
Abstract
The tremendous cost of drug development is often attributed to the long time interval between identifying lead compounds in preclinical studies to assessing clinical efficacy in randomized clinical trials. Many candidate molecules show promise in cell culture or animal models, only to fail in late stage in human investigations. There is a need for novel technologies that allow investigators to quickly and reliably predict drug safety and efficacy. The advent of microtechnology has made it possible to integrate multiple microphysiologic organ systems into a single microfabricated chip. This review focuses on three-dimensional engineered skin, which has enjoyed a long history of uses both in clinical treatments of refractory ulcers and as a laboratory model. We discuss current biological and engineering challenges in construction of a robust bioengineered skin and provide a blueprint for its potential utility to model dermatologic disorders such as psoriasis or cutaneous drug reactions.
Keywords
Introduction
The main function of the skin is frequently described as one of a physical barrier or thermoregulation. However, a more nuanced understanding has emerged over the past decade that has demonstrated numerous other roles, including immunologic, endocrine, metabolic, neurosensory, and psychosocial functions. 1 These involve coordinated functions between the epidermis and dermis, as well as other skin components which include hair, sensory nerves, the immune system, eccrine glands, apocrine glands, and sebaceous glands. This milieu forms a microenvironment where one appendage may unexpectedly influence the functions of others. For instance, the role of the hair follicle (HF) in re-pigmentation and wound re-epithelialization is observed clinically in patients with vitiligo or leg ulcers, and supported by studies in mouse model.2,3 More recently, transplanted bioengineered mouse pelage follicle germs have been shown to form HFs that respond appropriately to acetylcholine stimulation (i.e. piloerection), suggesting that they form autonomous connections with the arrector pili muscle and the autonomic nervous system. 4 Unexpectedly, the HF may itself also guide innervation both in vitro and in vivo. 5
The increasing demand for skin grafts coupled with the complex three-dimensional (3D) architecture of the skin has led to the development of sophisticated tissue engineered skin equivalents. Apligraf®, which is Food and Drug Administration (FDA) approved for the treatment of refractory venous and diabetic ulcers, is comprised of bi-layered “dermis” and “epidermis” derived from human foreskin fibroblasts and keratinocytes. 6 Further refinements in 3D printing have now led to the development of bioprinted skin, where stem cells or keratinocytes and fibroblasts can be printed directly on wounds in a spatially defined manner.7–9 These novel biological dressings have been shown to improve wound healing in mouse models, but human data are not yet available.9,10 While these novel methods offer significant technical advances, the incorporation of cell types other than fibroblasts and keratinocytes into skin equivalents is an area of active research.
One of the driving forces behind the development of an integrated organ on a chip system is to facilitate the detection of adverse drug reactions and to model clinical diseases. In order for bioengineered skin to realize its full potential for these purposes, a more biologically robust model is necessary. The skin is particularly challenging in this regard since it is comprised of heterogeneous cell populations whose interactions vary both in time and space. In this review, we first define the cellular components that are necessary to model skin diseases in biologically meaningful ways. Next, we outline some engineering challenges involved in building a complex organ from these components. Finally, we provide a blueprint for constructing engineered skin for modeling inflammatory skin disorders and adverse drug reactions.
Biological challenges in building “skin on a chip”
Early efforts to develop engineered skin were based on normal human keratinocytes (NHKs) that proliferated and differentiated on de-epidermized dermis.
11
This approach soon gave way to NHKs cultured supported on membranes, and eventually, on a dermal equivalent comprised of fibroblasts suspended in collagen matrices (Figure 1(a)). This two-components model proved to be adequate for their original uses which were: (i) improving wound healing, (ii) assessment of percutaneous absorption and metabolism of drug candidates, and (iii) screening for potential irritants and allergens.
12
However, as more became known about skin function it also became clear that a two-component model of the skin is not sufficient to accurately recapitulate the complex pathophysiology of skin diseases.
A two-component model of engineered skin. (a) Keratinocytes cultured on a fibroblasts/collagen matrix formed stratified squamous epithelium with an air–liquid interface. (b) Hematoxylin and eosin (H&E) staining of engineered skin. (c) H&E histology of normal skin, showing rete ridges (RRs) and a hair follicle (HF). (d) H&E histology of tuberculoid leprosy, demonstrating epitheloid granulomas (G) near peripheral nerves (PN). (e) H&E staining of chronic cutaneous lupus erythematosus (CCLE) demonstrating lymphocytic lichenoid infiltrate (lymph). Inflammatory cytokines milieu in CCLE results in vacuolar interface changes, basement membrane thickening, and follicular plugging (FP). (f) Proliferation of spindle cells in Karposi’s sarcoma forming slit like vascular spaces. (Images in Figure 1(c) to (f) reprinted with permission from New Zealand Dermatological Society Incorporated, published online at: http://www.dermnetnz.org)
Histopathologic comparison between engineered skin (Figure 1(b)) and normal human skin (Figure 1(c)) showed that engineered skin does not contain rete ridges (RR) or other appendages such as hair follicles (HFs). Histopathologic examination of the skin in systemic diseases resulting from infectious (Figure 1(d): leprosy), inflammatory (Figure 1(e): lupus erythematosus), or neoplastic (Figure 1(f): Kaposi’s sarcoma) etiologies further highlights the need for a more sophisticated skin equivalent. In tuberculoid leprosy (Figure 1(d)), granulomatous infiltrates of histiocytes and macrophages are seen surrounding peripheral nerves. In chronic cutaneous lupus erythematosus (Figure 1(e)), lymphocytes are often seen at the dermal–epidermal junction. Some sequelae of this inflammatory reaction include follicular plugging, thickening of basement membrane, and increased mucin deposition in the dermis. In Karposi’s sarcoma, HHV-8 infection leads to proliferation of spindle-shaped cells forming slit-like vascular spaces with extravasated red blood cells. Although it is difficult to ascribe a cause and effect relationship to any particular histologic features in these diseases, it is clear that cell types beyond fibroblasts and keratinocytes are essential for interfacing between the skin with other organ systems.
