
Abstract
Diabetic nephropathy is a serious complication of diabetes mellitus with a pressing need for effective metabolic markers to detect renal impairment. Of potential significance are the inositol compounds, myo-inositol (MI), and the less abundant stereoisomer, D-chiro-inositol (DCI), which are excreted at increased levels in the urine in diabetes mellitus, a phenomenon known as inosituria. There is also a selective urinary excretion of DCI compared to MI. As the biological origins of altered inositol metabolism in diabetes mellitus are unknown, the aim of this study was to determine whether the diabetic kidney was directly responsible. Kidneys isolated from four-week streptozotocin-induced diabetic rats were characterized by a 3-fold reduction in glomerular filtration rate (GFR) compared to matched non-diabetic kidneys. When perfused with fixed quantities of MI (50 µM) and DCI (5 µM) under normoglycemic conditions (5 mM glucose), GFR-normalized urinary excretion of MI was increased by 1.7-fold in diabetic vs. non-diabetic kidneys. By comparison, GFR-normalized urinary excretion of DCI was increased by 4-fold. Perfusion conditions replicating hyperglycemia (20 mM glucose) potentiated DCI but not MI urinary excretion in both non-diabetic and diabetic kidneys. Overall, there was a 2.4-fold increase in DCI urinary excretion compared to MI in diabetic kidneys that was independent of glucose ambience. This increased urinary excretion of DCI and MI in diabetic kidneys occurred despite increased renal expression of the inositol transporters, sodium myo-inositol transporter subtype 1 and 2 (SMIT1 and SMIT2). These findings show that the diabetic kidney primarily mediates inosituria and altered urinary partitioning of MI and DCI. Urinary inositol levels might therefore serve as an indicator of impaired renal function in diabetes mellitus with wider implications for monitoring chronic kidney disease.
Introduction
Diabetic nephropathy is a serious complication arising from diabetes mellitus which is a serious metabolic disease characterized by renal impairment. 1 Although end-stage renal failure occurs relatively infrequently in the diabetic population, its predecessor, renal impairment, is more common with diabetic nephropathy representing the largest proportion of chronic kidney disease. 2 The key to preventing end-stage renal failure in diabetic patients is early diagnosis so that medical treatment can be instituted. Urinary albumin measurement is commonly utilized to identify risk for kidney disease. However, its utility as a prognostic biomarker for diabetic nephropathy is increasingly debated because early-stage kidney disease can develop independently of detectable changes in albumin excretion.2,3 These observations together with the rising prevalence of diabetes and diabetic kidney disease underpin a pressing current need for new potential biomarkers to detect early-stage diabetic kidney disease and monitor development of ensuing later stage nephropathy.
Of potential interest in this regard are the inositol polyol compounds, myo-inositol (MI) and D-chiro-inositol (DCI). MI plays various physiological roles in membrane structure, signal transduction and as an organic osmolyte. 4 Circulating MI blood concentrations in humans are normally maintained at low micromolar levels (30–50 µM), and typically much lower than the intracellular concentrations of MI in the millimolar range. 5 By comparison, DCI is less abundant with circulating concentrations approximately 1–2% of MI in humans and displays insulin-like actions.5,6
Normally, both MI and DCI are freely filtered through the glomeruli of the Bowmans capsule and almost completely reabsorbed through the proximal tubules and short loop of Henle. However, in both type-1 and type-2 diabetes mellitus their urinary excretion are markedly elevated, a phenomenon known as inosituria.5,7,8 This abnormal inositol excretion is also associated with a selective 4- to 7-fold increase in DCI urinary excretion compared to MI, which is inversely correlated with measures of insulin sensitivity.5,7 The multiplied product of DCI and MI urinary concentrations has even been suggested to have potential utility as a predictive biomarker for type-2 diabetes mellitus in humans. 8
The biological mechanisms leading to inosituria in diabetes are unknown and the possibility that kidney function may play a direct role has not been previously explored. Potentially, inosituria could arise through several mechanisms including: (1) increased uptake of dietary-derived inositol leading to increased renal clearance; 9 (2) competition of inositol reabsorption in the proximal tubules by high glucose ambience; and (3) reduced tubular re-absorption of inositol as a direct consequence of impaired kidney function. The aim of the present study was to distinguish these possibilities. Accordingly, we compared inositol-handling capacities in kidneys isolated from four-week streptozotocin (STZ)-induced diabetic and non-diabetic rats under conditions of normal and high glucose ambience.
