
Abstract
The coronary artery disease is a leading cause of death and morbidity worldwide. This disease has a complex pathophysiology that includes multiple mechanisms. Among these is the oxidative/nitrosative stress. Paradoxically, oxidative/nitrosative signaling plays a major role in cardioprotection against ischemia/reperfusion injury. In this context, the gas transmitter nitric oxide may act through several mechanisms, such as guanylyl cyclase activation and via S-nitrosylation of proteins. The latter is a covalent modification of a protein cysteine thiol by a nitric oxide-group that generates an S-nitrosothiol. Here, we report data showing that nitric oxide and S-nitrosylation of proteins play a pivotal role not only in preconditioning but also in postconditioning cardioprotection.
Introduction
Nitric oxide (nitrogen monoxide, NO) is synthesized by various nitric oxide synthase (NOS) enzymes or can be produced by other reactions in the biological systems, which are collectively called “non-NOS” processes. The latter includes “non-enzymatic” reactions which are favored by acidic conditions, such as reduction of nitrite to NO, and reactions catalyzed by “non-NOS enzymes,” such as cytochrome c, hemoglobin, and xanthine oxidoreductase.1–3
There are three known isoforms of NOSs; besides endothelial NO-synthase (eNOS), cardiac myocytes constitutively express neuronal NO-synthase (nNOS). These two NOSs are Ca++/calmodulin-controlled isoenzymes and within the organ and cells they are localized to different microdomains, and are linked to selective signaling that is further impacted by a broad array of key processes in health and disease4,5 (see also below). Moreover, the cardiovascular system can express an inducible isoform of NOS (iNOS), which can produce in immune and pathological processes a large amount of NO independently of the level of Ca++ in the cell. Besides the differences in Ca++ dependency the three NOSs bind calmodulin with different affinities: the general order of affinity is iNOS≫eNOS≫nNOS, so that calmodulin can be considered a subunit of iNOS.6,7 Whether mitochondria also contain a specific NOS contributing to overall NO generation is a matter of debate. 8
NO is considered a free radical with primary regulatory functions in the heart. It is well known that NO modulates cell function by the activation of soluble guanylyl cyclase (sGC) to form cyclic guanosine monophosphate (cGMP). However, several recent studies suggest that, in addition to activating cGMP-dependent signaling pathways, NO generates redox/nitrosative processes, including the posttranslational modification known as protein S-nitrosylation (SNO), which, as we will see in more detail, consists in the covalent attachment of an NO-moiety to a nucleophilic protein sulfhydryl resulting in S-nitrosothiol formation9,10 (Figure 1).
Nitric oxide-based signaling
The effective targeting of oxidative/nitrosative process in the cardiovascular system critically depends on identifying the primary intracellular regulators. As a matter of fact, both reactive oxygen and nitrogen species (ROS/RNS) are localized signaling molecules, at least in physiological conditions.9–18 In particular, targets of NO and ROS/RNS are involved in several processes of cardioprotection, including preconditioning (PreC) and postconditioning (PostC) phenomena.
Both PreC and PostC have been demonstrated to significantly attenuate ischemia/reperfusion injury. While PreC can be obtained with one or more brief coronary occlusions of a few minutes (from 2 to 10 min) each before the infarcting ischemia,19,20 PostC may be performed with one or more brief occlusions of a few seconds (from 5 to 60 s) each, starting very early in reperfusion, i.e. a few seconds after the end of the infarcting ischemia.21,22 These two cardioprotective strategies not only reduce infarct size and affect all form of cell death but they also attenuate postischemic contractile dysfunction, arrhythmias, and endothelial dysfunction. Specifically, both PreC and PostC limit endothelial cell dysfunction by increasing eNOS activity and NO bioavailability in neighboring cells.23–27 The formed NO may be responsible for nitrosative signaling, including nitration and nitrosylation of proteins in these two cardioprotective mechanisms (see below).
From the concepts described above can be inferred that the protein SNO is primarily a protective reaction. However, we must keep in mind that SNO may also have deleterious effects on cardiovascular function in some specific conditions. For instance, iNOS may be part of pathological processes, such as mitochondrial dysfunction and septic shock.28–30 In these conditions, an excessive SNO can have deleterious effects on cardiac and endothelial functions (see also below).31,32 Yet within the heart iNOS acts as a potent protective enzyme against ischemia/reperfusion injury in the so-called second window of preconditioning (SWOP). 33 In this setting, it has been reported that atorvastatin may reduce infarct size by increasing the activity of iNOS and cyclooxygenase (COX-2). Of note, it has been demonstrated that COX-2 is activated by iNOS-dependent SNO. 34 Actually, the SWOP, which occurs 12–24 h after the preconditioning stimuli, is primarily mediated by gene induction and protein synthesis. For the mechanisms involved in this window of protection, the reader is referred to other excellent reviews on this topic.20,35–37
Our minireview will focus on acute cardioprotection mechanisms (PreC and PostC), which are primarily mediated by activation of signaling pathways and post-translational modification of proteins. In particular, in this minireview we focus on the role of SNO of proteins in cardioprotection. The distinction of nitration from one side and nitrosation or nitrosylation from the other side will be first considered. Then, the biological conditions that can favor the reaction of SNO will be analyzed in the context of preconditioning and postconditioning.
What is protein SNO?
The biology and chemistry of NO are quite complex, for example (a) NO plays a plethora of regulatory roles in normal physiology and is involved in several pathophysiological and pathological processes; (b) it can be generated by enzymatic and non-enzymatic processes; and (c) it is involved in several processes of both pro-oxidation and antioxidation. In this respect, NO is involved in a cornucopia of redox reaction leading to processes of nitration or alternatively to processes of nitrosation/nitrosylation (for reviews see also Sun and Murphy 9 , Pagliaro et al. 10 , Espey et al. 38 , Simon et al. 39 , Zhang et al. 40 and Wink et al. 41 ).
Nitrosation or nitrosylation: Are they synonyms?
Although nitrosation has been defined as the formation of a covalent bound between a nitrosonium equivalent (NO+) and a nucleophilic amine, whereas nitrosylation can be defined as the addition of NO without change in the formal charge of the substrate, 38 nitrosation and nitrosylation are often used interchangeably to refer to the same substrate modification. As such, nitrosation/nitrosylation of proteins (i.e. the incorporation of NO-moieties by covalent bonding to various protein groups) is chemically possible not only in the case of cysteine thiols, which leads to the formation of a thionitrite, but also in the case of tryptophan indols, and amines (lysine and N-terminal).39,40 Therefore, from a terminology viewpoint, the incorporation of NO/NO+ moiety to a thiol can be clearly individuated because of the prefix “S,” referring to the incorporation of the moiety to a sulfur atom to form the S-NO bond.
The S-nitrosylating species
Nitrogen oxidized species are the most likely candidates for the modification of a reduced thiol. Actually, NO2 and N2O3 have been considered the typical S-nitrosylating species. They are the higher oxidation state (one-electron oxidation) of nitrogen oxides and are formed by the reaction of NO with molecular oxygen (O2) or with peroxynitrite (ONOO−), or, alternatively, from acidified nitrite. The reaction of NO with the superoxide anion (O2−) will lead to ONOO−, which, however, may react again with NO (the so-called secondary reaction) 41 forming the nitrosylating species N2O3.