Epidermis components
A full thickness model of the skin contains the epidermis, the dermis, and the hypodermis which comprised primarily of adipocytes and the vasculatures. A simplified model of these layers is illustrated in Figure 2. The epidermis is a stratified squamous epithelium that contains four layers: stratum basale, stratum spinosum, stratum granulosum, and stratum corneum. The keratinocyte stem cells are located in the stratum basale, and each layer represents increasingly greater level of differentiation. The stratum corneum does not typically contain living cells, but protein and lipid components that form permeability barrier (e.g. preventing free water loss), and immune functions (e.g. preventing entry of pathogenic bacteria, virus, or allergens).
13
Recent work has shown that the epidermal barrier may also regulate metabolic functions, as increased transepidermal water loss in mice deficient in acyl-CoA-binding protein led to increased lipolysis and lipid accumulation in the liver.
14
A simplified “part list” for engineered skin construct. The epidermis is a stratified squamous epithelium that also contains Langerhans cells and melanocytes. In addition to fibroblasts and collagen, the dermis also contains dermal dendritic cells, mast cells, vascular plexus, nerves, hair follicles, eccrine glands, apocrine glands, and sebaceous glands. The hypodermis is comprised predominantly of adipose tissue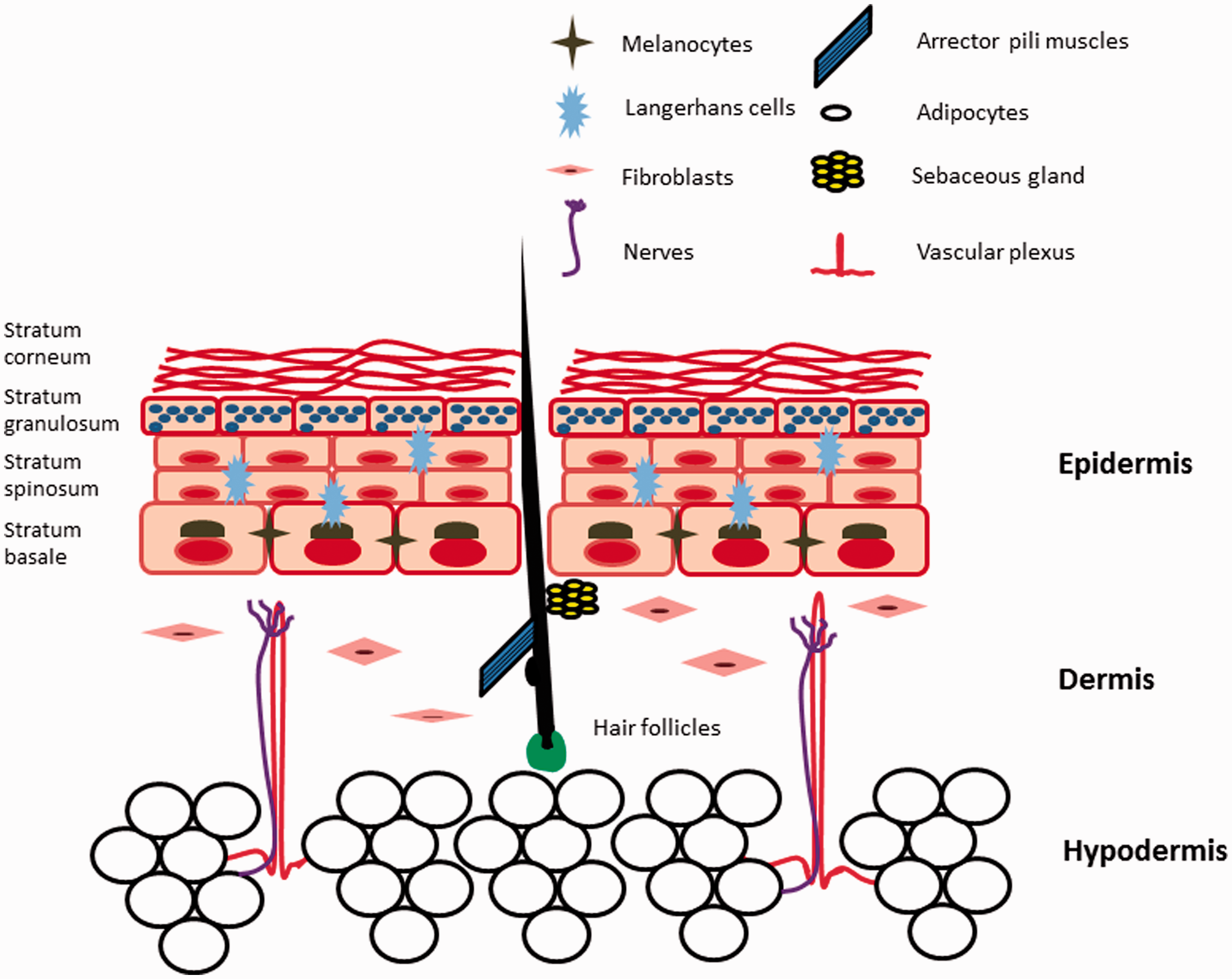
In addition to keratinocytes, the epidermis also contains melanocytes, Langerhans cells, and nerve endings. Melanocytes are primarily located in the stratum basale, where their dendrites facilitate transfer of melanin to keratinocyte stem cells that are also located in this layer (Figure 2). A melanin cap is thought to surround the keratinocyte nucleus and protect the chromosomes of these stem cells against the mutagenic effects of ultraviolet radiation. 15 Also present in the epidermis are Langerhans cells, a type of antigen-presenting cells which play a major role in immune surveillance. Upon stimulation by pathogenic stimuli such as bacteria or contact allergens, these cells emigrate to the lymph nodes, where they present antigens to naïve T cells and instruct them to become memory T cells. Through their modulation of regulatory T cells (Treg), Langerhans cells are also of critical importance in both maintaining tolerance to self-antigen in normal skin and activating protective memory T cells upon infectious challenges. 16
Finally, various types of sensory and autonomic nerve endings can be found in both the dermis and epidermis. It has long been observed that many skin disorders including atopic dermatitis, psoriasis, and acne are exacerbated by psychological stress. 17 A growing body of literature now suggests that neurotransmitters and neuropeptides that are released by these cutaneous nerves modulate the activities of the skin immune cells. For instance, many Calcitonin Gene-related Peptide (CGRP)-containing nerve fibers in the skin are in direct contact with Langerhans cells and keratinocytes that express CGRP receptors. 18 The neuropeptide inhibited the antigen-presenting activities of Langerhans cells (LCs), as well as the differentiation of and cytokine production by T helper cells in vitro, 19 suggesting that it may be a link between the nervous system and inflammatory skin diseases such as contact dermatitis or psoriasis.
Dermis components