Methods
All experiments were approved by the University of Auckland Animal Ethics Committee. Male Wistar rats at weaning received standard normal chow (Teklad TB 2018; Harlan, Madison, WI). Male rats aged between 6 and 8 weeks and between 200 and 250 g in weight were kept under a 12-h light/dark cycle (50–70% humidity, 22 ± 2℃) with ad libitum access to food and water. Body weight and blood glucose were measured weekly. Induction of diabetes was achieved by a single dose of intravenous injection of STZ (55 mg/kg of body weight) or saline as Sham controls. Blood glucose level was monitored daily for the first three days. Rats with two consecutive readings of blood glucose >11 mM were categorized as diabetic. Animals were kept for four-week postinjection then randomly assigned into four treatment groups: (1) normal rats with normal glucose perfusate; (2) normal rats with high glucose perfusate; (3) STZ-rats with normal glucose perfusate; and (4) STZ-rats with high glucose perfusate. All surgical procedures and measurements were performed between 07:00 and 09:00 am to avoid an underlying circadian variation and only kidneys removed with a surgical removal duration of less than 30 min were used in the study.
Isolated perfused kidney
Rats were anaesthetized with 4% isoflurane in 2 L/min oxygen and maintained on 2.5% isoflurane in 2 L/min oxygen The surgical technique was performed as described. 10 Following removal of the kidney, a 40-min stabilization period was included after which samples were collected for the next 20 min. Aliquots of both perfusate and urine were analyzed for sodium (blood-gas analyzer, Bayer), glucose (YSI 2300 STAT plus Glucose and lactose Analyzer, YSI Inc., USA), and creatinine (creatinine assay kit, Cayman, Ann Arbor, MI).
Isolated perfused kidneys from four-week diabetic and matched normoglycemic saline injected control rats were used to investigate MI and DCI urinary excretion. Perfused kidney viability was assessed using several criteria including glomerular filtration rate (GFR), glucose re-absorption, sodium ion re-absorption, urinary flow rate, and perfusion pressure.
10
The perfusion system comprised a single pass pump-driven forced perfusion (20–40 mL/min) where diabetic and non-diabetic kidneys were perfused with Krebs-Henseleit Buffer (118.5 mM NaCl, 1.18 mM MgSO4, 1.18 mM KH2PO4, 24.8 mM NaHCO3, 4.75 mM KCl, 2.54 mM CaCl2), bovine serum albumin (4% w/v), dextran (1.67% w/v), 20 amino acids (13 mM), creatinine (200 mg/L), MI (50 µM), DCI (5 µM), and glucose (either 5 mM or 20 mM). GFR was calculated from the clearance of creatinine using the following equation: GFR = (UCr × UFR)/PCr where UCr is the concentration of creatinine in the urine (mg/mL), UFR is the urinary flow rate (mL/min), and PCr is the concentration of creatinine in the perfusate (mg/mL). The concentration of creatinine was determined using a creatinine assay kit (Cayman). The glomerular filtration fraction was calculated using the following formula:
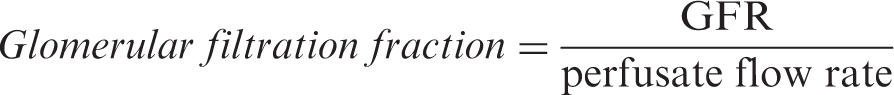
Inositol quantification
MI and DCI were quantitated using scyllo-inositol (Sigma Aldrich) as an internal standard. Buffer samples were lyophilized and following re-suspension in water (100 µL), the solutions were passed through anion exchange resin columns (Sigma IR120) followed by cation exchange resin (Sigma IRA400) to remove salt and glucose. Samples were then lyophilized overnight.
