
Abstract
Indiscriminate use of over-the-counter antibiotics has led to the rapid emergence of resistant genes in bacteria, with the ultimate crisis to global health. One of the prominent sectors with the antimicrobial resistance (AMR) concern is the farm animals that exist in close contact with humans where the environmental conditions are favorable for the rapid dissemination of pathogenic organisms and resistance genes. Hence, to understand the threat with environmental AMR, a detailed molecular insight is very important. In this study, fecal samples from both poultry and associated humans were studied by metagenomics analysis. From the results, a primary understanding on the microbial diversity difference could be generated from the selected samples. Here, the poultry samples were identified to have more microbial diversity. At the same time, several pathogens were found to be shared commonly between the hosts. Upon detailed examination, several AMR genes were also observed to be common between the poultry and human samples. The results of the study are highly relevant in light of the “One Health” concept where an integrated approach is targeted.
Introduction
Antimicrobial resistance (AMR) has emerged as one of the most critical global health threats affecting humans, animals, and the environment within the “One Health” framework (Walsh, 2018). The alarming prevalence of AMR is largely due to the use of potential antibiotic drugs for the treatment and management of infectious diseases. The dissemination of AMR is not limited to hospital environments, as multiple interconnected factors across human, animal, and environmental sectors contribute to its spread, which further aggravate the risk of resistance emergence (Salam et al., 2023). Livestock and poultry production systems have received increasing attention as potential reservoirs for AMR. In poultry farming, antibiotics are commonly used for the prophylaxis, and studies have reported previously that these drugs are available over-the counter for disease management (Mor-Mur and Yuste, 2010). However, these have been restricted significantly during the last few years. Furthermore, poultry sectors facilitate the dissemination of AMR genes through multiple transmission pathways, posing potential risks to both animal and human health. The identified transmission pathways are (i) the direct contact between infected poultry and the farm workers, (ii) through the environment with the use of manure, litter, and wastewater (Sana et al., 2025), and (iii) consumption of infected meat or products (Lammie and Hughes, 2016).
In India, poultry is the leading source of meat, accounting for 65% of total meat consumption with a significant increase from 23% two decades ago (Sajish et al., 2025). The rapid expansion of poultry production has been associated with increased antibiotic use, which ultimately leads to the emergence of AMR. Considering the widespread of AMR in the poultry sector in India, identifying the emergence and the evolutionary dynamics of AMR has become essential.
Comprehensive resistome profiling to determine the prevalence and distribution of AMR genes at the human-poultry interface cannot be effectively achieved using conventional culture-based approaches. Advances in sequencing technologies, particularly shotgun metagenomic sequencing, provide powerful tools for investigating microbial communities and AMR genes directly from complex samples (Boukerb et al., 2021). This approach identifies the microbial taxa and resistance genes within poultry and farm workers, allowing comparative analysis of shared and unique microbial communities. Use of shotgun metagenomics can provide valuable molecular insights into the prevalence and distribution of AMR within poultry farming sector. Therefore, the current study used shotgun metagenomics to profile the AMR genes from fecal samples collected from broiler chickens and farm workers from poultry farms. These findings would provide preliminary insights into AMR in poultry farm environments and support the need for future large-scale surveillance and prevention of AMR.
Methodology
Sample collection
A total of 12 poultry fecal samples were collected from the two farms located in the Ernakulam district, Kerala, India. The farms were selected based on the accessibility and the willingness of the farm owners to participate in the study. In addition to poultry samples, human participants were recruited from farm workers who had regular occupational exposure to poultry. Two workers, one from each farm were also selected based on predefined inclusion and exclusion criteria. Eligible participants were adult workers (≥18 years) who had been working on the farm for more than 1 week prior to sample collection and who had daily exposure to poultry for approximately 8 h. Individuals below 18 years of age, those with a history of infectious diseases, individuals who had used antibiotics within the previous 1 month, and workers reporting chronic alcohol consumption were excluded from participation. Based on these criteria, two male farm workers were included in the study, and both reported no antibiotic use during the 1 month preceding sample collection. Fecal samples were collected using sterile containers under aseptic conditions, transported to the laboratory, and processed for DNA isolation.