In many models of engineered skin, the dermis is constructed as a matrix of collagen with embedded fibroblasts (Figure 1(b)). While this may suffice for modeling the structural function of the dermis, it is not sufficient for capturing the mechanistic complexities of many skin diseases. In human skin, the dermis is a tightly integrated microenvironment that accommodates not only fibroblasts, but also nerve endings, vascular and lymphatic networks, macrophages, dermal dendritic and mast cells, and skin appendages (Figure 2). The development of a vascularized skin equivalent is particularly important for modeling inflammatory skin disorders, as it both provides a platform for physiologic integration with other organ systems and aids in the histopathologic diagnoses of skin diseases. Inflammatory skin diseases are often defined by the composition of the inflammatory infiltrates (e.g. neutrophils, lymphocytes, eosinophils, or histiocytes), as well as their characteristic distribution in the skin (e.g. small venules or arteries). However, the mechanistic link between these histopathologic findings and development of clinical diseases is neither well understood, nor are these interactions captured in simple 3D skin models.
Perhaps the biggest biological challenge in developing robust engineered skin is the incorporation of skin appendages such as HFs, sebaceous glands, eccrine, and apocrine sweat glands. Among these, the HF is perhaps the most extensively investigated, as it appears to play both homeostatic and pathogenic roles. For instance, epithelial stem cells and melanocytes stem cells in the follicular bulge may contribute to re-epithelialization and repigmentation during wound healing.20,21 Conversely, abnormal keratinization, leading to plugging of the HFs, contributes to the pathogenesis of acne. 22 Recent investigations have demonstrated that chimeric engineered skin comprising of human keratinocytes and neonatal murine dermal papillae (DP) cells can form rudimentary hair, whereas those that contain human DPs do not.23,24 Our group has recently demonstrated that 3D architecture may be important for maintenance of hair inductive capacity of human DPs, as growth of DP cells in spheroids partially restored both gene signatures of intact DPs, and their hair inductive capability. 25
Engineering challenges and approaches in development of engineered skin
In order to build a skin on a chip that accurately mimics native skin physiology and pathology, many of the complex skin features mentioned earlier must be incorporated. While advanced cell culture and microfabrication approaches are beginning to make this more feasible, significant engineering challenges must still be addressed. An integrative approach to skin-on-chip development must consider cell sources, biomaterial fabrication methods, cell culture and differentiation protocols, and integration with the abiotic interface in order to meet the design goals laid out earlier, as summarized in the following sections.
Cell source selection
As outlined earlier, most current skin constructs are made using normal cells derived from neonatal or adult skin. However, the limited supply and heterogeneity of such tissues prohibits their broad use for building consistent and reproducible skin platforms for drug testing and disease modeling. Induced pluripotent stem cells (iPSC) are an attractive source of starting material for skin-on-a-chip models, as iPSCs can be expanded to create a vast uniform cell bank for larger scale production and can be derived from patients with skin diseases for disease modeling. However, as the skin is a heterogenous tissue comprising of a large number of cell types, it is challenging to develop and validate the numerous differentiation protocols. Further, while biomarker-based validation of skin cell differentiation may be useful, functional tests incorporating cells in 3D engineered skin constructs are the gold-standard assays to validate differentiation of skin cell types. These tests may include homeostatic functions, such as maintenance of skin permeability barrier, or pathogenic functions, such as recruitment of inflammatory cells in response to irritants.
