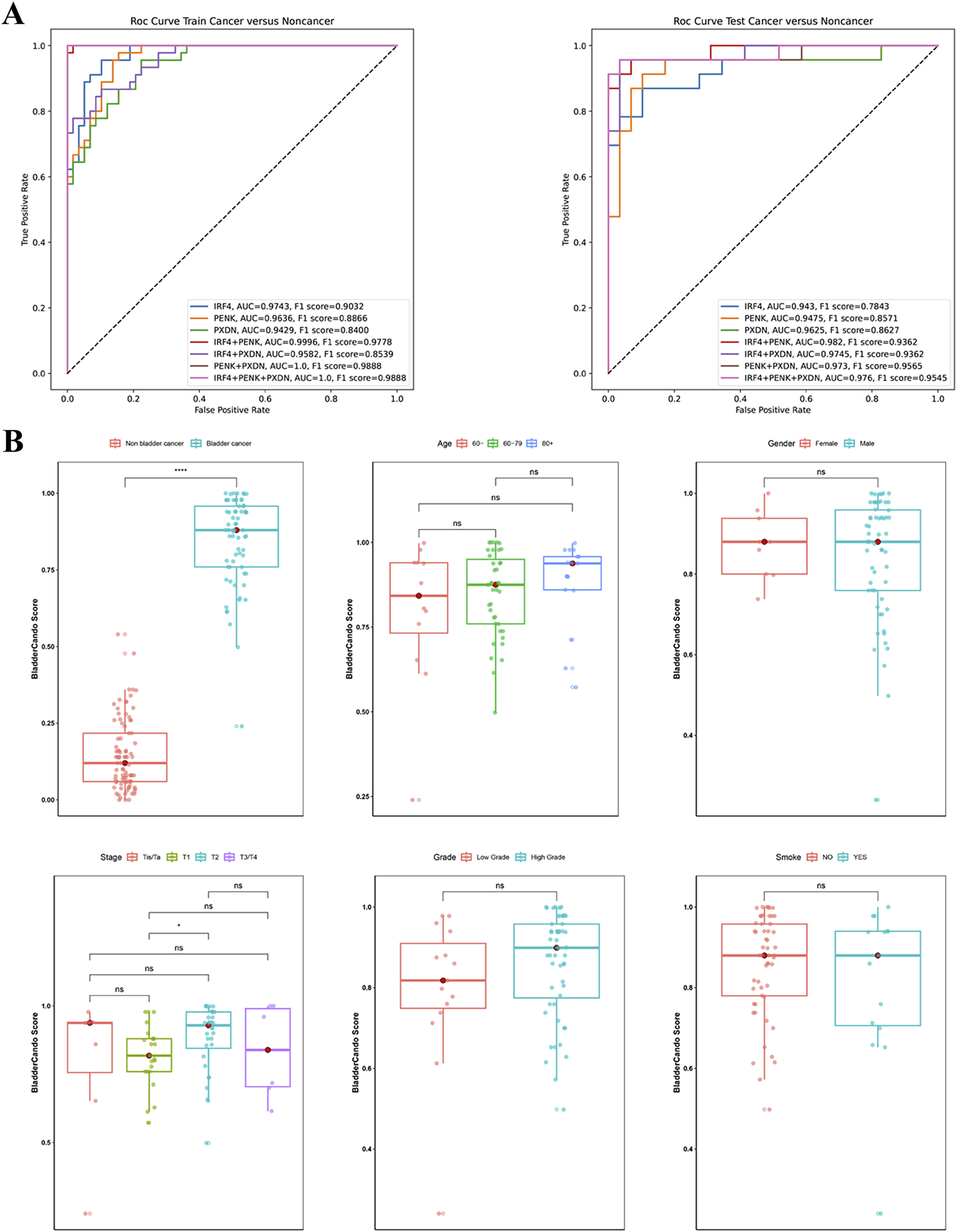
Model Performance
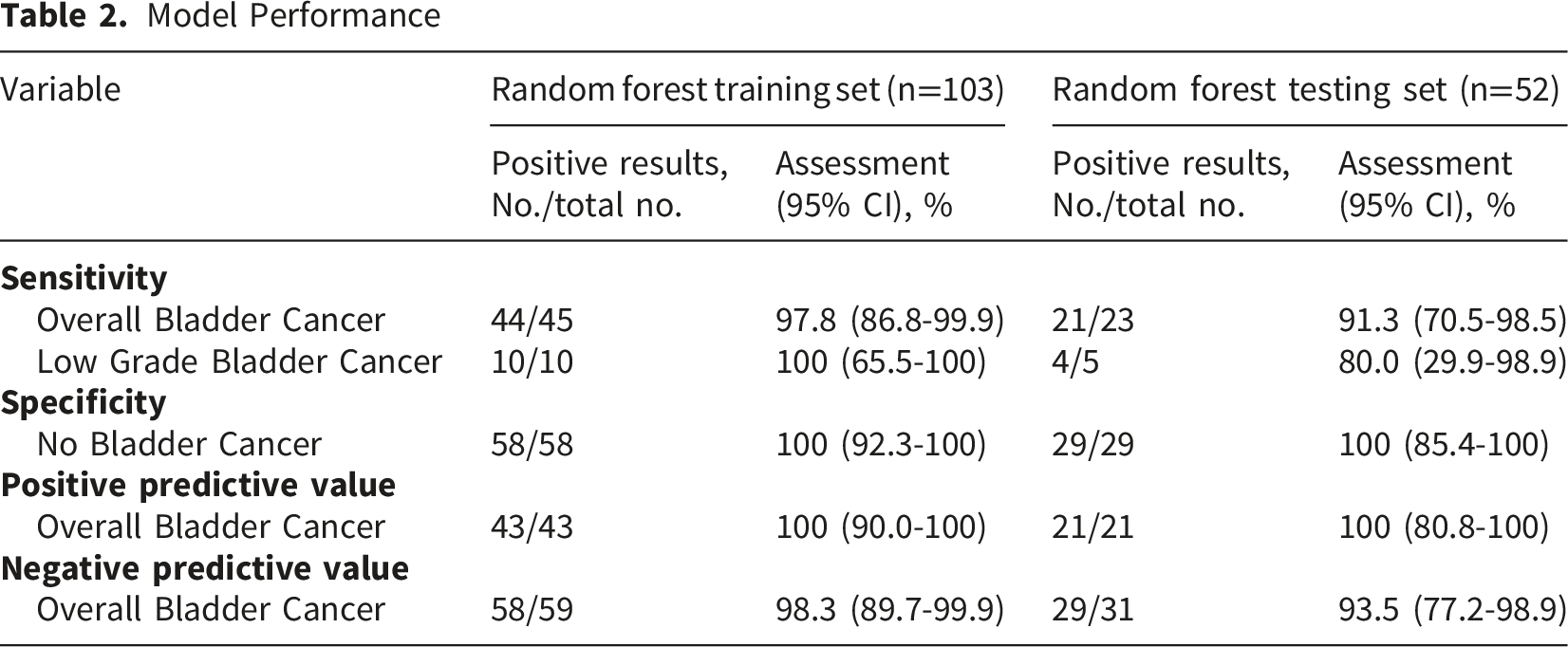
To assess the impact of clinical variables on the BladderCando score (a methylation-based diagnostic index ranging from 0 to 1), we compared scores across age groups (<60 vs. 60∼79 vs.≥80 years), smoking status (smoker and non-smoker), genders, tumor stages (Tis/Ta, T1, T2, and T3/T4), and grades (LG vs. HG). No significant differences were observed between smoking status (P>0.05, Mann-Whitney U test) and genders (P>0.05, Mann-Whitney U test), across age groups (P>0.05, Kruskal-Wallis test), or among tumor stages (P>0.05, Kruskal-Wallis test). However, LG tumors exhibited significantly lower scores than HG tumors (P<0.05, Bonferroni-adjusted) (Figure 3B).
Models using smoking status and age as additional features showed no improvement of performance of our final model (Figure S1), indicated that smoking status and age had limited impact on the construction of the final random forest model. The BladderCando scores of different age groups and smoking status illustrated no significant statistical difference as well (Figure 3B).
DCN as a Central ECM Regulator in IRF4/PXDN/PENK-Associated Co-Expression Networks
According to the GeneCards database, IRF4 regulates immune responses and may influence tumor immunogenicity. PXDN is linked to fibrosis, potentially contributing to tumor-associated stromal changes. PENK modulates pain perception but could also play a role in the tumor microenvironment by affecting signaling pathways related to pain and inflammation, influencing cancer progression (Supplemental Table 2).