These species reacting with thiol groups of cysteine modify proteins. This post-translational modification can be referred to as S-nitrosation which, in analogy with the phosphorylation of proteins, has become popular as protein SNO or protein SNO (or SNO of proteins). From here on, we will prefer either the term “nitrosylation” to refer in general to substrate modification by this kind of reaction or the term “SNO of proteins” to refer to this specific type of reaction observed in biological system, which involves cysteine thiols.
Nitration
Before considering the biology of protein SNO, let us consider nitration to distinguish it from nitrosylation. In fact, nitration must not be confused with nitrosylation/nitrosation. Nitration involves an electrophilic addition of a nitrotriatomic group, such as an NO2+ equivalent (nitronium, an electron acceptor), to an aromatic ring (the site of electron density). 38 In the biological system, the term nitration refers to the incorporation of the nitrotriatomic group at position 3 of the phenolic ring of tyrosine residues (nitration of tyrosine at its ortho position, which is known as 3-nitration of tyrosine or 3-NT). This protein modification is clearly related to an oxidative stress by the formation of ONOO−, from the reaction of NO with O2− , in the pathophysiology of the cardiovascular system.42–44
However, it must be stressed that nitrosylation and nitration, though they are two different types of reactions, are not mutually exclusive events, but are related in a continuum or Yin-Yang process and can lead in several circumstances to identical final products. 29 Even ONOO− can lead to the chemistry of the nitrosylation pathway; for instance, it has been suggested that only when the ratio between NO and O2− is close to 1 : 1 the formation of ONOO− can lead to nitration, whereas a small excess of NO will lead to nitrosylation, perhaps via the secondary reaction. 38 For example, it has been suggested that 3-NT formation is a step of cardioprotective pathways.45,46 We have recently suggested that after an initial increase of 3-NT, during cardioprotective maneuvers, a subsequent SNO of proteins may prevail in the cardioprotective scenario when NO may be in excess with respect to O2−.47–49 We suggest avoiding the use of nitrotyrosilation to indicate tyrosine nitration, since the use of nitrotyrosilation may make confusion with nitrosylation that is a different reaction, as above reported.
Denitrosylation
Clearly SNO is a labile modification, and the levels of nitrosothiols in the biological system are low, due to a rapid turnover. Its transience makes it a typical reaction of nitrosative signaling. Therefore, the concept of denitrosylation has been introduced. It consists in the removal of the nitroso-group and is an important aspect of SNO signaling. In fact, denitrosylation may limit the amount of protein SNO in order to avoid “excessive” SNO, as can occur, for instance, when iNOS is produced in pathological processes comprising nitrosative stress.30–32
Denitrosylation was firstly considered as a spontaneous and non-regulated process. Recently, several non-enzymatic and enzymatic mechanisms of denitrosylation have been described in vitro and in vivo. Non-enzymatic processes include reactions mediated by ascorbate, nucleophilic compounds and transition metal ions that have the possibility to also occur in vivo (for reviews see Martinez-Ruiz et al. 18 and Stamler and Toone 50 ). Enzymatic processes may be attributed to denitrosylases, whose actions would be analogous to those of the phosphatases in kinase signaling. Several denitrosylases may contribute to the compartmentalization of the action of SNO of proteins. Although S-nitrosoglutathione reductase and the thioredoxin system have emerged as the two main enzymatic processes of denitrosylation in vivo, several other enzymes with denitrosylase capability have been observed in vitro and/or in vivo, including carbonyl reductase-1, disulfide isomerase, glutathione (GSH) peroxidase, superoxide dismutase (SOD), and xanthine oxidase.47,51–58 With respect to thioredoxin system, it has been observed that both cytosolic and mitochondrial isoforms of thioredoxin (Trx1 and Trx2, respectively), as well as the reductases that recharge them, TR1 and TR2, are involved in the process of denitrosylation in an NADPH-dependent manner.56,59 Yet, the removal of the NO -moiety by glutathione reductase may depend on the transnitrosation of SNO with GSH to form GSNO (S-nitrosoglutathione), which will be enzymatically removed. That is, GSH can remove SNO from proteins to generate GSNO which is then converted back to GSH by S-nitrosoglutathione reductase. Similarly, thioredoxins can denitrosylate proteins and thioredoxin is then regenerated by thioredoxin reductase. Alternatively, a disulfide bond and a concomitant release of NO-moiety can occur by nucleophilic attack of vicinal protein thiols. The importance of dynamic SNO/denitrosylation reactions are essential in cardiovascular regulation as demonstrated, for instance, in genetically modified models of increased or decreased activity of S-nitrosoglutathione reductase. In fact, in these models the sepsis-induced myocardial depression is positively influenced by denitrosylation.32,58–60 For reviews on denitrosylation see Martinez-Ruiz et al. 18 , Anand and Stamler 57 and Benhar et al. 60
Biology of SNO
NO is produced by many cell types and has many diverse biological functions. In the cardiovascular system, NO is a key regulator of vascular tone,61,62 where it is known to mediate its effects, in part, by binding to the heme moiety of its effector, soluble GC, with the formation of cGMP and subsequent activation of cGMP-dependent signaling. The effects of cGMP are mainly mediated by cGMP-dependent protein kinases, cGMP-regulated ion channels, and by cGMP-dependent phosphodiesterases 63 (Figure 1).
There are many additional cGMP-independent actions of NO that rely on modifications of biomolecules, including the post-translational modification of proteins, such those that are considered here, i.e. reactions with a reactive cysteine residues to lead to SNO or with tyrosine to lead to 3-nitrotirosine formation.
Clearly, SNO is the most important and, perhaps, the most frequent process of nitrosation in ischemia/reperfusion and cardioprotection scenarios. In fact, NO is clearly involved in cardioprotection and within tissues there are many proteins with cysteine/thiol groups, especially within mitochondria membrane. Moreover, because of NO’s high reactivity the occurrence of SNO in biological systems is quite probable and it is likely that this will influence many protein functions. In fact, SNO has emerged as an intriguing signaling modality, effectively acting as a reversible molecular switch analogous to phosphorylation.