Dried samples were derivatized by incubation with 80 µL of methoxyamine hydrochloride in pyridine (2 g/100 mL, Sigma Chemical Co.) at 30℃ for 30 min and then incubated with 80 µL of N-methyl-N-(trimethylsilyl) trifluoroacetamide (Sigma Chemical Co.) at 37℃ for 90 min. Derivatized samples (1 µL) were injected on a ZB1701 column (Zebron, 30 m × 250 µm i.d. × 0.15 µm Wlm thickness, Phenomenex, Torrance, CA, USA) connected to a Shimadzu GCMS-QP2010 GC/MS system equipped with a quadrupole mass selective detector with an electron impact (EI) mode operating at 70 eV. The MS was operated in scan mode (start after 4.5 min, mass range 40–650 amu at 0.15 s/scan). The oven temperature was initially held at 70℃ for 5 min. The temperature was raised with a gradient of 10℃/min to 179℃. The gradient was then increased to 180℃ at 0.5℃/min followed by a 2-min hold. The temperature was raised with a gradient of 10℃/min to 220℃. The temperature was held for 1 min and raised again to 265℃ with a gradient of 2.5℃/min followed by a 1-min hold. The temperature was then raised to 280℃ with a gradient of 10℃/min and held for 1 min, and again raised to 290℃ with a slow gradient of 1℃/min. The oven temperature was raised to 300℃ with a gradient of 10℃/min. The flow through the column was held constant at 1 mL He/min. The injection volume was 1 µL, and the split ratio was 1:25. The temperature of the inlet was 230℃, the interface temperature was 250℃, and the quadrupole temperature was 200℃. The GC column was equilibrated for 6 min prior to the injection of next sample.
Quantitative real time polymerase chain reaction
Quantitative polymerase chain reaction (qPCR) was used to measure the renal expression of sodium myo-inositol transporter subtype 1 and 2 (SMIT1 and SMIT2). Kidney total RNA was extracted with TriPure Reagent (Roche). Total RNA was DNase-digested with DNA-free (Ambion) prior to cDNA construction with transcriptor first strand cDNA synthesis kit (Roche) using random hexomer primers. cDNA samples were diluted 5-fold and 1 µL of cDNA was mixed with 1 µL of both forward and reverse primers (1.25 µM), 5 µL of SYBR Green Mastermix (Roche), and 2 µL of PCR-grade water in a 384 multiwell plate. qPCR was performed using a LightCycler 480 (Roche). Amplification of SMIT1 and SMIT2 was performed using the following primers: SMIT1: 5′-ATGTGTCTGTCATCCTGCTC-3′ and 5′-TTCCTCAAACCCTCCAA CCT-3′, SMIT2: 5′ GCTGCCTATGAGCTTAATGGCTTG-3′, and 5′-CCAGACCTCCTGC AACAGTG-3′. Gene expression was normalized to the geometric mean of a combination of reference genes selected namely β-actin (ACTB, with primers 5′-GGGAAATCGTGCGTGACATT-3′ and 5′-GCGGCAGTGGCCATCTC-3′), ubiquitin C (UBC, with primers 5′-GGC AAAGATCCAGGACAAGGAG-3′ and 5′-GCCTGCAAAGATCAGCCTCT-3′) and hypoxanthine-guanine phosphoribosyltransferase (HPRT, with primers 5′-GCGAAAGTGGA AAAGCCAAGT-3′ and 5′-GCCACATCAACAGGACTCTTGTAG-3′).
Western blotting
To measure the protein expression of SMIT1 and SMIT2, whole kidney samples were homogenized in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 0.1% (w/v) SDS, 1% (v/v) Triton X100, 0.25% (w/v) deoxycholate) at 20 mg of wet weight/mL by TissueLyzer II (Qiagen). For western blotting, membranes were incubated with goat anti-SLC5a3 (SMIT1) IgG (Santa Cruz Biotechnology Inc., Santa Cruz, USA), rabbit anti-SLC5a11 (SMIT2) IgG (Aviva System, San Diego, USA) or mouse anti-β-actin IgG (Santa Cruz Biotechnology Inc., Santa Cruz, USA) followed by incubation with QDot 655 rabbit anti-goat IgG, QDot 655 goat anti-rabbit IgG or QDot 655 goat anti-mouse IgG (Invitrogen, Carlsbad, USA), respectively. Protein band intensities were visualized using a Fujifilm detection system and normalized to ACTB.
Statistical analyses
Statistical analyses were performed by unpaired two-tailed student’s t-test or two-way ANOVA followed with post hoc Tukey-Honestly significant difference or Student’s t-test. All data were expressed as mean ± SEM and statistical analyses were performed using GraphPad Prism version 5.02 (GraphPad Software, San Diego, CA) and JMP software (SAS Institute Inc., USA). P < 0.05 was considered statistically significant.
Results
Diabetic kidneys are characterized by impaired function
STZ is selectively toxic to pancreatic islet β-cells and is used routinely to generate experimental insulin-dependent diabetes mellitus by induction of pancreatic β-cell death.