To further elucidate the mechanisms of IRF4, PENK, and PXDN in bladder carcinogenesis, we performed co-expression analysis using the LinkedOmics database, which identified 777, 383, and 1,084 genes statistically significantly correlated with IRF4, PENK, and PXDN expression, respectively (false discovery rate <0.05). Intersection analysis via a Venn diagram revealed 63 overlapping genes among these co-expressed gene sets (Figure 4A, Supplemental Table 3). PPI network analysis of the 63 overlapping genes identified decorin (DCN) as the hub protein exhibiting the highest degree centrality, connecting to the three target genes (Figure 4B). GO enrichment analysis of the 63 genes revealed significant enrichments across all three domains: Biological Processes (BP) predominantly involved extracellular matrix and structure organization, external encapsulating structure formation, wound healing, and collagen fibril assembly; Cellular Components (CC) were enriched in collagen-containing extracellular matrices, collagen trimers, and presynaptic active zone membrane constituents (including intrinsic and integral membrane components); Molecular Functions (MF) featured extracellular matrix structural constituents, serine-type endopeptidase/peptidase/hydrolase activities, and structural molecules conferring tissue elasticity (Figure 4C, Supplemental Table 4). Gene association and immune infiltration analysis. (A) Venn diagram of differentially expressed genes related IRF4, PENK, PXDN. (B) WGCNA. WGCNA, weighted gene co-expression network analysis. (C) The GO outcomes are displayed with bar plots. GO, Gene Ontology; BP, biological process; CC, cellular component; MF, molecular function. (D) Comparison of 3 immune checkpoint gene expression in high-expression and low-expression groups of IRF4, PENK, PXDN and DCN, representing via split violin plots. (E) Immune infiltration analysis of StromalScore, ImmuneScore and ESTIMATEScore. (F) Correlation with immunecytes via TIMER2.0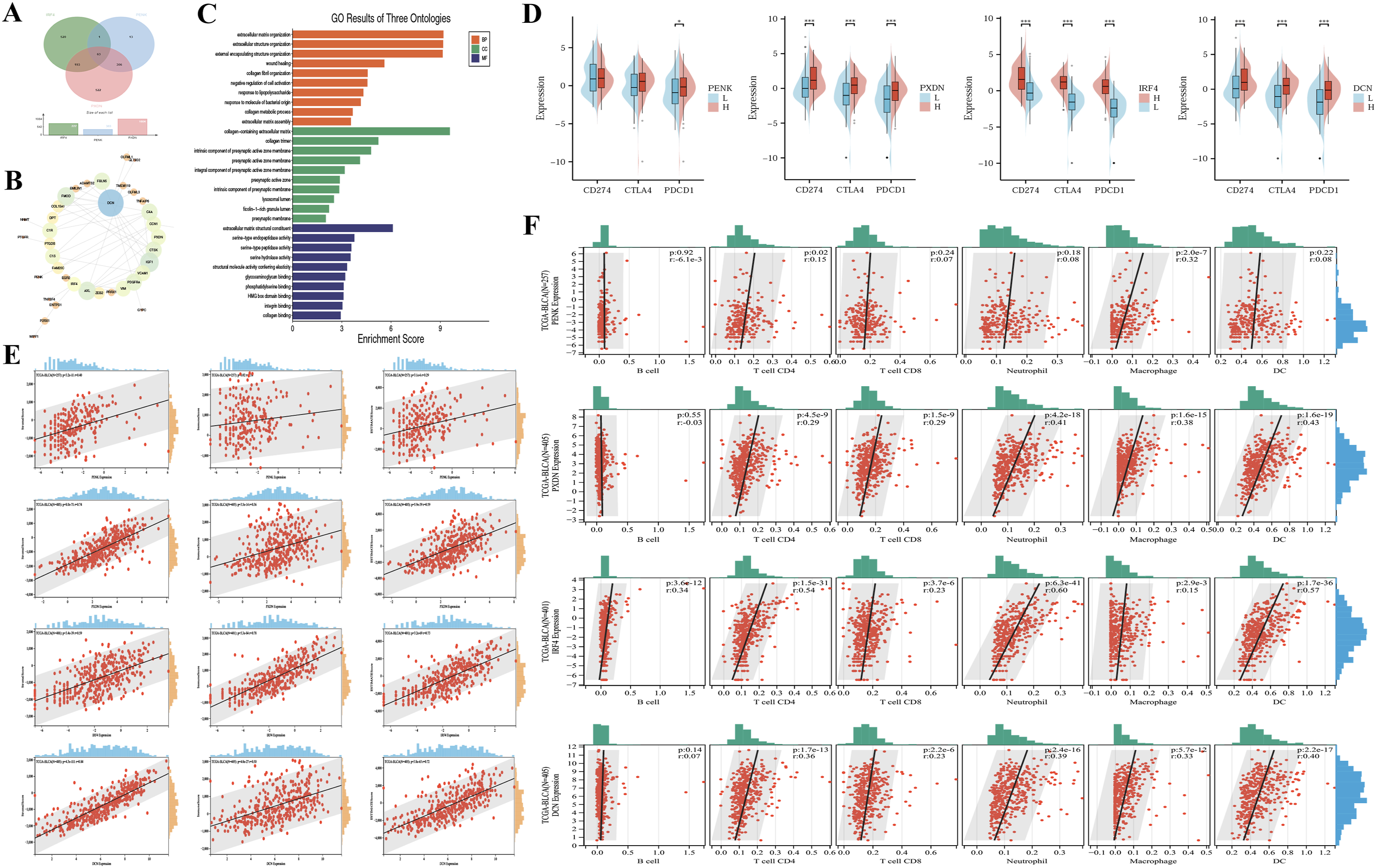
IRF4, PXDN, DCN, and PENK Link Immune Checkpoint Expression to Tumor Immune Infiltration
Pathway analysis revealed that IRF4, PENK, and PXDN, along with their co-regulated genes, are predominantly involved in extracellular matrix (ECM) organization and collagen-related biological processes, both of which are closely associated with immune regulation. To investigate the potential immunomodulatory implications, we further evaluated the correlation between core immune checkpoint genes (CD274, CTLA4, PDCD1) and the expression levels of IRF4, PENK, PXDN, as well as the hub gene DCN. In the TCGA-BLCA cohort, patients with high expression of PXDN, IRF4, or DCN exhibited significantly elevated mRNA levels of CD274, CTLA4, and PDCD1 compared to low-expression subgroups (P < 0.001, Figure 4D). Notably, no such association was observed for PENK.