Extraction of meta-DNA and sequencing
The meta-DNA was extracted from all the collected fecal samples (2 human and 12 poultry) using NucleoSpin DNA Stool, Mini kit (M&N-740472.50) as per the manufacturer’s instructions. The purified DNA was quantified, and the six samples collected from the poultry farm were pooled together to represent the two sets of farm microbiome. The pooled poultry metagenomic DNA samples from each farm along with the corresponding human fecal metagenomic DNA samples from farm workers of the same farm were used. The results were labeled as Human 1 and Poultry 1 (H1–P1) and Human 2 and Poultry 2 (H2-P2). Standard paired-end DNASeq library preparation was performed for all four samples by using Illumina-recommended protocols. Quality control measures were taken using Qubit to ensure that the libraries met the necessary standards for sequencing (Meyer and Kircher, 2010). The genome was sequenced with 151 base-paired end sequencing with Illumina HiSeq X Ten. The microbial reads were assembled using MetaSpades (Nurk et al., 2017) with default parameters. The assembled contigs were classified taxonomically by using Kaiju against the NCBI nonredundant protein database with default parameters (Menzel et al., 2016). MetaErg stand-alone framework was used to assess the functional capacities of microbial communities present in the samples.
Resistome, virulence, and plasmid screening
The resistome of the fecal metagenome samples was identified by ABRicate using the comprehensive antibiotic resistance database (CARD; https://card.mcmaster.ca/) (McArthur et al., 2013). The bacterial virulence genes and plasmids from the metagenome samples were identified by ABRicate using Virulence Factor Database and PlasmidFinder. The summarized data were visualized using a heatmap constructed by TBtools (Carattoli et al., 2014; Chen et al., 2005).
Ethical considerations
This research followed the Declaration of Helsinki. The study was approved with ethical clearance granted by the Institutional Review Board (IRB) of Government Medical College, Kottayam (IRB no: 99/2021). All human participants gave their informed consent expressly as written before they were included in the study. No personal details of any participant were presented in this publication, and therefore, no explicit publication consent was required.
Results
The metagenomic DNA extracted from the pooled poultry and human fecal samples was sequenced using an Illumina sequencing platform. The raw reads obtained were subjected to quality control, and the filtered reads were analyzed to determine the total number of sequences and bases, the minimum sequence length, the N50 length, and the GC content. Functional metagenomic annotation identified 58,673 predicted coding sequences in H1, 125,408 in H2, 308,722 in P1, and 251,022 in P2. These values represent the number of predicted genes from the assembled contigs and correspond to the sequencing output and assembly characteristics of each dataset.
The taxonomic classification of the assembled metagenomic contigs revealed a diversity of microbial communities within the samples. The majority of the bacterial population in the human microbiome was Faecalibacterium (H1-13.62% and H2-11.06%). Prevotella were the second most abundant bacteria in H1 samples, while Roseburia constituted 9.67% in H2. Various diverse populations were also predicted for both H1 and H2, which include Mitsuokella multacida, Sutterella wadsworthensis, Roseburia inulinivorans, and Gemmiger formicilis. In the poultry sample P1, Candida tropicalis accounted for 4.60% of the classified reads. Several additional microbial taxa were detected in this dataset, including Corynebacterium variabile (0.98%), Corynebacterium stationis (0.89%), Escherichia coli (0.69%), Enterobacter hormaechei (0.20%), Candida viswanathii (0.15%), Citrobacter freundii (0.11%), Campylobacter jejuni (0.11%), Enterococcus durans (0.11%), Pseudomonas aeruginosa (0.05%), Shigella sonnei (0.04%), Klebsiella pneumoniae (0.018%), and Campylobacter coli (0.0062%). The P2 samples were also found to harbor the pathogens C. stationis (2.07%), E. coli (1.26%), Helicobacter pullorum (1.00%), Corynebacterium casei (0.93%), S. sonnei (0.04%), Corynebacterium diphtheriae (0.02%), Clostridioides difficile (0.021%), and K. pneumoniae (0.017%).