11
Rats that received a single injected dose of STZ showed a rapid increase in blood glucose (>15 mM) on the first day postinjection and these levels were maintained throughout the entire experiment (Figure 1(a)). A STZ dosage at 55 mg/kg body weight generated a moderate diabetic phenotype where blood glucose ranged typically between 18 and 30 mM whereas blood glucose of the control group remained between 4 and 7 mM. Following four weeks of STZ-induced diabetes and prior to kidney removal, mean plasma glucose levels were elevated by approximately 5-fold (27.8 ± 0.9 vs. 5.2 ± 0.1 mM, P < 0.001) whilst mean body weight was 19% less compared to age-matched non-diabetic controls (353 ± 11 g vs. 435 ± 7 g, P < 0.001).
Diabetes progression and characteristics of isolated four-week diabetic vs. non-diabetic kidneys. (a) Blood glucose concentrations of non-diabetic (○) STZ diabetic rats (•) (n = 14 animals/group); (b) urinary flow rates; (c) venous flow rates; (d) GFR calculated using creatinine; (e) glomerular filtration fraction. Kidneys were perfused with perfusate containing either 5 mM (white bars) or 20 mM (black bars) glucose. Data represent mean ± SEM. n = 6 kidneys/group. Statistical analyses were performed using two-way ANOVA. **P < 0.01, ***P < 0.001
Arterial pressure and perfusion flow rates in non-diabetic and diabetic kidneys perfused with either 5 mM or 20 mM glucose. Data represent mean ± SEM (n = 6 kidneys/group)

For the measurement of GFR, we initially considered either inulin or creatinine as potential markers. 12 Inulin is increasingly preferred as some limitations with creatinine have been reported. 12 Nevertheless, we utilized creatinine in this instance as we found that high glucose concentrations (20 mM) interfered with inulin quantitation whereas creatinine measurement was unaffected. 10 The observed GFR values for non-diabetic kidneys utilizing creatinine as a clearance marker were similar to those reported by other studies using the isolated perfused kidney.13,14 By comparison, GFR was decreased in diabetic kidneys by approximately 3-fold (P < 0.001) showing an intrinsic impairment in renal function (Figure 1(d)). This reduction in GFR in four-week diabetic kidneys occurred irrespective of whether the perfusion was carried out with 5 mM or 20 mM glucose and was responsible for the corresponding reduction in glomerular filtration fraction from approximately 2.5% to 0.8% (Figure 1(e)).
Isolated diabetic kidneys replicate inosituria and increased DCI/MI urinary excretion
To distinguish renal-specific effects from possible circulating metabolic factors with regard to inositol handling, kidneys were perfused with fixed concentrations of MI (50 µM) and DCI (5 µM). These inositol concentrations are similar to physiological circulating levels reported in non-diabetic rat plasma.15,16 For the experiments where diabetic kidneys were perfused with 5 mM glucose, we first ensured that any potential changes in inositol excretion were not due to any residual glucose present in the STZ-diabetic kidney. A 40-min pre-perfusion was therefore undertaken and as indicated by the arrow in Figure 2(a) there was a dramatic reduction in urinary glucose from diabetic kidneys over the 40 min time course. For all experiments, MI and DCI analyses were taken from total pooled urinary samples collected between 40 and 60 min where glucose levels were similar in both diabetic and non-diabetic kidneys (shaded bar).
Renal handling of MI and DCI in isolated kidneys perfused with 5 mM (white bars) or 20 mM (black bars) glucose. (a) Urinary excretion of glucose from non-diabetic (○) and diabetic (•) kidneys (n = 3 separate kidneys for each group). Inositol analyses were taken from pooled samples collected between 40 and 60 min. (indicated by the bar); (b) MI venous concentrations; (c) DCI venous concentrations; (d) urinary excretion of MI normalized to GFR; (e) urinary excretion of DCI normalized to GFR; (f) DCI/MI urinary excretion ratio. Data represent mean ± SEM. n = 6 kidneys/group. Statistical analyses were performed by two-way ANOVA. *P < 0.05, ***P < 0.001
Following perfusion with either 5 mM or 20 mM glucose, venous concentrations of MI (Figure 2(b)) and DCI (Figure 2(c)) were not altered significantly from the arterial concentrations of 50 µM and 5 µM for either non-diabetic or diabetic kidneys. These levels were not surprising given the observed glomerular filtration fraction (Figure 1(e)) indicating that greater than 98% of the perfusion buffer was not passed through the glomeruli. However, despite fixed concentrations of MI and DCI in the perfusion buffers, analyses of the pooled urinary fractions showed that GFR-normalized MI urinary excretion was increased by 1.4-fold (5 mM glucose) and 2-fold (20 mM glucose) in diabetic kidneys vs. non-diabetic kidneys (Figure 2(d), P < 0.001). By comparison, DCI urinary excretion was increased approximately 4-fold in diabetic kidneys vs. non-diabetic kidneys (P < 0.001) (Figure 2(e)).