Subsequent immune microenvironment analysis demonstrated that high-expression groups of PENK, PXDN, IRF4, and DCN displayed significantly enhanced stromal, immune, and ESTIMATE scores relative to low-expression counterparts (Figure 4E, P < 0.01). Timer Immune cell infiltration profiling further revealed that elevated expression of PXDN, IRF4, and DCN correlated positively with increased infiltration of CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells (DC). Specifically, IRF4 expression additionally showed a positive association with B cell infiltration, while PENK expression was uniquely linked to CD4+ T cell and macrophage infiltration (P < 0.01, Figure 4F).
Concordant Cell-type-specific Expression Patterns in Tumor and Urine Samples
To investigate the differential expression of PENK, PXDN, IRF4, and DCN within the tumor microenvironment (TME), we analyzed scRNAseq data from dataset GSE267718, which comprises paired tumor tissue and urine samples from eight bladder cancer patients. UMAP dimensionality reduction and clustering were performed on the eight tumor tissue samples, and cell clusters were annotated using canonical marker genes. Identified cell populations included T cells, B cells, erythroid-like and erythroid precursor cells, macrophages/monocytes, endothelial cells, fibroblasts, neutrophils, epithelial cells, cancer cells and unknown (Figure 5A-B). Cell-type-specific expression analysis revealed that PXDN is predominantly expressed in fibroblasts and endothelial cells, DCN is enriched in fibroblasts, IRF4 is significantly enriched in T cells, while PENK shows relatively low expression across the annotated cell types (Figure 5C-D). Single-cell scRNA sequencing data analysis in tissue samples. (A) Stacked bar plots of distribution of various cell types in GEO datasets. (B) UMAP plots show the cell clonal clusters. UMAP, Uniform Manifold Approximation and Projection. (C) Gene expression levels of various cells based on the landscape of UMAP plots. (D) Distribution of gene expression levels in the different cells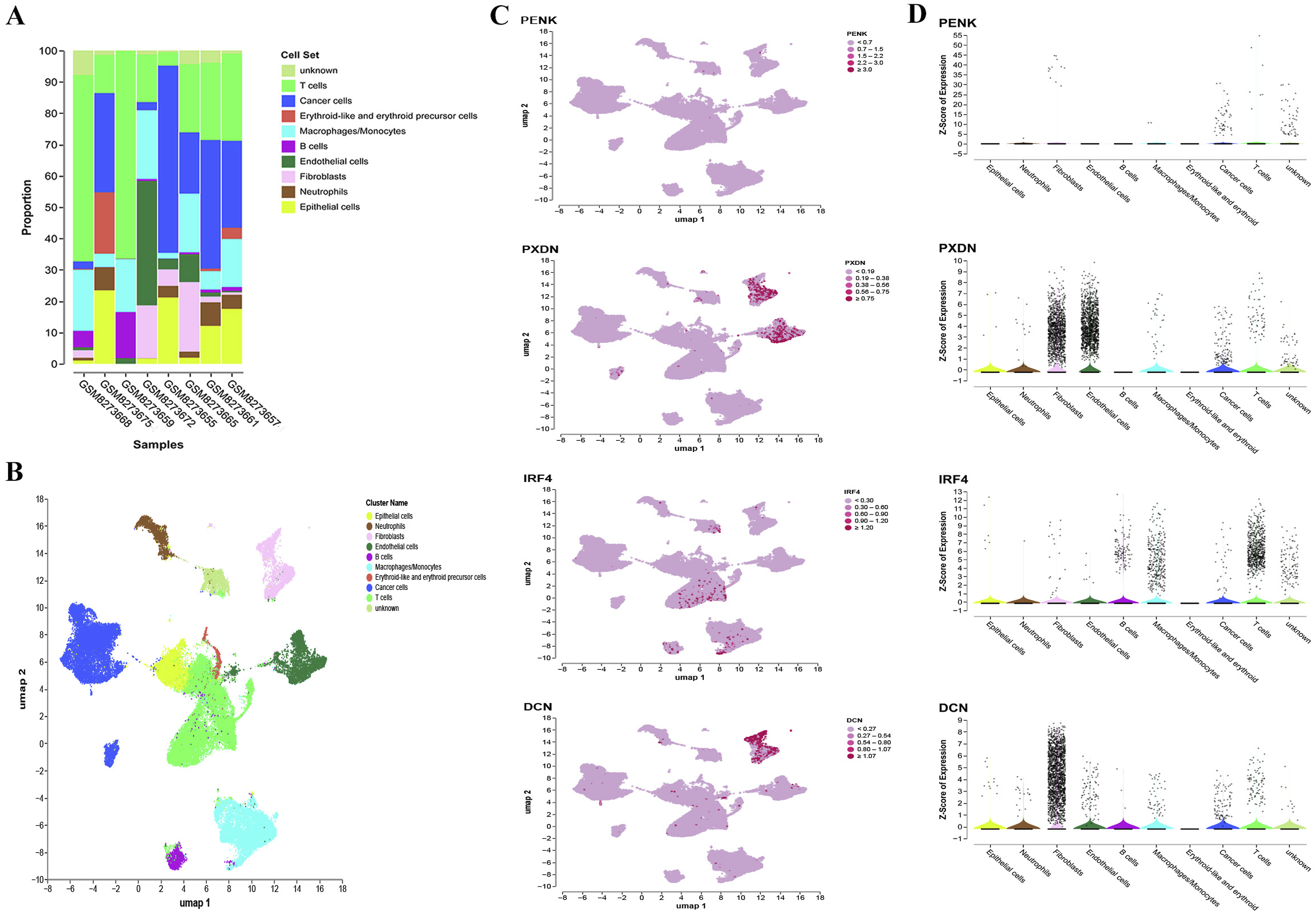