Importantly, these changes (e.g. SNO or tyrosine nitration) have each been shown to occur with physiological and/or pathophysiological levels of NO. Moreover, these modifications have only been found on a limited number of residues in a subset of proteins in in vivo and in vitro studies, suggesting that the modifications do not occur randomly and, therefore, may constitute a signaling event, akin to phosphorylation.9,10,64,65 As said, mitochondrial proteins would be preferably S-nitrosylated. In fact, it has been reported that within the hydrophobic environment of biological membranes the reaction between NO and O2 is accelerated, thus favoring protein nitrosylation66,67; it has also been suggested that the high level of reactive-cysteines in mitochondrial proteins, and the increased stability of N2O3 in the hydrophobic milieu of the mitochondria, would favor SNO.9,68
Several studies have shown that nitrosative protein modifications can alter protein function. In particular, there is evidence that SNO is reversible, as described above. On the contrary, tyrosine nitration is considered mainly as an irreversible modification that can impact on some signaling pathways associated with formation of ONOO− and NO2.69,70 As said, NO may react with O− to form ONOO−, and it is widely accepted that enhanced ONOO− formation is cytotoxic via nitrosative stress. Its toxicity is also in line with the irreversibility of the reaction. However, we should keep in mind that physiologic levels of ONOO− may contribute to regulation of normal cellular functions via SNO of mitochondrial and non-mitochondrial proteins.69–71 Nevertheless, several NO-mediated positive signaling appears to derive from SNO of proteins. In fact, many physiological cellular functions in which NO is involved, such as excitation–contraction coupling, G protein-coupled receptor signaling, cardioprotection and regulation of cell death processes (i.e. apoptosis and/or autophagy), require directly protein SNO and/or are mediated by process that follow SNO of proteins (see also below). Although autophagy may be a death process (especially when apoptosis is disabled), it is a pro-survival process that has been seen operative in several mechanisms of cardioprotection.72,73
For some authors, SNO of proteins may be considered the typical reaction occurring in redox signaling. In fact, a number of S-nitrosylated proteins have been identified in cardiomyocytes57,61–63,65,71,74,75 and cardiomyocyte mitochondria in physiological conditions and in cardioprotection.47,65,76 Yet, SNO can be reversed by intracellular reductants such as glutathione or ascorbate,9,77 as well as by several enzymes, including SOD9,52 (see also below). Therefore, dynamic SNO/denitrosylation reactions, which are reminiscent of phosphorylation/de-phosphorylation processes, seem essential in cardiovascular regulation. 58
How is protein SNO protective?
There is a large and growing list of proteins for which SNO has been shown to alter activity and/or function.9,64,65,78,79 SNO of proteins fulfills the requirements for a signaling post-translational modification: SNO is stimulus mediated and is initiated by processes that favor NO formation, either involving or not NOSs. NO formed by NOSs or by “non-NOS” processes can directly (or indirectly, see above) lead to SNO of proteins, which can bind to other proteins and can lead to SNO of additional proteins by tranSNO.
Similar to other post-translational modifications, SNO can alter the structure, activity, localization, and/or stability of proteins, thus altering cell function. Moreover, the nitrosylated cysteine residues result protected from irreversible oxidation. Similar to phosphorylation, SNO appears to be not only reversible but also targeted to specific cysteines. In fact, target proteins may have thousands of cysteine residues, but only a few are reversibly targeted for SNO, as it occurs for ryanodine receptor 1 (RyR-1).80,81 The denitrosylation processes are important for the role of SNO, which can put a stop to the signaling, as described above.
SNO has been shown to modify the activity of proteins in virtually all cellular pathways. However, a large number of ion channels seem preferred targets by SNO80,82–86; this is in line with the observation that SNO formation is favored by membrane microdomains and it is especially important in the context of cardioprotection. For instance, Ca++ channel SNO may reduce calcium overload in I/R (see also below).
Although there are no doubts that SNO modifies protein and cell function, the protective effects of these modifications can be gleaned from specific modifications only. For example, protein kinase B (Akt) is a kinase that when phosphorylated plays an important role in signaling cardioprotection and PTEN (phosphatase and tensin homologue deleted on Chromosome 10) is a phosphatase that dephosphorylates Akt. Both Akt and PTEN have been reported to be subjected to the process of SNO. While SNO of Akt prevents the formation of a disulfide bond and attenuates the activity of the enzyme, 87 SNO of PTEN results in its ubiquitination and degradation, thus limiting Akt dephosphorylation. 88 These modifications may be a sort of brake booster, so that Akt activity is permitted, but its exaggerated activation is avoided. In fact, prolonged/exaggerated Akt activation has been proposed to be deleterious. 89
As stated above, SNO has also been suggested to shield thiol groups from irreversible oxidation. In fact, data suggest that S-nitrosylated thiols are protected from irreversible oxidation90–92 and this aspect may play a very important role in cardioprotection.93,94 Redox stress and redox signaling are very important in the genesis of cardiac injury and cardioprotection, respectively.10,95 Moreover, SNO can also favor other redox modifications, such as disulfide bond formation or glutathiolation that may also regulate function. For example, a disulfide bond between the subunits of protein kinase A (PKA) is formed after treatment with nitrocysteine, an NO donor, thus leading to PKA activation, 96 which has been also involved in cardioprotection.97–99 Moreover SNO, by enhancing S-glutathiolation of aldose reductase or mitochondrial complex II increases the activity of these enzymes.100,101 Therefore, S-glutathiolation favors the reduction of aldehydes and the electron transfer, respectively. Interestingly, S-glutathiolation of both complex II and aldose reductase is decreased during reperfusion following ischemia100,101; it might be interesting to determine whether cardioprotective maneuvers reverse the loss of S-glutathiolation in reperfusion.
Preconditioning and postconditioning
As stated above, PreC and PostC are two cardioprotective strategies against ischemia/reperfusion injury.
Postischemic reperfusion may result in exaggerated ROS generation, Ca++ overload, and reduced availability of NO. These modifications together with swift pH recovery may favor prolonged opening of mitochondrial permeability transition pore (mPTP), and other processes contributing to cell death, myocardial infarction, stunning (a transient postischemic contractile dysfunction), and arrhythmias.26,102–104
Ischemic preconditioning can be obtained with brief periods (a few minutes) of intermittent ischemia and reperfusion. These maneuvers trigger two periods of cardioprotection: one immediately after the preconditioning maneuvers that elapse 2–3 h (early preconditioning also known as first window of protection), and a second period of protection (SWOP, also known as late preconditioning) that starts 12–24 h after the preconditioning maneuvers and elapse 48–72 h.22,35–37,105 Here, we consider the first window of protection, which is applied immediately before the infarcting ischemia and exerts the most potent protection against infarct size. Recent data suggest that early preconditioning cardioprotection is also operative during reperfusion (i.e. in the postischemic phase) and limits much of the damage due to ischemia and to reperfusion106–109 (see also below).
Postconditioning can be defined as brief (a few seconds) intermittent cycles of reperfusion alternating with coronary re-occlusion applied immediately after the infarcting ischemic event. It has been shown to reduce ischemia/reperfusion damage, in some cases equivalent to that observed with preconditioning.
Either pre- or postconditioning phenomena can be triggered by pharmacological interventions, including exogenous NO-donors; namely pharmacological PreC or pharmacological PostC.110–115
After the discovery of PreC and PostC phenomena, reperfusion injury has been appreciated as a reality from which protection is feasible, especially with PostC, which is under the control of physicians. Some potentially cooperative protective signaling cascades are recruited by both pre- and postconditioning, namely the RISK (Reperfusion Injury Salvage Kinase), the SAFE (Survival Activating Factor Enhancement), and the cGMP/protein kinase G (PKG) pathways (Figure 2; these pathways have been described in several recent reviews, for example27,116–122). In brief, several extracellular factors produced endogenously (e.g. adenosine, bradykinin, opioids, etc.) bind to cell surface receptors promoting the activation of kinase signaling pathways. The extent of interaction between different pathways and the precise sequence of elements in these pathways are unclear. Nevertheless, a pivotal role has been attributed to the activation of phosphatidyl inositol 3 kinase (PI3K)/Akt and p42/p44 extracellular signal-regulated kinase (ERKs).116–118 This pathway, known as the RISK pathway, leads to the inhibition of mPTP opening at reperfusion, via downstream components of the cascade which include the inhibition of glycogen synthase kinase 3 ß (GSK3ß). A pivotal role is also played by NO and NO. Although the extent to which cGMP/PKG pathway contributes to protection is not clearly established at the present, several studies support the involvement of NO/cGMP/PKG activation in cardioprotection via the attenuation of Ca++ overload, which may be responsible for mPTP opening in reperfusion.