Culture time and medium composition
Although the challenge of maintaining multiple cell types in culture is an issue with any model organ/tissue on chip, the potential large number of cell types in skin presents particularly significant challenges for developing optimized media and culture protocols. Current protocols for making two-component skin constructs consisting of fibroblast seeded-collagen dermis and NHKs epidermis are already fairly complex. 26 First, the fibroblast-seeded collagen dermis is cast and cultured submerged in a “cast feed” medium for 1 week. NHKs are then added to the top, allowed to attach, and then cultured submerged in an “epidermalization” medium for 1 week. The epidermal surface is then exposed to an air interface, while the dermal surface is exposed to a “cornification” medium for 1 week. The constructs reach maturity after 3 weeks of culture and are finally cultured in a “maintenance medium.” Each of these media types includes a different basal media formulation and concentrations of serum and other factors in order to ensure appropriate development of the skin construct at each step.
For each additional component added to the skin construct, including blood vessels, HFs, melanocytes, or immune cells, additional media components or differentiation protocols may be necessary. It is challenging to design experiments that can efficiently assess whether specific media components support or interfere with the development of additional skin structures. However, in some cases the presence of multiple cell types can promote enhanced function of engineered tissues, as illustrated in multicellular engineered liver where parenchymal cells can enhance viability and function of hepatocytes. 27 Thus, supportive heterotypic cell interactions can potentially simplify the media and culture protocols. Furthermore, the timing of adding each cell type/component is also critical. In two-component skin, preculture of the fibroblast seeded dermis before adding NHK is critical for proper epidermalization. The optimal time for incorporating other cell types such as endothelial cells (ECs) has yet to be determined.
Structural complexity
Typical skin constructs are fabricated through casting a single uniform dermal scaffold, which can recapitulate the basic dermal/epidermal structure, but has so far failed to faithfully replicate native skin architecture, including appendages such as HFs and intricate microstructural features of the basal lamina. Tissue microfabrication approaches, which can control the structure of biomaterial scaffolds and positioning of cells to high resolution, are powerful tools for engineering more complex features in skin constructs. For example, micromolding has been used to recreate the complex topology of skin basal lamina, which resulted in improved epidermalization of engineered skin equivalents.
28
Incorporation of blood vessels can enhance integration and survival of engineered tissue grafts following implantation and can enable connection of a skin on chip to other organs. Skin was one of the first tissues to be vascularized in vitro.
29
ECs seeded in a collagen or fibrinogen can form capillary-like structures which could eventually be connected to a host and become perfused with blood after implantation.30–33 However, blood vessels formed by de novo vasculogenesis from ECs can take up to a week to form, are difficult to interface with an abiotic flow system connecting to other on-chip organs, and the resulting vascular network architecture is uncontrolled. An alternative approach to vascularizing tissues in vitro is to create microfluidic biomaterial scaffolds, in which an open channel network is preformed in a tissue scaffold during fabrication.34,35 ECs can then be cultured in a monolayer on the channel surfaces to more rapidly create perfusable vessels in vitro. Methods to precisely control vascular network architecture will be necessary for control of perfusion and tissue oxygenation. Figure 3 illustrates a prototype of vascularized skin equivalent. However, there is no current standard for how each organ chip can be integrated with one another, and creation of a universal interface might facilitate “plug-and-play” integration of the skin with other organ chips.
A prototype of vascularized skin equivalent. Engineered skin equivalent with liquid–air interface is perfused from the “dermis” with vascularized channels lined with endothelial cells. The cartridge module could be linked in a “plug-and-play” manner with other organ chips. (A color version of this figure is available in the online journal).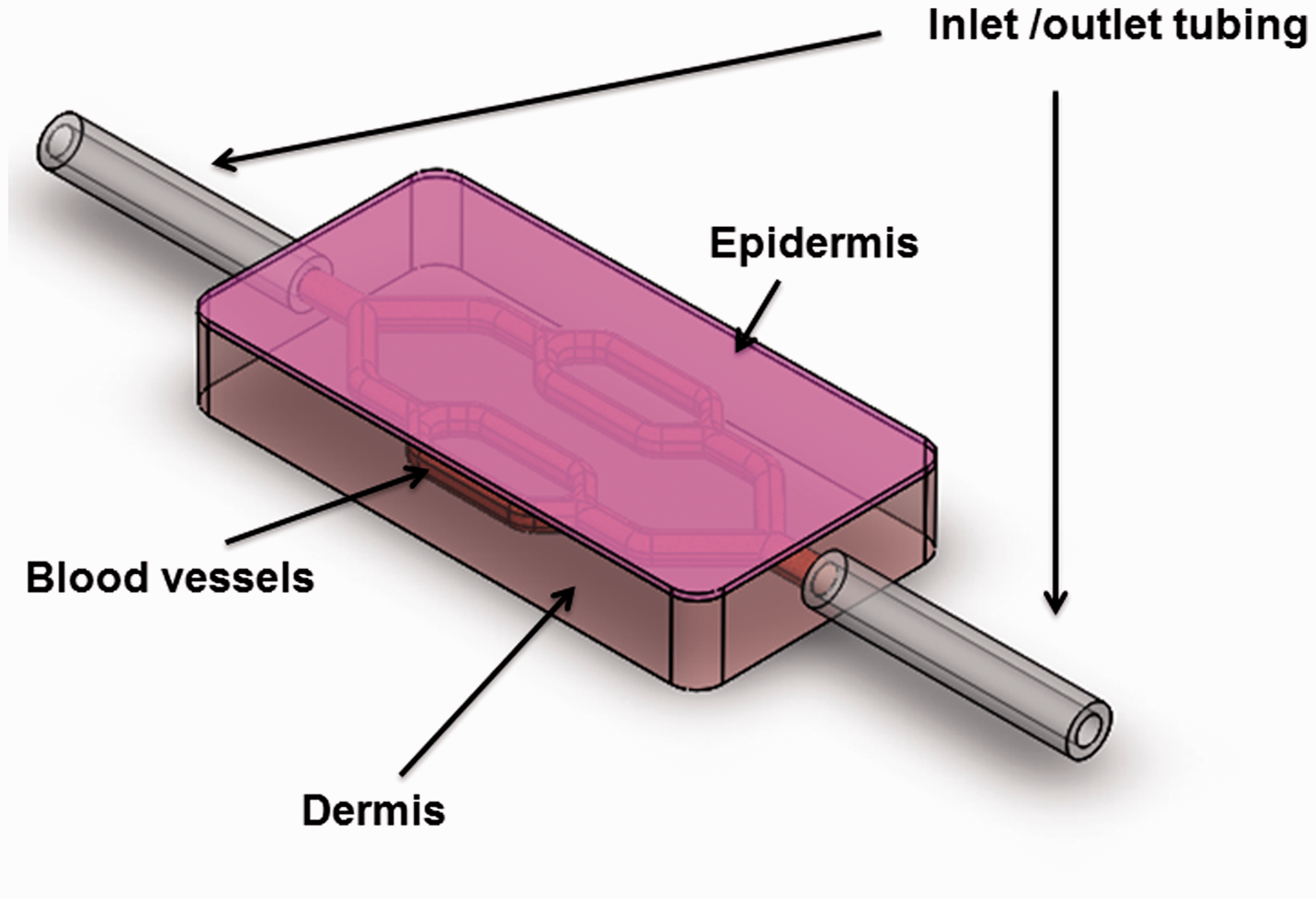
Creating the abiotic–biotic interface