Additionally, we analyzed the paired urine samples from these eight bladder cancer patients using scRNA-seq data. Based on the expression of marker genes, we categorized the cells in the TME into T cells, B cells, erythroid-like cells and erythroid precursor cells, macrophages/monocytes, endothelial cells, fibroblasts, neutrophils, epithelial cells, and cancer cells (Figure 6A-B). Notably, the proportion of cancer cells was higher in urine samples compared to tissue samples, which highlights the feasibility of using urine samples for diagnostic testing (Figure 6A). Cell-type-specific expression analysis in urine samples revealed results similar to those observed in tissue samples (Figure 6C-D). In conclusion, the single-cell analysis further supports our previous assumptions, suggesting that the observed cell-type-specific expression patterns may be the underlying reason for the exceptional accuracy of the 41-CpG-site random forest classifier targeting IRF4, PENK, and PXND in distinguishing bladder cancer from non-BC individuals. Single-cell scRNA sequencing data analysis in urine samples. (A) Stacked bar plots of distribution of various cell types in GEO datasets. (B) UMAP plots show the cell clonal clusters. UMAP, Uniform Manifold Approximation and Projection. (C) Gene expression levels of various cells based on the landscape of UMAP plots. (D) Distribution of gene expression levels in the different cells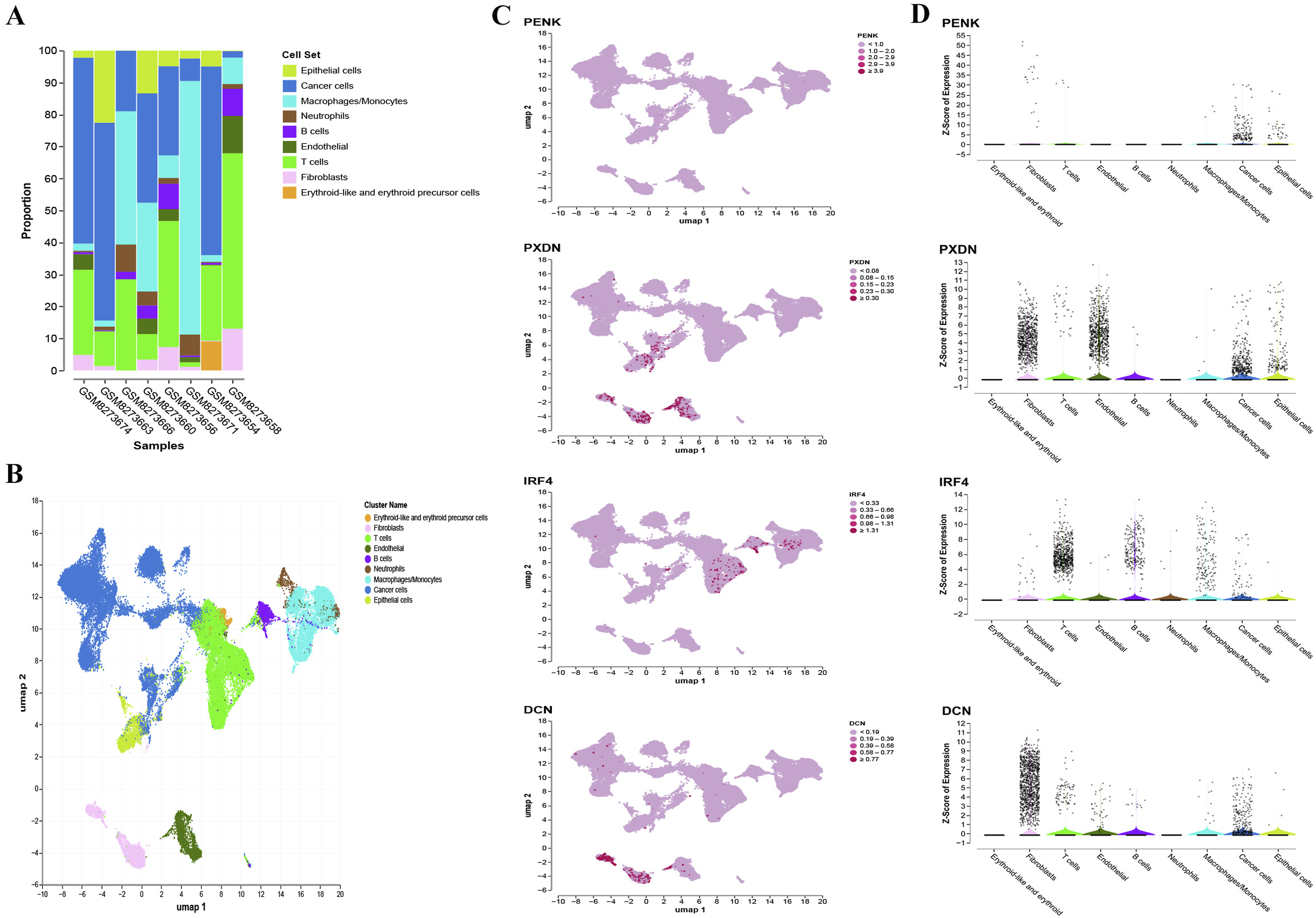
Discussion
In this study, we propose a novel targeted methylation detection methodology combining dual-stage multiplex PCR enrichment with next-generation sequencing (NGS). Then we constructed machine learning models to discriminate bladder cancer and non-bladder cancer based on the DNA methylation sequencing data. 201 participants were initially screened, of whom 46 were excluded according to predefined exclusion criteria and 25 declined cystoscopy. Although participant refusal reflects the real-world acceptance of invasive diagnostic procedures, it may introduce potential selection