A comparative analysis was also carried out to identify the species that were common between the samples. Here, P2 was observed to have a greater number of species (2432) that were unique to it, followed by P1 (2219). At the same time, H1 and P1 were found to have 183 shared species, while H2 and P2 shared 24 species. The opportunistic pathogens identified from the shared microbiome, included Klebsiella oxytoca, Enterobacter kobei, and Enterobacter ludwigii. The epidemiological triad framework was used as a conceptual model to illustrate possible host-microbe-environment interactions associated with the detected microbial profiles (Fig. 1).
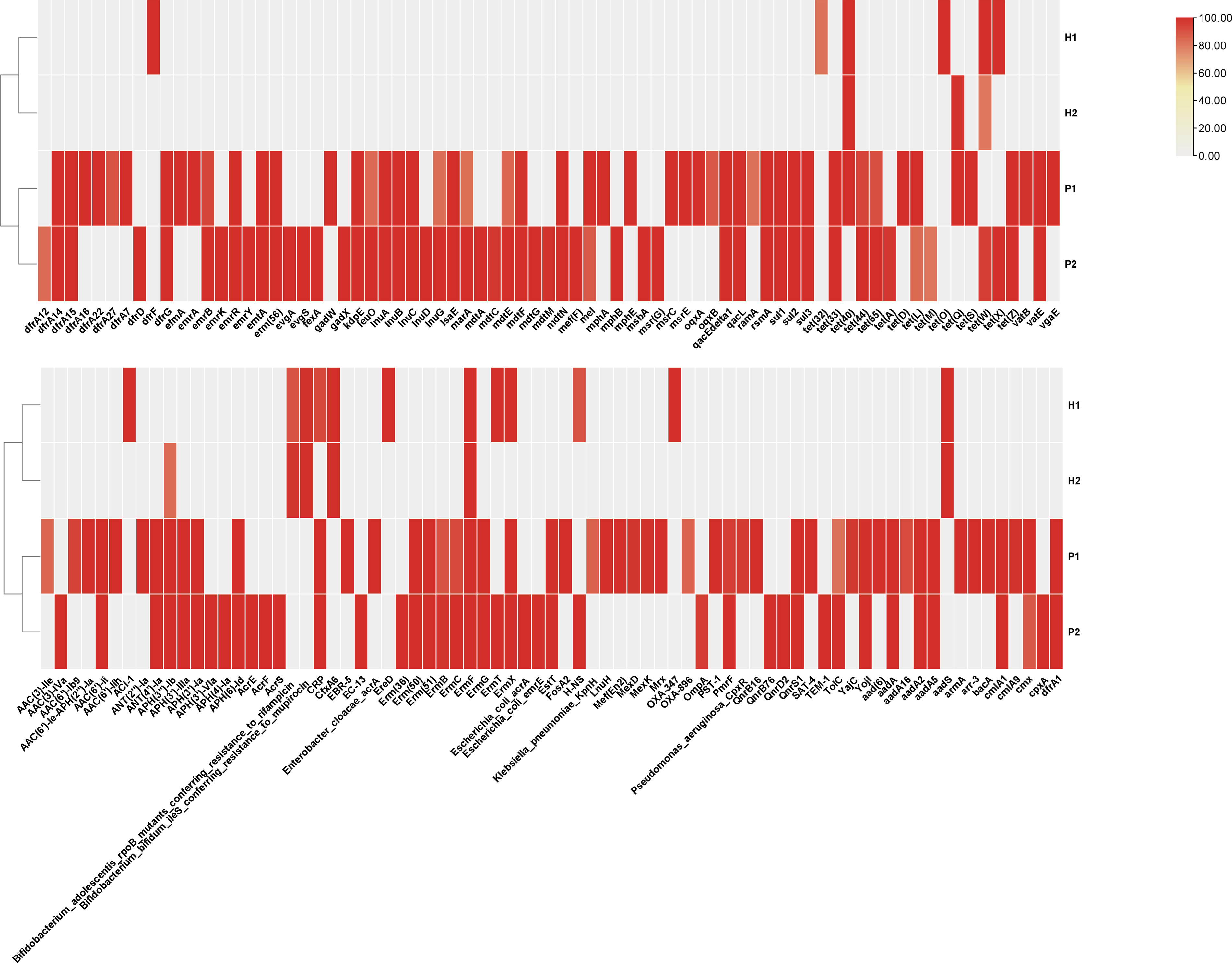
Comparative heatmap representing the AMR gene distribution in the metagenome of the tested human and poultry fecal samples. AMR, antimicrobial resistance.