117
Moreover, it has been suggested that the activation of intramitochondrial protein kinase C epsilon (PKCɛ) may cause opening of the mitochondrial KATP channel (mKATP), resulting in a slight increase in ROS, which together with NO may favor SNO formation which, in turn, may favor mPTP inhibition (Figure 2).
119
An additional pathway, the so-called SAFE pathway, has been proposed to play a role in cardioprotection. The principal components of the SAFE pathway are tumor necrosis factorα, the Januse kinase (JAK) which phosphorylates the transcription factor signal transducer and activator of transcription 3 (STAT3).
121
Besides the nucleus, STAT3 translocates to the mitochondria where it phosphorylates critical components conferring cardioprotection (Figure 2). However, the actions of several components of these pathways need to be finally proven.
Cardioprotective pathways activated by preconditioning and postconditioning
As said, the cardioprotective signaling pathways are thought to converge on mitochondria, and various mitochondrial proteins have been identified as targets of post-transitional modifications in both pre- and postconditioning. In these protective pathways, phosphorylative/dephosphorylative processes are widely represented. However, cardioprotective modalities of signal transduction also include redox signaling by ROS, SNO by NO and derivative, S-sulfhydration by hydrogen sulfide, as well as O-linked-glycosylation with beta-N-acetylglucosamine.10,123,124 All these modalities can interact and regulate an entire pathway, thus influencing each other. For instance, enzymes can be phosphorylated and nitrosylated in specific and different site(s) with consequent increase or decrease of their specific activity. For example, ERKs may be S-nitrosylated, thus inhibiting its phosphorylation and activation. 125 Another protein that may undergo NO-mediated SNO and phosphorylation is the regulator protein phospholamban, which is involved in the control of cardiac contractility and protection.9,126,127
Both pre- and postconditioning may be triggered by endogenous and exogenous NO.9,21,26,27,128 The relative importance of classical cGMP/PKG pathway and non-classical processes, such as nitrosylation are under intense investigation.
There are several lines of evidence that SNO of critical proteins plays a pivotal role in cardioprotection by preconditioning34,129–132 (see also below). Whether SNO is involved in cardioprotection by PostC is going to be clarified in recent years (see also below). In fact, a PubMed search for the words “nitric oxide and preconditioning” gives out thousands of articles, whereas a search for the words “nitric oxide and postconditioning” returns hundreds of items. Moreover, a PubMed search for the words “S-nitrosylation and cardioprotection” gives out 16 items,9,34,47,64,65,129–140 six of which34,64,129,130,134,137 are found also with the words “S-nitrosylation and preconditioning” that gives out in total 17 references. 10,34,64,129–132,134,137,141–149 However, four items only are found with the words “S-nitrosylation and postconditioning.”10,47,141,149 Nevertheless, with a more accurate search we could found few other articles that discuss the role of SNO of proteins in postconditioning. Some of these studies used NO-donors in reperfusion (pharmacological PostC) as protective agents and analyzed the role of protein SNO.28,46,150–153 Intriguingly, in a recent Editorial by Schulz and Ferdinandy, 144 which was written as a comment to an interesting article of Sun et al., 154 the authors wonder whether or not “nitric oxide signaling differ in pre- and postconditioning” and in particular they wonder whether “S-nitrosylation is involved in postconditioning’s protection.” Below we will see that SNO is involved in both ischemic and pharmacological postconditioning.
Protein SNO in preconditioning
Several reports have shown that PreC maneuvers induce NO production by activation of NOSs. While the iNOS is involved in the second window of protection,36,37 both constitutive isoforms of NOS, namely eNOS and nNOS, are implicated in the first window of protection.99,155,156 Nevertheless, inhibition of NOS not always abolishes the cardioprotection induced by PreC.156–159 Therefore, the “non-NOS” production of NO may also play a role in PreC scenario. 160 It has been suggested that PreC may provide an environment that favors NO production and SNO of proteins. Among pro-SNO conditions are included favorable ion content, redox equilibrium, and acidic pH. 129 In particular, acidic conditions favor non-enzymatic nitrite reduction to NO and render protein cysteine residues with the pKa lower than intracellular pH more sensitive to SNO. However, S-NO bonds are unstable in acidic conditions, so that the observation that pH is transiently acidic in early reperfusion of protected hearts (see also below) is in line with an important role of protein SNO for protection against reperfusion injury. In fact, it has been suggested that transient acidic pH in early reperfusion “triggers” SNO formation and the subsequent recovery of pH may avoid S-NO bond instability. 47
The “classical” protection induced by NO in PreC is dependent, at least in part, by the activation of GC/cGMP/PKG, which in turn may lead to the opening of the mKATP channel in cooperation with intramitochondrial PKCɛ13,33 (Figure 2).
Recently, Sun et al., have reported that the protective effect of NO is not related primarily to activation of the sGC/cGMP/PKG signaling pathway, but rather through SNO signaling in PreC-induced early cardioprotection (Figure 1). 154 In fact, the infusion of the inhibitor of sGC, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), did not abolish completely the cardioprotection induced by PreC. Intriguingly, hearts treated with ODQ displayed enhanced protection concomitant with a higher SNO level. These results suggest that NO-mediated cardioprotection is regulated by SNO of proteins rather than through activation of the sGC/cGMP/PKG signaling.154,161 With an elegant experimental approach, the same group 131 has demonstrated in isolated heart that caveolin-3-associated eNOS/NO trafficking between plasma membrane and mitochondria provides an important signaling pathway regulating SNO of mitochondrial proteins. It seems that the NO/SNO signaling induced by PreC maneuvers is transported to mitochondria starting from the caveolae. In fact, the caveolae are important for many signaling pathways and the disruption of caveolae could inhibit protection by blocking a number of signaling pathways.162,163 This is in line with the observation that blockade of the internalization of signaling molecules associated with G protein-coupled receptors inhibits cardioprotection afforded by PreC, 164 and it is in line with the central role of mitochondria in cardioprotection.165,166 However, as said above, a role for sGC in cardioprotection has been demonstrated several times, including studies which used a specific sGC activator.167–169 This suggests that both effects (sCG activation and protein SNO) may take place in cardioprotection, with, perhaps, the prevalence of one or the other effect in the different phases of the cardioprotective signaling cascade.