bias. To minimize this risk, the inclusion and exclusion criteria were carefully defined to avoid being overly restrictive while ensuring clinical appropriateness. For example, patients with benign urinary diseases were retained to reflect real-world diagnostic challenges, whereas individuals with concurrent malignancies or prior treatments were excluded to avoid confounding effects. Notably, patients with low-grade and early-stage bladder cancer were adequately represented, which is critical given the known diagnostic difficulty in this subgroup. Consequently, their inclusion was essential for rigorously evaluating the diagnostic accuracy and robustness of the model.
Our approach synergistically integrates the advantages of existing techniques: initial PCR amplification captures low-abundance targets, while subsequent NGS achieves site-specific sequencing depths exceeding 1000×at individual CpG loci. Large-scale interrogation of CpG sites in a single analytical workflow improves the specificity and sensitivity of bladder malignancy detection; however, the associated cost escalation may limit clinical applicability. To optimize the equilibrium between diagnostic precision and economic feasibility, we developed a targeted amplification strategy. Promoter regions of 3 genes, previously implicated in bladder carcinogenesis were prioritized for analysis. These CpG-rich genomic segments, each spanning approximately 150 base pairs, underwent two sequential rounds of multiplex PCR amplification for targeted enrichment. Subsequent bisulfite conversion and next-generation sequencing (NGS) enabled high-resolution quantification of methylation levels across these loci. This methodology enhances cost-efficiency by focusing on compact, functionally relevant regions, thereby reducing sequencing burden while improving depth-of-coverage and signal-to-noise ratios through selective amplification. The resultant customized panel, comprising 3 genes and 44 CpG loci, provides a robust framework for subsequent translational investigations in bladder cancer epigenetics.