Most current attempts at creating human-on-a-chip models link different tissues/organs through an abiotic interface, such as through microfluidic channels. However, making secure fluidic connections between abiotic materials and 3D tissue constructs is not trivial, particularly in the case of tissues such as skin in which cells can drastically remodel the initial geometry of the scaffold through contraction and matrix degradation. Well-established protocols for skin construct fabrication use varying methods to control how the scaffold remodels. 26 For example, by first casting an acellular layer of collagen on a microporous membrane before layering on fibroblast-seeded collagen, the lateral contraction of the dermal scaffold is constrained by attachment to the membrane. When the matrix contracts, the constructs become thinner but the intended surface area is maintained. The morphological change of fibroblasts and scaffold contraction seems to be critical for proper epidermalization by NHKs. Most current microfluidic tissues have been fabricated within manifolds that have static fluidic inputs and use scaffolds where contraction by cells is negligible. However, significant contraction of engineered skin can disconnect the tissue from a static abiotic fluidic interface, causing leaks. Flexible connections that can be directly embedded within microfluidic tissue scaffolds may help maintain the interface between abiotic flow connections while still permitting the natural remodeling process of the skin constructs.
Access to different skin compartments (i.e. the epidermis and vasculature) is also desirable for a skin on chip in order to mimic delivery of topically or systemically delivered drugs. The design of the skin chip should allow for independent delivery of compounds to the epidermal surface and needs to prevent leaks such that compounds only go through the epidermis and not around it. Lastly, the skin on chip should be readily removable from the device to allow for standard tissue processing and histology, or potentially even for grafting studies to assess function in vivo.
Towards development of organomimetic models for skin disorders
Organomimetic microphysiologic systems are now increasingly being used as surrogates for modeling whole organs. Recently, a proof of concept “lung-on-a-chip” system was not only shown to replicate the pathophysiology of pulmonary edema, but it also accurately predicted the toxicity of the cytokine interleukin-2, which causes pulmonary edema in cancer patients, and the ability of a novel compound, GSK2193874, to reverse this toxicity. 36 In this system, a flexible porous membrane separated human alveolar epithelial cells exposed to air on one side, and EC-lined vascular channel exposed to dynamic flow condition on the other, mimicking the microstructure of an alveolus. Such studies have shown that not only are the biochemical niches critical for cellular functions, but further, that biophysical forces such as cyclic mechanical stretch or extracellular matrix play a tremendous role in driving physiologic responses. 37
With regard to skin, the use of 3D organomimetic model has several key advantages compared to the typical tissue culture counterpart. First, it is well known that cellular functions are altered when skin cells are cultured in 2D as compared to 3D. 38 Second, skin equivalents allow evaluation of functional properties such as transepidermal water loss and permeability that are not possible to evaluate in a tissue culture model.
As discussed previously, 3D skin equivalents have already enjoyed clinical uses in the treatment of chronic ulcers. More recently, the development of 3D inkjet and laser printing technology has facilitated the development of more complex and heterogeneous tissues. Atala and colleagues, for instance, used inkjet printing to construct tissues comprised of human amniotic fluid-derived stem cells, canine smooth muscle cells, and bovine aortic ECs. 39 The ability to pattern different cell populations in a precise way is especially advantageous for modeling the skin, which contains many different cell types and appendages (Figure 2). Efforts to construct more complex engineered skin by bioprinting are now underway. Recently, it was shown that 3D skin equivalents comprised of alternating layers of fibroblasts and keratinocytes suspended in collagen can form skin-like tissue. 7 Furthermore, when these constructs were transplanted onto full thickness wound in mice, they can be integrated with surrounding mouse skin, raising the possibility that it may one day be possible to print new skin directly onto sites of trauma or burn wounds. 9 In order to provide a blueprint toward modeling complex skin diseases with engineered skin, we discuss two common dermatologic disorders, psoriasis and cutaneous drug eruption. These disorders often have extracutaneous manifestations, and thus, present good case studies to illustrate the challenges in integrating engineered skin with other microphysiologic organ systems.