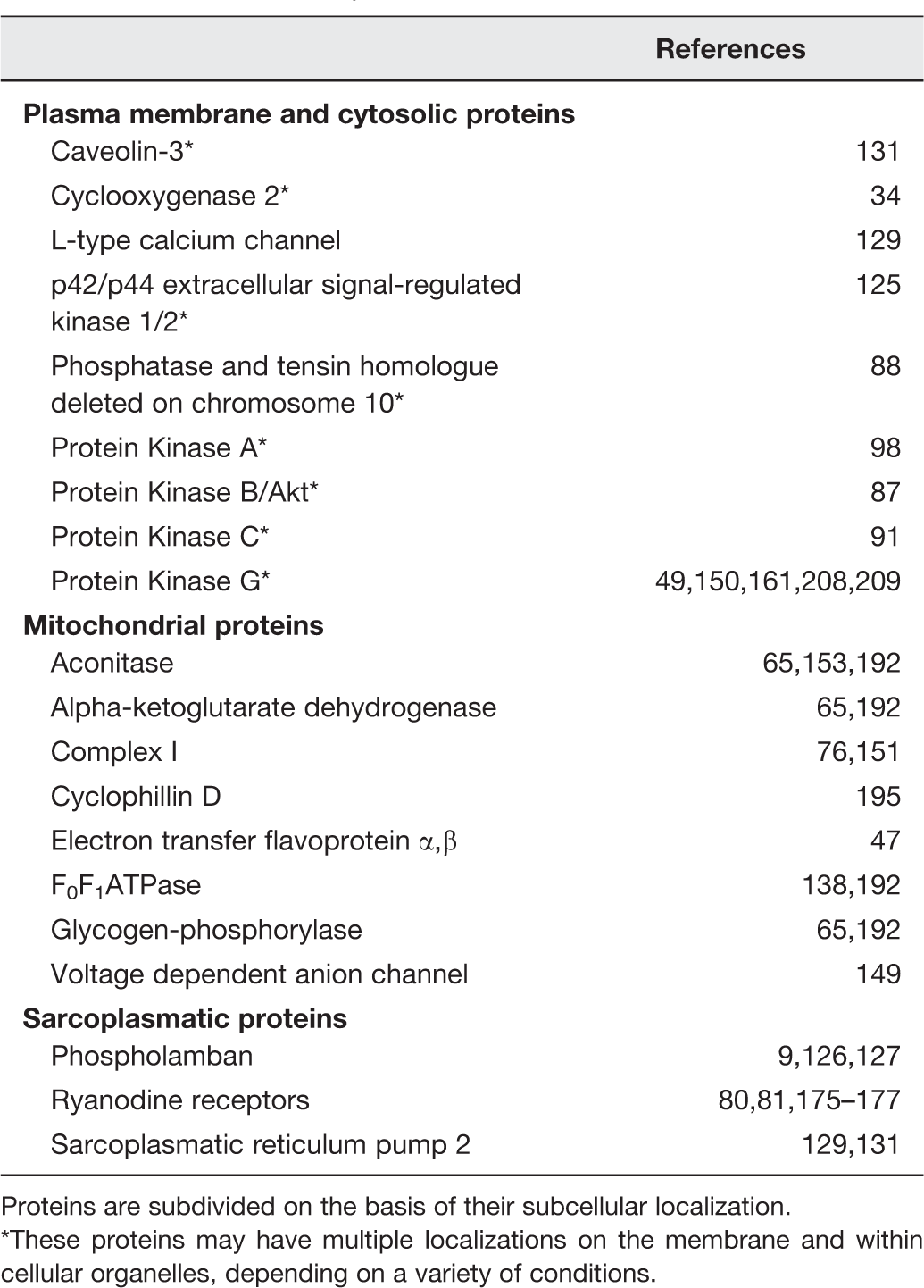
Several nitrosylated proteins have been somehow involved in cardioprotection (Table 1).
65
Here, we consider some of them involved in preconditioning only. The action of SNO during PreC is important for many enzymes and cellular structures. In fact, SNO induces modification of the activity of both metabolic enzymes
155
and proteins involved in the mechanisms for calcium control,
177
which are particularly important for cardioprotection (Figure 3). In fact, in cardiac cells the levels of Ca++ are regulated by different mechanisms, including Ca++ handling by trans-membrane calcium channels,129,178 mitochondrial transporters, and sarcoplasmatic components, such as ryanodine receptor 2 (RyR-2) and sarcoplasmatic reticulum pump (SERCA2)129,177 (Figure 4). The relative contribution of each of these mechanisms to the intracellular Ca++ levels is different in preischemic, ischemic, and postischemic conditions with also gender-based differences.130,179
S-Nitrosylated proteins during preconditioning (PreC) Control of Ca++ Handling by S-nitrosylation S-nitrosylated proteins reported to be subjected to S-nitrosylation and involved in cardioprotection. Proteins are subdivided on the basis of their subcellular localization. These proteins may have multiple localizations on the membrane and within cellular organelles, depending on a variety of conditions.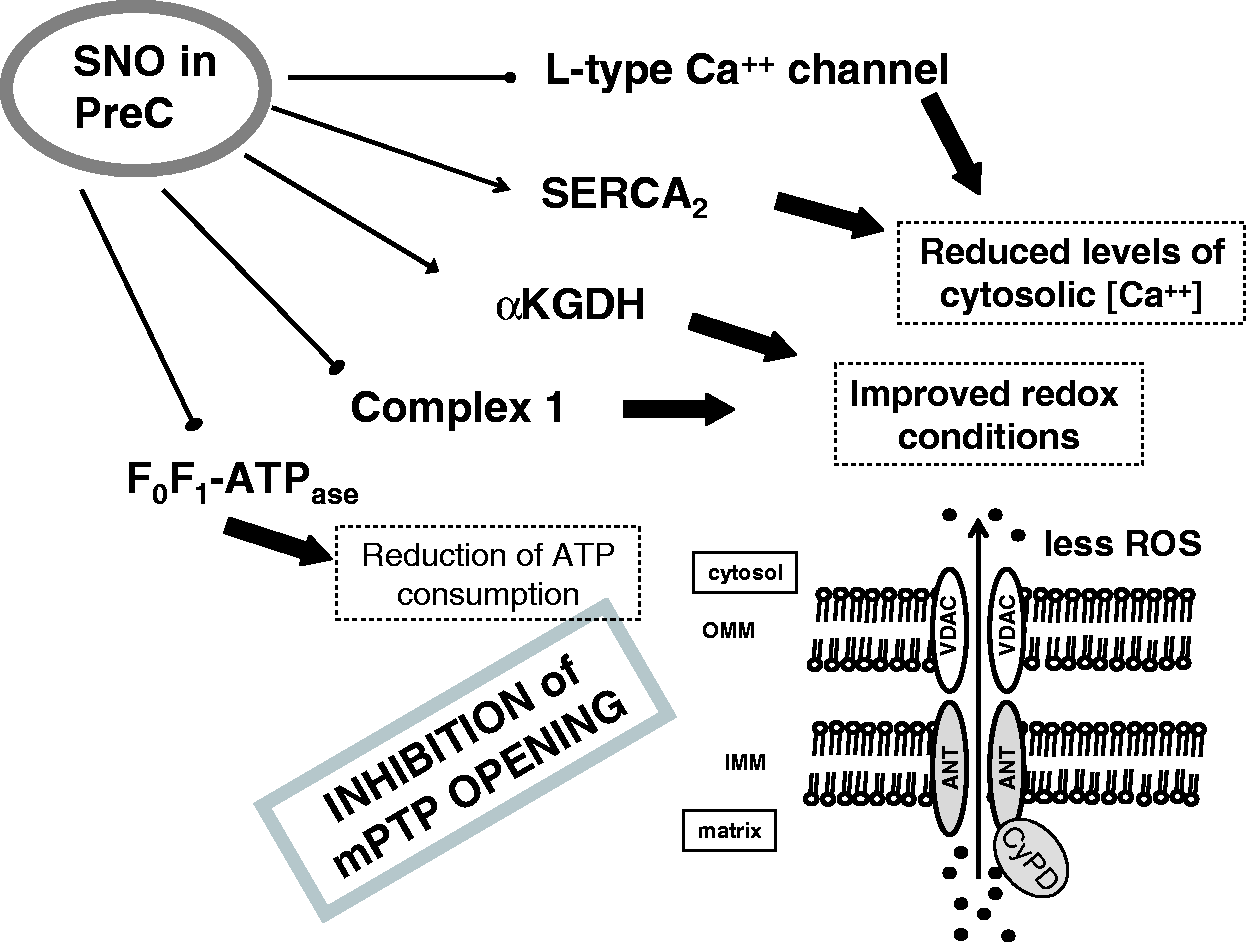
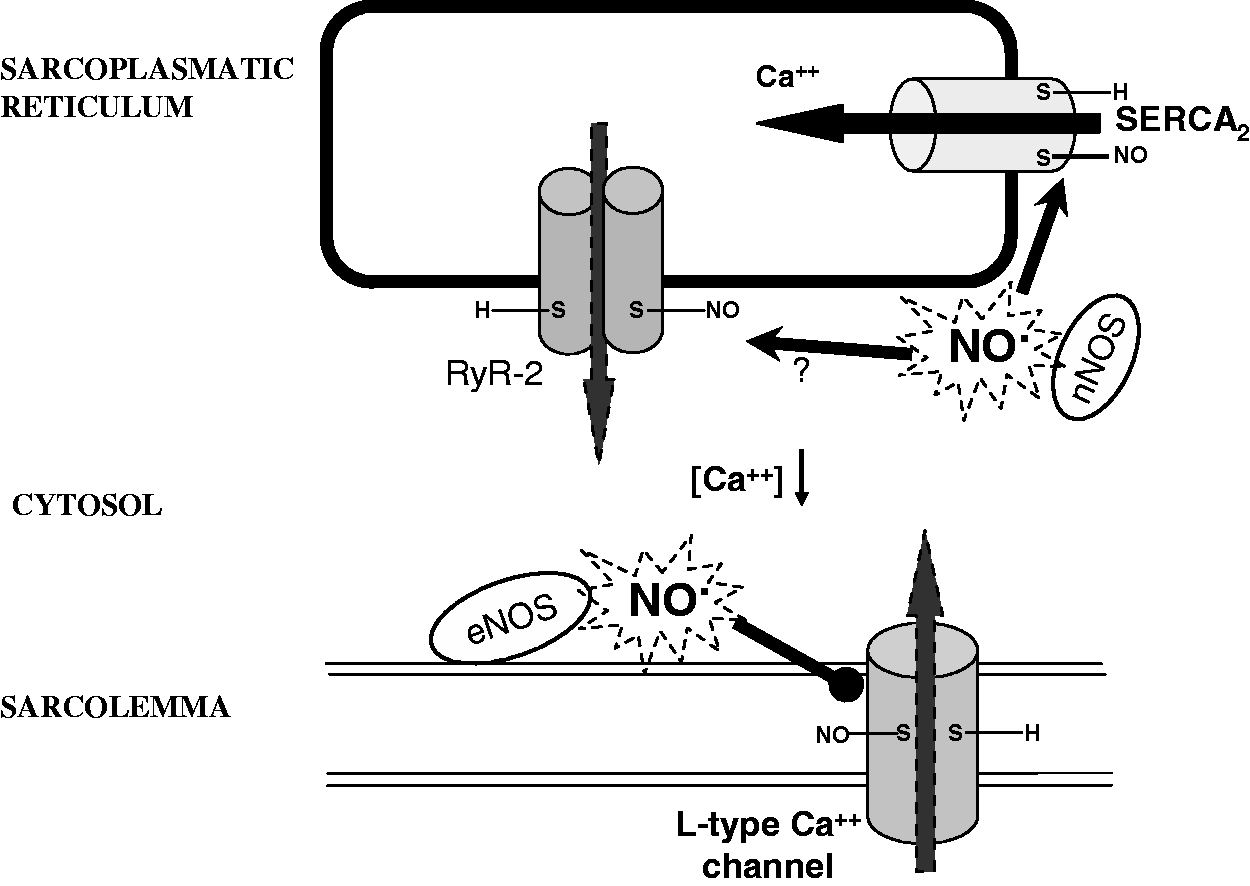