Another outstanding advantage is that we construct a RF model to distinguish bladder cancer and non-bladder cancer, this algorithm improves the sensitivity and outputs robust predictions due to the characteristics of its own, to be specific, the errors occurred at several individual loci will not interfere with the final result. Meanwhile, the traditional MS-PCR methods distinguish cancer and non-cancer merely based on the cutoff values of methylation loci, which is susceptible to interference and not rigorous. Moreover, it is convenient to apply the random forest algorithm to clinical practice because no complicated software is needed, basic version of Python or R is sufficient to convert the methylation data to the results we desire.
This study included a total of 44 CpG sites across three genes, of which 41 CpG sites were retained for model training after preprocessing. The BladderCando diagnostic model, established using a random forest classifier, demonstrated robust discriminatory performance based on the methylation profiles of these loci in urine samples. These findings suggest that the selected CpG markers may provide a feasible basis for the development of a simplified and cost-effective MS-PCR-based detection strategy in future studies, with the potential to enhance clinical applicability. The sensitivity of current standards urine cytology for detecting bladder cancer ranges from 13% to 75%, with a specificity of 85% to 100%. 21 Sensitivity is positively correlated with tumor grade. 22 Factors such as urinary tract infections, stones can affect the results of urine cytology. Our method represented a promising alternative, as the protocol demonstrated high sensitivity and specificity while remaining robust to variations in tumor stage and less affected by benign urinary diseases. The three-group comparison further supported the robustness of the model, as samples from the Interference group exhibited methylation profiles and BladderCando scores similar to those of healthy controls. Notably, for low-grade bladder cancer patients, the model achieved 80% sensitivity in the validation cohort which was an incredible improvement compared with the current golden standard methods urine cytology and cystoscopy.23,24 Moreover, because low-grade tumors shed fewer cells into urine, 25 FDA approved urine-based biomarkers lack sensitivity for low grade bladder cancer.5,7,26 Our model outperformed these commercial tests at this stage, although only 5 low grade patients enrolled in the validation cohort. Additionally, the overall performance is remaining at a relatively high level with 91.3% sensitivity and 100% specificity, 93.5% negative prediction value and 100% positive prediction value, only in terms of specificity is inferior to the urine cytology test according to the previous reports. Besides, the machine learning model remains consistent predictive abilities. Clinical information, include age, gender and tumor stage, have little impact on the BladderCando score. Statistical difference only has been observed in grade, which could be attributed to the similarity of methylation patterns among several particular low-grade bladder tumor samples and normal tissues.
We analyze the pathway of the 3 core genes, GeneCards database indicates that these genes are related to immune activities. It reminds us that methylation pattern changes in bladder cancer cells may occur in genes associated with cytokine-related (IRF4), TME-related (PXDN) and fibroblast-related (PENK) immune activities and these changes might be the prominent factors assisting cancer cells escaped from the immune surveillance in the critical period for bladder cancer formation. IRF4 was reported as a molecular program controller responsible for immunosuppression in tumors, especially expressed by a subset of intratumoral CD4+ effector Tregs, in accordance with our findings in the scRNA-seq analysis. 27 PXDN involves in extracellular matrix organization and collagen chain trimerization. In upper urinary tract urothelial carcinoma, it is observed that upregulation of genes related to extracellular matrix implicating activated immune reactions.28,29 Vice versa, it infers that downregulation of these genes would mitigate the corresponding immune activities. Moreover, lack of CD8+ T cells in the tumor parenchyma but enriched in the fibroblast- and collagen-rich peritumoral stroma is the reason causing poor immune therapeutic effects in urothelial carcinoma. Transforming growth factor β(TGF-β) signaling in fibroblasts shapes the tumor microenvironment to restrain anti-tumor immunity by restricting T cell infiltration. 30 It is worth mention that PENK is also a member of TGF-β pathway, which implies PENK might have similar functions as TGF-βin affecting fibroblasts in the immune microenvironment.