Psoriasis vulgaris
Psoriasis is a chronic autoimmune disease that affects approximately 1–3% of total population. 40 Inflammatory arthritis was the first extracutaneous finding linked to psoriasis, and between 6 and 42% of all psoriasis patients are estimated to have joint disease. 41 More recently, a significant number of studies have linked psoriasis to obesity and metabolic syndrome. In a meta-analysis of 2.1 million patients (201,831 of whom have psoriasis), the odds ratio (OR) for developing obesity among psoriasis patients was 1.66 (95% confidence interval [CI] 1.46–1.89) compared with those without psoriasis. 42 Conversely, in the pediatric population, the OR for developing psoriasis is much higher (OR 1.78, CI: 1.49–2.14) in extremely obese children. Furthermore, the mean total cholesterol, low-density lipoprotein cholesterol, triglycerides, and alanine aminotransferase level were all higher in children with psoriasis compared to those without the skin disease. 43 Thus, psoriasis is increasingly being conceptualized as a skin manifestation of systemic immune-mediated inflammatory disease processes that share genetic susceptibility, environmental factors, and inflammatory pathways as other chronic inflammatory disorders such as atherosclerosis or inflammatory arthritis. 44
The pathogenesis of psoriasis is complex and discussed in greater detail elsewhere.
44
Both laboratory investigations and clinical experience with targeted therapies have suggested dysfunction of the interleukin-17 (IL-17) and IL-22 axes.45,46 Briefly, dendritic cells (DCs) from psoriatic lesions polarize T cells toward T helper -1 (Th1) and T-helper 17 (Th17) phenotypes. Th1 cells elaborate interferon gamma, which induces keratinocytes to produce an array of pro-inflammatory chemokines including CXCL9, 10, and 11. These cytokines recruit more Th1 cells into the lesion, setting up a positive feedback loop.
47
In contrast, IL-17 induces the expression of chemokines that are involved in neutrophil recruitment into the site of inflammation. The characteristic thick, red, scaly plaques seen in psoriasis (Figure 4(a)) are attributed to IL-22, a cytokine which strongly induce epidermal hyperplasia (Figure 4(b)) and is produced by both Th17 and the newly identified Th22 cell subset.
46
Psoriasis. (a) Clinically, psoriatic lesions are well-demarcated thickened red scaly plaques with silvery scale. (b) Histologically, the thickened plaque is seen here as regular epidermal hyperplasia, often with neutrophil abscesses in the epidermis. The dermis contained inflammatory infiltrates comprising of Th1, Th17, and Th22 subsets. (c) Normal engineered skin equivalent control compared to (d) skin equivalent stimulated with IL-22 demonstrating epidermal thickening, but no parakeratosis of elongation of rete ridges. (Images in Figure 4(a) and (b) reprinted with permission from New Zealand Dermatological Society Incorporated, published online at: http://www.dermnetnz.org; images in Figure 4(c) and (d) reprinted with permission from Boniface et al.,
49
Copyright 2005. The American Association of Immunologists, Inc.)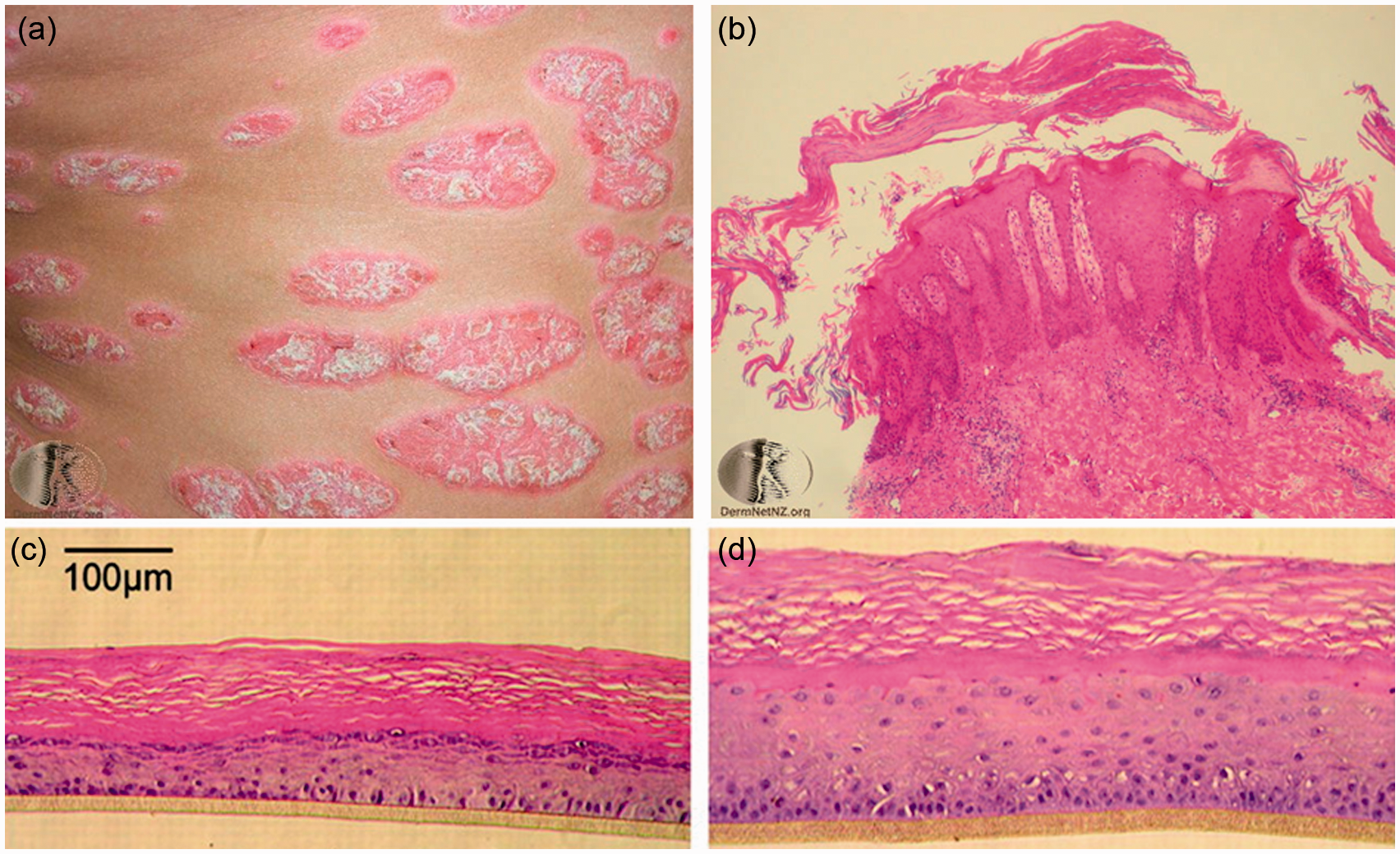