Similar to RyR-1 in skeletal muscle,174,175 RyR-2 in cardiac cells 176 is endogenously S-nitrosylated, suggesting that SNO is a physiological modulator of both skeletal and cardiac cell excitation–contraction coupling. In particular, a number of studies have reported that NO-donor concentration, oxygen tension, membrane potential, the presence of RyR agonists and other sulfhydryl modifying reagents may influence the effect of SNO on RyR activity.174–176,180–183 The majority of studies report an increased Ca++ release following SNO of RyR-1 in skeletal muscle, whereas the effect of RyR-2 SNO is less clear in cardiac muscle (question mark in Figure 4). Some evidences suggest that RyR-2 SNO increased Ca++ release while other data suggest that RyR-2 SNO reduced Ca++ release. In fact, it has been reported that like RyR-1, RyR-2 activity is dependent on oxygen tension. However, unlike RyR-1, RyR-2 was not effectively S-nitrosylated and activated by NO-donor. Yet, RyR-2 was modified and activated by GSNO, ONOO−, and HNO (one-electron reduction product of NO).176,181,184,185 Either ischemic PreC or application of GSNO, HNO or NO donors resulted in cardiac protection against I/R injury and elicited a similar pattern of protein SNO.129,184,186 This suggests that SNO of protein protects cells from I/R damage. Although it is likely that RyR-2 is modified in the ischemic/reperfused and conditioned hearts, the redox modifications and activity are unclear.