Except for the mechanism exploration of these 3 genes, in some extent, they had been emphasized their clinical evaluation according to the published research. High IRF4 expression was believed to serve as a reliable predictor of non-small cell lung cancer (NSCLC) immunotherapy outcomes. 31 A novel IRF4 risk score was designed as a robust prognostic biomarker and could benefit in immunotherapy among hepatocellular carcinoma (HCC) patients. 32 Evidence supports PXDN’s role as a valuable diagnostic and prognostic biomarker across diverse cancers. Critically, specific mutational features of PXDN are significantly associated with patient survival in several malignancies, most notably uterine corpus endometrial carcinoma (UCEC). 33 A multicenter clinical study in the Republic of Korea illustrated that PENK methylation test was a promising method in early detection of bladder cancer. 34
This study also exhibits some limitations. Firstly, under conditions of oxidative stress, cytosine has the potential to convert into uracil, leading to the possibility of false positive errors in bisulfite sequencing. Second, due to the single-center study design and lack of an independent external cohort to assess the model’s generalizability, the clinical applicability and robustness of the BladderCando model remain unsubstantiated and there is a substantial risk of overfitting and optimism bias. In addition, due to the shortage of the fund, the ages of healthy volunteers were below 40. Therefore, further prospective multi-center studies with a large sample-size should be undertaken to expand the understanding of the ability of BladderCando to distinguish between non-BC individuals and malignant bladder tumor patients and to validate the robustness and generalization of the model, especially among low grade bladder urothelial carcinoma. Third, our diagnostic model was built on urine methylation data, whereas the TME analyses relied on public tissue and urine single-cell datasets. We acknowledge that direct integration of matched urine methylation and single-cell/TME data from the same cohort would be more robust. Future prospective studies with paired profiling are therefore needed to further validate the biological relevance of the markers and avoid potential misinterpretation. Last, technical interventions incorporated into the methodology are required in further investigation to mitigate the risk of cytosine unintended conversion into uracil. 35
Conclusion
We developed a cost-effective urine-based diagnostic strategy—a 41-CpG-site random forest classifier targeting IRF4, PENK, and PXDN (BladderCando)—that demonstrated high diagnostic performance in distinguishing non-BC individuals from malignant bladder tumors, with notably improved detection in low-grade disease, a clinically challenging subgroup. Nevertheless, the robustness and generalizability of the model require further validation in independent, multi-center cohorts. By enabling accurate and noninvasive assessment, this approach may help mitigate biopsy-related risks and discomfort, while providing potential clinical value for patient stratification.
Supplemental Material
Supplemental Material - Urine IRF4/PENK/PXDN Methylation Signatures Enable Machine Learning-Driven Bladder Cancer Detection and Microenvironment Dissection
Supplemental Material for Urine IRF4/PENK/PXDN Methylation Signatures Enable Machine Learning-Driven Bladder Cancer Detection and Microenvironment Dissection by Yi He, Wei Chen, Shengjie Dai, Xintao Wang, Xiaokai Zhao, Wenhua Xie, Siyu Lei, Wei Zhu, Yi Qian, Jinpeng Feng, Ziying Gong, Jieyi Li, Pengmin Yang Xinyu Xu, Wenjie Fei, Daoyun Zhang, Yifang Cao, Jing jin in Technology in Cancer Research & Treatment
Footnotes
Acknowledgments
We specifically acknowledge Huiyu Fu, Jiayu Peng, Lijun Zhang, Hao He, Xinyun Wang (Department of R&D, Jiaxing Yunying Medical Inspection Co., Ltd., Zhejiang, China) for performing targeted bisulfite sequencing experiments. Additionally, this manuscript was language edited by ChatGPT (https://chat.openai.com) and DeepSeek (![]() ).
).
Ethical Considerations
This study was conducted according to the principles of the World Medical Association Declaration of Helsinki of 1975, as revised in 2024. Ethics approval was obtained from the Ethics Committee of the First Hospital of Jiaxing (Approval No. LS2020-355, Date: 2021-01-06).
Consent to Participate
All participants provided written informed consent for this study.
Author Contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Zhejiang Provincial Natural Science Foundation of China [LMS25H050003], the Key Research and Development Program of Zhejiang province [2023C03057], the Jiaxing Municipal Key Technology Industrial Development Project [2025AC014], the Jiaxing Science and Technology Program-Social Development Special Project [2025BS009], the Intramural Research Grant of Jiaxing First Hospital [2024-YB-075].
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Xintao Wang, Xiaokai Zhao, Jieyi Li, Pengmin Yang, Xinyu Xu and Wenjie Fei are employed by the Jiaxing Yunying Medical Inspection Co., Ltd. and Shanghai Yunying Medical Technology Co., Ltd. Ziying Gong and Daoyun Zhang are employed by the Jiaxing Yunying Medical Inspection Co., Ltd., Shanghai Yunying Medical Technology Co., Ltd. and Shanghai Yunying Biopharmaceutical Technology Co., Ltd. Besdies, Ziying Gong and Daoyun Zhang hold shares of these companies. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data Availability Statement
Data is provided within the manuscript or supplementary information files which can be found at ![]() . The raw data is available from the corresponding author upon reasonable request.
36
. The raw data is available from the corresponding author upon reasonable request.
36
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.