The resistome prediction
The identification of AMR genes in the metagenome samples was carried out by using ABRicate tool. Here, the poultry metagenome was observed to contain the highest number (P1-92 and P2-85) of AMR genes when compared with that of the human metagenome (H1-18 and H2-9). In the current study, 22 AMR genes were detected in the analyzed metagenomic datasets (Supplementary Data). These include 11 genes for antibiotic inactivation (mph (B), fosK, aadA16, blaEBR-1, ant (6)-Ia, lnu (D), lnu (A), lnu (B), lnuC, vat (E), and aadD), 3 genes for target protection (qnrD2, tet (32), and vgaE), 3 genes for target replacement (dfrD, dfrA14, and tet (44)), and 2 genes for efflux pumps (fexA and tet (Z)). In the comparative analysis of the resistomes of human and poultry fecal samples, four AMR genes (crp, ermF, ermX, and tet (40)) were found to be shared between H1 and P1 samples. Similarly, three genes (tetW, ermF, and APH (3′′)-Ib) were observed to be shared between H2 and P2 (Fig. 2).
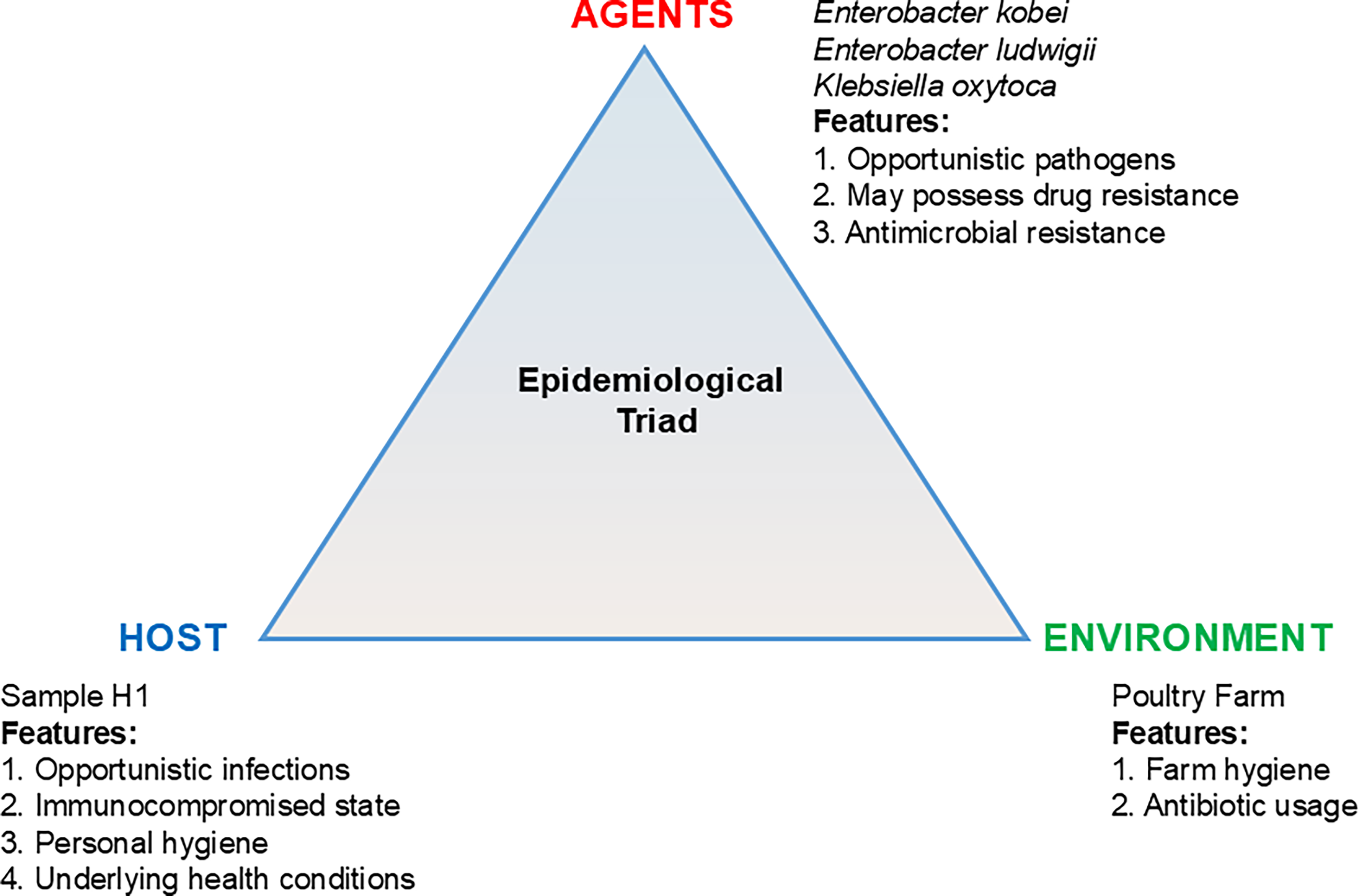
The anticipated epidemiological triad (hypothetical) of the farm worker H1 showing the possible risk associated with the shared pathogen from the poultry farm environment. H1, Human 1.
Discussion
Shotgun metagenome sequencing helps to understand the presence of various pathogens, virulence factors, and the AMR genes in the sample (Li et al., 2015). In this study, metagenomics revealed the diverse microbial taxa and distribution of AMR genes across both human and poultry datasets. The microbial profile obtained from the poultry included several microbial taxa with pathogenic potential, such as E. coli, S. sonnei, and K. pneumoniae. The presence of such taxa in poultry-associated microbes has been reported earlier (Mor-Mur and Yuste, 2010; Wang et al., 2021; CDC, 2023). These potential pathogens may pose a risk to humans through direct contact, contamination of water bodies or vegetable crops and livestock product supply chains. S. sonnei is associated with foodborne diseases, whereas E. coli and K. pneumoniae are associated with various infections, including urinary tract infections and sepsis, in humans (Cho et al., 2014). The presence of C. jejuni is highly alarming since the bacteria is well-known as a foodborne pathogen. C. tropicalis was observed to be dominant in P1 poultry sample. Antibiotic usage in poultry farming has been implicated in the development of Candidiasis in poultry, as it can disrupt the normal microflora. The use of antibiotics can also lead to bacterial dysbiosis, where the environment becomes more conducive for the growth of Candida (Cafarchia et al., 2019).