The effects of NO-related activity and cellular thiol redox state on basal L-type calcium current, (ICa,L), have been studied using the patch clamp technique. It has been found that both cGMP-dependent and redox-dependent mechanisms (i.e. SNO and/or thiol oxidation) are involved in inhibition and stimulation of ICa,L, respectively. These findings suggest that direct redox modulation of sarcolemmal L-type calcium channels is a physiologically relevant mechanism for modulation of cardiac mechanical function. 187 However, it has been reported that nitrosothiols induce inhibition of L-type calcium channels (Figure 4), and that this inhibition involves both a reduction of the open probability of the single channel and a reduction of the conductance.129,178,188,189 Importantly, it has been reported that α1-subunits of the Ca++ channels are constitutively S-nitrosylated in the mouse heart. Yet, their nitrosylation increases after ischemia leading to the inhibition of the channels in female hearts only, supporting gender differences in postischemic Ca++ overload in cardiac cells, which can be at the basis of smaller infarct size in females. 84 In particular, the SNO of α1 subunit is increased during PreC maneuvers. Moreover, the pharmacological PreC with GSNO (a NO donor) reduces the damage after I/R and is associated with a decreased Ca++ entry through L-type calcium channels. 129 Similarly, the pharmacological preconditioning induced by platelet-activating factor leads to the SNO of calcium channels and consequent reduction of postischemic Ca++ overload.132,190
Another important protein that is S-nitrosylated is the SERCA2 pump, 129 which is also involved in calcium handling and is associated to infarct size reduction and improved cardiac function following myocardial ischemia.129,191 In fact, during ischemia and early reperfusion, SERCA2 pump activity may reduce calcium overload, also improving diastolic cardiac relaxation. 192 Importantly, PreC and GSNO pretreatment (concentration-dependently) increase SERCA2 activity. Moreover, it has been suggested that SERCA2 pump SNO is responsible for the increased activity, which is associated to a reduced open probability of mPTP (Figures 2 to 4). In fact, as said, the pore opening depends on calcium levels.
Besides single protein SNO, such as Ca++ channels and SERCA2, multiple S-nitrosylated proteins have been shown by proteomic studies after PreC.64,193 Many of these proteins have been found within mitochondria. In fact, PreC increases the SNO of different proteins responsible for mitochondrial metabolism (e.g. alpha-ketoglutarate dehydrogenase, glycogen phosphorylase, aconitase, etc.), with modification and preservation of their activity.65,138,172 During PreC was also observed the inhibition of F0F1-ATPase by SNO, with consequent reduction of adenosine triphosphate (ATP) consumption by reserve mode of F0F1-ATPase, which typically occurs in ischemic conditions138,172 (Figure 3). The inhibition of F0F1-ATPase preserves ATP levels and reduces the mitochondrial membrane potential, thereby reducing the driving force for calcium uptake into the mitochondrial matrix, thus increasing tolerance to ischemia. 160 Another important component that is subjected to SNO during PreC, is the mitochondrial respiratory complex I, which is reversible inhibited when nitrosylated. 151 An irreversible inhibition of this complex can occur when it is subjected to nitrosation by ONOO−.9,194
As stated above, an important effector of cardioprotection is the inhibition of mPTP opening.25,165,195 In fact, this pore is regulated by ROS, calcium, and mitochondrial membrane potential, which are regulated by SNO of proteins. Not only the decrease of calcium loading by increased reuptake by SNO of SERCA2 but also the SNO of F0F1-ATPase reduces indirectly the opening of mPTP, which reduces the breakdown of glycolytic ATP and the acceleration of the fall in the mitochondrial membrane potential. Moreover, SNO of CyPD (cyclophillin D) 173 and/or of voltage dependent anion channel, 149 two putative components of mPTP rich in thiol groups, may occur in cardioprotection.
Recently, Kohr et al. 134 using two different methods to measure protein oxidation have shown that preconditioning leads to SNO of many proteins and that a large majority of these proteins are protected from oxidation.
Altogether, these data support the view that SNO of mitochondrial proteins serves as an important mechanism of preconditioning cardioprotection.
Is SNO important for postconditioning’s protection?
Several lines of evidence and experimental data suggest that SNO is involved in postconditioning’s protection.
As stated, NO can derive from enzymatic and non-enzymatic processes and the latter are favored by acidosis,1,196 a condition that is of paramount importance in inducing postconditioning’s cardioprotection. 128 As reported by several authors, PostC attenuates endothelial cell dysfunction by increasing eNOS activity and NO bioavailability.21,197 Activation of PKC plays a central role in cardioprotection 26 and postconditioning,198,199 and, importantly, PKC activation via redox-sensible SNO process has been also suggested. 91 We should recall that PKC is also a target of PKG, thus further supporting a protective role of sGC/cGMP/PKG signaling. Whether PKG may target directly or indirectly PKC is a matter of debate.13,149,200,201
We have found that PostC discretely changes the activity of antioxidant enzymes in early reperfusion, slightly decreasing SOD and increasing catalase activity.
47
Since SOD may be a de-nitrosylating enzyme,
9
these effects may favor SNO thus reducing injury due to oxidative stress (Figure 5).
Protein S-nitrosylation during intermittent reperfusion in postconditioning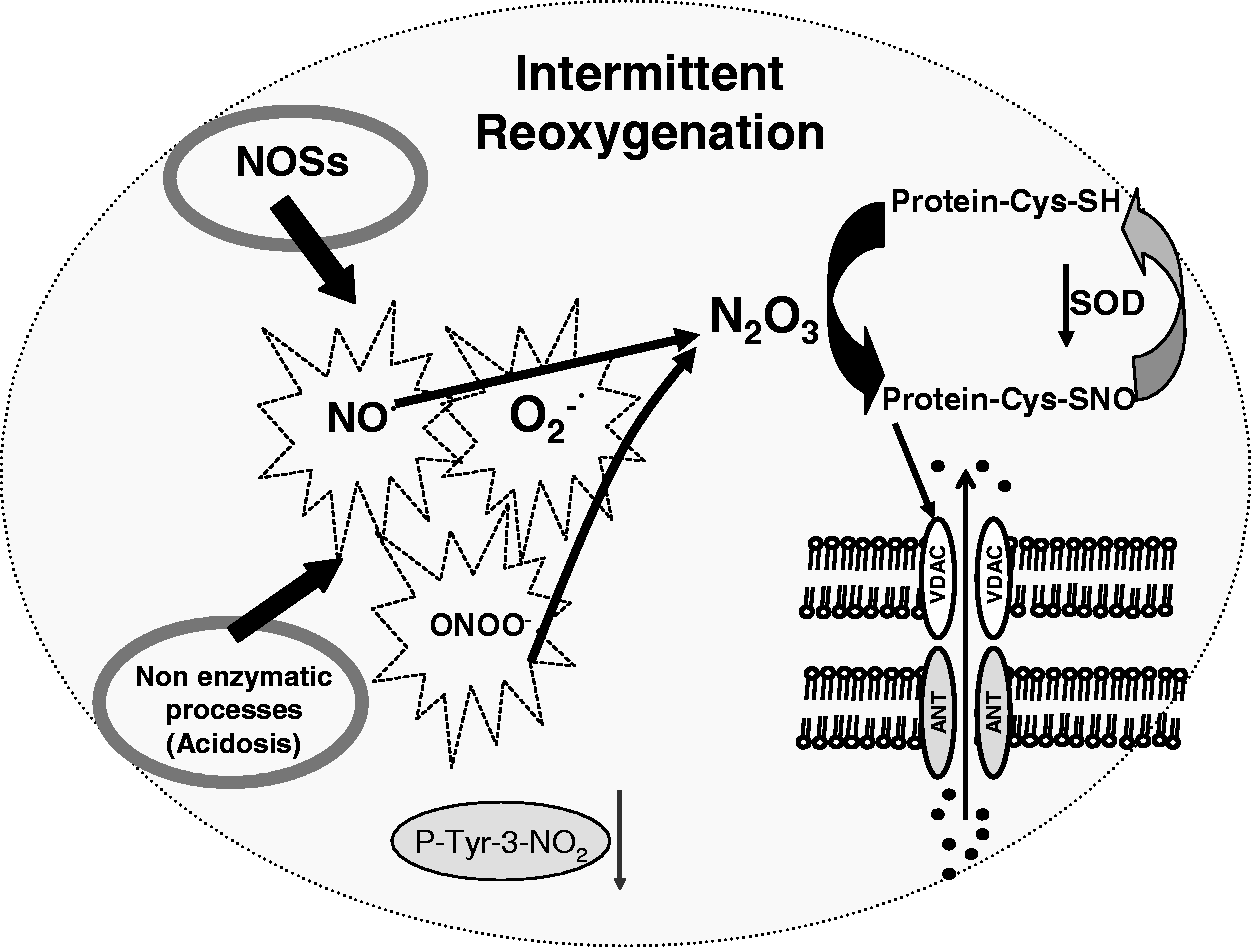
The instantaneous redox state and ultrastructural accessibility of cysteine residue(s) under low-oxygen tension, such as hypoxia, ischemia and postconditioning intermittent ischemia/reperfusion may determine whether a particular thiol in a given protein is subjected to SNO. 202