Previous efforts to model psoriasis using skin equivalents have included (i) constructing engineered skin with fibroblasts and keratinocytes derived from psoriatic and non-psoriatic skin 48 or (ii) stimulation of engineered skin with pro-inflammatory cytokines.49,50 Skin equivalents constructed from psoriatic lesions derived fibroblasts and keratinocytes showed increased proliferation, as measured by Ki67 positivity. Compared to engineered skins derived from normal skin, psoriasis-derived skins showed increased expression of inflammatory cytokines such as IL-6 and IL-8. 48 However, these differences were not persistent and decreased dramatically after 21 days in culture. A complementary approach is to mimic the cytokines milieu of psoriatic skin with a cocktail of pro-inflammatory cytokines. Stimulation of engineered skin derived from normal fibroblasts and keratinocytes with cytokines such as Tumor necrosis factor (TNF)-α, IL-1α, and IL-6 resulted in increased expression of psoriasis markers, including human beta defensing-2 and elafin. 50 Compared to normal skin equivalent (Figure 4(c)), stimulation of engineered skin with IL-22 (Figure 4(d)) resulted in epidermal hyperplasia similar to that seen in psoriatic plaques. 49 However, unlike true psoriatic skin lesions (Figure 3(b)), parakeratosis (retention of nuclei in the stratum corneum) and elongation of RR were absent. As there were no other cell types in these skin equivalents, Th17 infiltrate, neutrophilic abscesses, and vascular dilation were also absent. To better recapitulate these pathologic mechanisms, engineered skins were co-cultured with Th cells that had been polarized in vitro into Th1 and Th17 phenotypes. 51 Histologic analyses showed that these in vitro polarized T cells indeed migrated into skin equivalents, induced epidermal hyperplasia, and increased expression of psoriasis markers. Thus, the incorporation of immune cell types including T cells and DCs maybe the first step toward developing physiologically realistic model of psoriasis.
Another challenge in modeling clinical diseases such as psoriasis is the assessment of disease severity and responses to treatment. In the clinical settings, severity of psoriasis is often assessed using psoriasis area and severity index score, a parameter that takes into account both the body surface area involved as well as morphological variables such as redness, thickness, and scaling. However, these clinical variables are often subjective and observer dependent. Moreover, clinical scores may bear little relevance to tissue engineered skin which rarely shows appreciable redness or scale. Another testing approach to assess disease severity is based on histological appearance and/or expression of tissue and serum markers. One of the histopathologic hallmarks of psoriasis is regular epidermal hyperplasia (Figure 3(b)), and the thickness of the epidermis has been used to monitor the response of engineered skin equivalent to IL-22 stimulation. 49 Investigations are currently underway to identify genetic, serum, tissue, and transcriptional biomarkers that can accurately predict disease progression and response to treatment. However, none of them has been completely predictive of disease severity or have been incorporated into routine clinical practice. 52
Adverse drug reactions
Adverse drug reactions are one of the most common reasons for dermatology consultations and clinic visits. In a retrospective chart review of 271 inpatient consults, up to 10% of inpatient dermatology consults is related to evaluation of a “drug rash.” 53 Most drug eruption is morbiliform (also sometimes called “maculopapular”), comprising of coalescing erythematous macules and papules, with no systemic involvement. However, certain types of adverse drug reactions may be associated with severe systemic derangements. These include acute generalized exanthemous pustulosis, toxic epidermal necrolysis/Steven–Johnson Syndrome, and drug eruption with eosinophilia with systemic symptoms (DRESS). With some exceptions, these drug eruptions often resolve with discontinuation of the medication. However, other types of drug reactions, such as drug-induced hyperpigmentation, can be permanent and may not resolve after the medication is stopped.