The resistome prediction, as conducted in the study, has resulted in the identification of various AMR genes from the metagenome samples. Among these, 22 different genes were found to be detected in the study sample. The majority of this group is coding for the function related to the antibiotic inactivation. lnu (D), lnu (A), lnu (B), and lnuC are the genes coding for lincosamide nucleotidyltransferase (LNU), causing resistance to lincosamide antibiotics through inactivation. Clindamycin and lincomycin are the main antibiotics coming under this class (Petinaki et al., 2008; Almuzara et al., 2013). Here, the fosK gene can also inactivate antibiotics, such as fosfomycin, through the enzymatic action of fosfomycin thiol-transferase. Fosfomycin is a reserve antibiotic used for the urinary tract infection caused by Gram-negative bacteria (Kitanaka et al., 2014). At the same time, blaEBR-1 is a beta-lactamase enzyme causing resistance to various drugs, like carbapenem, penam, and cephalosporin, through antibiotic inactivation (Bellais et al., 2002). The mph(B) gene confers resistance to macrolide antibiotics, such as azithromycin, through the production of macrolide phosphotransferase (Chesneau et al., 2007). The aadD is an aminoglycoside adenylyl transferase gene that confers resistance to aminoglycoside antibiotics, such as gentamicin and tobramycin, and vatE is a streptogramin acetyltransferase coding gene that confers resistance to streptogramin A antibiotics, such as virginiamycin and quinupristin (Bozdogan et al., 2003). The comparative metagenomic analysis has also resulted in the identification of four AMR genes that are common between H1 and P1, which include crp, ermF, ermX, and tet.
To study the health risks with the human subjects, a conceptual model of epidemiological triad-based analysis has been performed (Fig. 2). This is an epidemiological model used to explain the incidence of infectious diseases (John and Kompithra, 2023). This helps to study the infection transmission through the populations and the strategies for survival as the triad is connected together by the agent, host, and environment. In this study, E. kobei, E. ludwigii, and K. oxytoca were identified among the shared microflora between the H1 and P1 samples. An anticipated epidemiological triad was constructed to understand the possible health risks associated with these findings. However, the triad presented in this study is intended only as a conceptual illustration rather than a quantitative epidemiological analysis. The detected taxa, such as K. oxytoca, can even cause life-threatening infections, including antibiotic-associated hemorrhagic colitis, bacteremia, and urinary tract infections (Singh et al., 2016; Yang et al., 2022). E. kobei and E. ludwigii are known for their role in various infections, including urinary tract, respiratory, soft tissue infections, and endocarditis. An overlap in microbial taxa was observed between the poultry farms and farm workers, illustrating potential host-microbe-environment interactions. Despite the limited sample size and restricted geographical coverage, comparative metagenomic analysis provides useful preliminary insights into microbial community composition and AMR gene distribution at the human-poultry interface. These findings also support the significance of the one-health approach to analyze the underlying risk factors associated with intensive poultry farming and the importance of timely actions required to avoid the health hazards.
Conclusion
In this study, metagenomic datasets obtained from two poultry farms and the respective farmworkers were analyzed to characterize the microbial composition and AMR gene distribution at human-poultry interface. The comparative metagenomics revealed a diverse array of microbial profiles between poultry and human samples, while a degree of overlapping microbial taxa was also observed in these samples. These observations indicate the potential presence of taxa with pathogenic risks in the sample datasets. This study provides preliminary insights into microbial community patterns and AMR gene distribution in poultry-associated environments from a One Health perspective. Further studies with larger sample sizes and individual sample sequencing would help to better understand microbial diversity and resistome profiles at the poultry-human interface.
Authors’ Contributions
S.S.: Conceptualization, experimentation, data analysis and writing; P.V.K.: Data analysis and writing; A.M.N.: Experimentation; M.P.: Data analysis, review and editing; S.P.S.: Revision and editing; P.P.R.: Review and editing; J.M.: Supervision and editing; R.E.K.: Supervision and editing.
Declaration of Competing Interest
None of the authors has any personal or financial relationships that could inappropriately influence the content of the paper.
Data Availability Statement
The data supporting this study will be made available upon request.
Footnotes
Disclosure Statement
This is to declare that the authors have no conflict of interest in this research work.
Funding Information
The authors acknowledge the funding provided by the Department of Animal Husbandry, Government of Kerala, India under Grant No. AHD/3158/2018-B, for the project and the instrumentation support from the Kerala State Plan Fund and the Kerala State Council for Science, Technology and Environment, KBC-YIPB Project. The authors acknowledge the support received from Kerala Antimicrobial Resistance Strategic Action Plan (KARSAP).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.