We have shown in rat hearts that after 7 min of reperfusion ischemic PostC induces a reduction in 3-nitrotyrosine levels and an increase in SNO of proteins, which persist for at least the 120 min of reperfusion. 47 The reduced levels of 3-nitrotyrosine in PostC have also been observed by Inserte et al. 49 Very recently, we have shown that protein SNO occurs mainly in mitochondria either after ischemic PostC or pharmacological PostC induced by Diazoxide 149 (a drug supposed to promote ROS-signaling through actions on mKATP channels and connexin 43).25,203–205 In another study, addition of a mitochondria-targeted SNO at the start of reperfusion (i.e., pharmacological PostC) has also been found to be cardioprotective. 206 The NO-donor used in this study is the so-called MitoSNO, which comprises the well known NO-donor S-nitroso-N-acetylpenicillamine conjugated to a triphenylphosphonium (TPP) moiety. The lipophilic TPP allows MitoSNO to pass rapidly through membranes driven by the membrane potential and therefore to accumulate several-hundred-fold within the mitochondria, where it generates NO and S-nitrosylates thiol proteins. 206 The nitrosylation of proteins by MitoSNO and other donors has been confirmed by other authors both in basal conditions and in the context of postconditioning cardioprotection. 206–209 Intriguingly, in a recent study, Methner et al. 150 have reported that the most abundant isoforms of PKG (PKGI) within cardiomyocyte is involved in cardioprotection against ischemia/reperfusion injury. However, after cardiomyocyte-specific ablation of the PKGI gene in the mouse, these authors have shown that it is possible to protect the hearts with several interventions, including PostC with intermittent ischemia or with the NO donor MitoSNO, via SNO of mitochondrial proteins. Therefore, the authors concluded that PostC may afford protection either by-passing PKGI or by acting independently or downstream of it. Using this conditional knockout approach to inactivate PKGI the authors suggested differences between cGMP/PKGI pathway in myocytes and other cardiac cell types during ischemic PostC’s protection in vivo. However, they cannot rule out that the exogenous and endogenous NO may act to protect the heart from reperfusion injury in a manner that depends on PKG in other cardiac cell types. In fact, PKG has been involved in PostC protection in different models by several authors.49,150,170,171
Therefore, for the reasons above reported, NO appears to be an important mediator in PostC and both cGMP/PKG-dependent signaling49,150,170 and mitochondrial protein SNO47,149 play a pivotal role in postconditioning’s cardioprotection (Figure 5).
Our recent finding that PostC with Diazoxide enhances SNO, supports the idea that an appropriate redox environment is necessary for SNO-mediated cardioprotection.25,149,203–205 Intriguingly, Diazoxide enhances protein SNO also in the absence of ischemia, thus suggesting that part of the preconditioning effect induced by this drug may be attributed to the SNO of critical proteins.
During the first minutes of reperfusion usually a typical large burst of ROS occurs in naïf (not protected) hearts. The ROS burst is well documented to result in the irreversible oxidation of a number of important proteins. These proteins are irreversibly modified and need to be degraded and re-synthesized to regain normal function, otherwise myocardial injury occurs. Because SNO is a transient readily reversed modification, the shielding effect of SNO must be timing. This could be of paramount importance during PostC maneuvers. In fact the ROS burst is attenuated by PostC maneuvers and SNO occurring during PostC may shield modified cysteines from more irreversible states of oxidation till the burst of ROS vanishes. This point of view is in line with the experimental evidence that a delay in performing PostC maneuvers results in a loss of protection.22,94,210
It has been found that protein nitration may be beneficial45,46 or deleterious49,211,212 in PostC. We have proposed that protein nitration may be a transient effect of PostC, which is suddenly followed by the prevalence of protein SNO. 47
Further studies are needed to fully explore the role of protein SNO in both cardiomyocytes and in non-myocyte cell populations as well as the signaling pathways involved in cardioprotection. In particular, it is necessary to individuate the specific S-nitrosylated proteins, their main localization and the role they have in PostC cardioprotection.
Conclusions
Coronary heart disease is a major cause of death and morbidity worldwide. Nitrosative and oxidative/reductive stress is among the major contributors to its varied pathophysiology. New insights into the sources of oxidative/nitrosative stress, its compartmentalized targets, and the understanding of endogenous and exogenous modulators “designed” to control this stress may pave the road for novel approaches and therapies. These can go further to oxidant-scavenging therapies for cardiovascular disease that have been disappointing to date. With no doubt SNO is gaining much attention in the “cardiovascular community” and the understanding of its precise role may open new perspectives in the field of ischemic heart disease. As evidenced in this minireview the nitrosative/oxidative signaling is a major player in the triggering of cardioprotection and among the plethora of reaction mediated by this signaling the covalent attachment of an NO-moiety to a nucleophilic protein sulfhydryl resulting in SNO is a fundamental step for cardioprotection. Here, we have highlighted experimental evidences of SNO contribution in both pre- and postconditioning and have considered the possible cooperation between nitration and nitrosylation in inducing postconditioning protection.
Footnotes
Author contributions
All authors participated in the design of the Minireview and selection of the articles to be quoted; CP and PP wrote the first draft of the manuscript, and CA made the figures and legends. All the authors have read and critically suggested revisions before the submission.
ACKNOWLEDGEMENTS
The authors thank Prof Donatella Gattullo and Dr Daniele Mancardi for invaluable suggestions. The authors received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors for writing this article. The researches of the authors were supported by funding from the Italian Ministry of the University and Research (MIUR) “Progetti di Rilevanza Nazionale” [PRIN, 2008, CP] and “Fondi per la Ricerca Locale” [Unito ex-60%, PP, CP].