The diversity in cutaneous drug reactions also means diversity in the underlying pathogenic mechanisms. For instance, drug-induced hyperpigmentation may result from either deposition of the medication itself in the skin, such as that found in amiodarone-induced hyperpigmentation, 54 or deposition of hemosiderin and melanin pigments secondary to tissue inflammation, such as that found in minocycline-induced hyperpigmentation. 55 Integration with other organ systems, especially the liver and the immune systems, is especially important in modeling cutaneous adverse drug reactions. For instance, in DRESS, polymorphisms in hepatic drug detoxification enzymes lead to accumulation of drug metabolites. These metabolites may interact with cellular proteins, which function as haptens, to induce autoimmune responses against liver and skin cells. 56 The immune system is almost certainly involved, and on skin biopsy, an inflammatory infiltrate comprising of lymphocytes, eosinophils, and histiocytes is often diagnostic. Furthermore, there are some well-characterized association between particular Human Leukocyte Antigen (HLA) allotypes, and drug hypersensitivities. The strongest associations reported thus far are with the reverse transcriptase inhibitor, abacavir and HLA-B*5701, the gout prophylactic allopurinol and HLA-B*5801, and the antiepileptic carbamazepine and HLA-B*1502. 57 In hypersensitivity reaction associated with these medications, binding of the drug to the HLA molecules alters the conformation of the peptide binding clefts, thereby changing the repertoire of peptides that are presented. 58
Engineered skin equivalents have long been heralded as alternative to animal-based screening for adverse cutaneous drug reactions. To date, their uses have been limited to screening cosmetics or allergens that may cause phototoxic or irritant dermatitis.59,60 Irritant or phototoxic dermatitis is unique in that the molecules themselves are directly toxic to the keratinocytes, and therefore, do not require sensitization of the immune system. In contrast, drug eruptions such as DRESS require sensitization of the immune system, and in many cases, metabolism by the cytochrome P450 system in the liver. Since existing platform for engineered skin equivalent do not contain immune cells, they have limited utility beyond screening for irritant or phototoxic potentials. Newer platforms such as Skimune™ (Alcyomics, New Castle, UK) improve on these screens by co-culturing of skin explants with autologous DCs and T-cells. When stimulated by the appropriate drug, DCs primed T cells to proliferate and cytokines elaboration ensues. Cutaneous drug eruptions could then be measured by histopathologic examination or cytokine secretion. However, the repertoire and concentration of cytokines generated in this manner are limited, and thus, may impact the sensitivity of this assay. More problematically, skin explants are significantly heterogenous, and its histology maybe affected by both host (e.g. preexisting dermatitis such as eczema) and environmental factors (e.g. smoking or sun exposure).
As was in the case of psoriasis, the assessment of cutaneous drug reaction is difficult to translate from the bedside to the bench. In laboratory studies, the extent of adverse drug reaction has been assessed either by histopathology or by tests of cellular compromises (e.g. dimethylthiazol diphenyltetrazolium bromide reduction, lactate dehydrogenase, and interleukin-6 releases). 61 In clinical practice, the most relevant parameters to monitor are often not the skin findings themselves, but the effect of drug reaction on other organ systems. For instance, DRESS may result in significant liver, kidney, or cardiovascular dysfunctions that persist long after the cutaneous manifestation resolves. 62 Therefore, as microphysiologic platforms for screening drug reactions integrate an increasing number of organ systems, a critical task will be to develop a panel of biomarkers that capture not only the pathogenic mechanism of drug reactions and its effect on the skin, but also its impact on the functions of other organs.
Summary
Virtually all organs in the body are functionally integrated in some way. To model complex skin diseases such as psoriasis or adverse drug reactions, a model of the skin will need to be integrated with those of other organs such as the liver, the kidney, and the heart. This integration presents significant scientific challenges on cellular, tissue, and organ system level. At the cellular level, a major challenge is the formulation of a single medium that allows stable long-term maintenance of different cell types. For instance, Transforming Growth Factor (TGF)-beta1 has been found to enhance the human cell line A549’s functions, but inhibit those of the cell line C3A. 63 By culturing these cell lines in different micro-compartments and limiting cross-talks among them, Zhang and colleagues were able to culture these two cell lines simultaneously. 63 However, as the number of cell lines used expands, the formulation of a single universal medium is likely to become exponentially more difficult.
At the tissue level, a major challenge will be in testing and evaluation of organ functions. As mentioned previously, many dermatologic diseases are frequently monitored by their clinical appearance and extent, and there are few functional or serum biomarkers that are in clinical uses, or are specific for dermatologic diseases. For the purpose of screening for adverse drug reactions, investigators may instead have to rely on markers of liver, heart, kidney functions rather than the appearance or histology of skin equivalents.
At the organ system level, the major challenge in developing such “human-on-a-chip” model is designing and engineering control systems to facilitate the communications between various microphysiologic organs. Fortunately, sophisticated microfabrication strategies now allow for precise dynamic control of fluid flow, structural mechanics, and chemical delivery at a subcellular scale. Recently, investigators have described the development of a microfluidic system that connected engineered “lung,” “liver,” and “adipose” tissues to investigate the pharmacokinetics of naphthalene metabolism. 64 With the incorporation of additional organs and circulating immune cells, perhaps derived from patient-specific inducible pluripotent stem cells, it may be possible for engineered skin to be used as a biosensor to predict the effects of drugs in the physiologic context of health and disease.
Footnotes
Author Contributions
All authors participated in review of the manuscript, and NJE, WL, CH, BG and AMC wrote the manuscript.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number 5U18TR000561-02, and in part by the Leona M. and Harry B. Helmsley Charitable Trust (grant number SALK CU11-0049). NJE is supported by the Marion Sulzberger Research Fellowship from the Ronald O. Perelman Department of Dermatology at New York University School of Medicine. We appreciate helpful discussions with members of the Christiano laboratory. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.