When expressed as a ratio these perfusion studies showed a selective 2- to 3-fold increase in DCI urinary excretion compared to MI (P < 0.001) (Figure 2(f)). Both MI and DCI concentrations were fixed in the perfusion buffers and quantitated simultaneously by GC/MS from the pooled urine samples using scyllo-inositol as an internal standard. This selective partitioning of DCI and MI in the urine therefore occurs independently of GFR reflecting a fundamental alteration in inositol handling in the diabetic kidney.
Hyperglycemia potentiates DCI but not MI urinary excretion
We surmised that hyperglycemia might contribute to inosituria as glucose can intrinsically compete for MI uptake through the proximal and late proximal tubule compartments.17,18 However, at glucose levels characteristic of the diabetic state (20 mM) there was no significant effect of glucose ambience on MI urinary excretion in either diabetic or non-diabetic kidneys (Figure 2(d)). By comparison, there was an additional effect of high glucose ambience on DCI urinary excretion whereby 20 mM glucose elicited a 1.4-fold increase compared to 5 mM glucose for both non-diabetic and diabetic kidneys (P < 0.05) (Figure 2(e)). These findings show that hyperglycemia has a minimal overall effect on inosituria as MI is the predominant circulating stereoisomer in vivo but it can potentiate urinary excretion of the less abundant DCI.
Inosituria occurs in diabetes despite increased SMIT expression
We next investigated whether decreased renal reabsorption of MI and DCI was associated with altered expression of the renal inositol transporters, SMIT1 and SMIT2, which facilitate inositol transport by utilizing a Na+ concentration gradient. 19 SMIT1 is expressed predominantly in the medulla and selectively transports MI, 20 while SMIT2 transports MI and DCI with similar affinities. 21 A third inositol transporter, H+-dependent inositol transporter, does not appear to play a significant role in the inositol transport in the kidney and was therefore not examined in these experiments. 17
Interestingly, despite a reduction in inositol reabsorption in the kidney, transcription of both SMIT1 (Figure 3(a)) and SMIT2 (Figure 3(b)) were elevated in four-week diabetic kidneys vs. non-diabetic controls by 92% (P < 0.05) and 48% (P < 0.05), respectively. For SMIT1, quantitative western blot analyses showed a 67% increase in protein expression (P < 0.05). This increased expression is consistent with the regulation of SMIT1 by hyperosmotic conditions via a tonicity-responsive enhancer binding protein (TonEBP).
19
These findings show that reduced renal reabsorption of MI and DCI in diabetes mellitus occurs despite increased SMIT1 and possibly SMIT2 expression.
Renal expression of SMIT1 and SMIT2 in diabetes mellitus. (a) SMIT1 transcription using qPCR; (b) SMIT1 protein expression quantified by Western blotting and representative blot with ACTB as a loading control; (c) SMIT2 transcription using qPCR; (d) SMIT2 protein expression quantified by Western blotting and representative blot with ACTB as a loading control. n = 7 kidneys/group. Data represent mean ± SEM. Statistical analyses were performed using Student’s unpaired t-test. *P < 0.05
Discussion
The occurrence of inosituria in diabetes mellitus is characterized by increased urinary excretion of inositols as well as a selective increase in the renal clearance of DCI compared to MI.5,7 Here, we considered three possible contributing mechanisms: (1) increased dietary uptake and circulating levels of MI and DCI; (2) inhibition of inositol re-absorption in the proximal tubules by high glucose ambience; and (3) impaired inositol handling due to impaired renal function.
To control for the first possibility we perfused diabetic rat kidneys with fixed concentrations of MI and DCI. Under these experimental conditions, the essential features of inosituria and urinary partitioning of DCI vs. MI were reproduced in diabetic kidneys. The potential effect of high glucose ambience was investigated by perfusion of non-diabetic and diabetic kidneys with glucose concentrations mimicking hyperglycemia. From these experiments there was a selective effect by high glucose resulting in increased DCI urinary excretion compared to MI. However, this hyperglycemic effect was secondary to the overall observed increase in inositol urinary excretion in diabetic kidneys. Overall, these findings support the third mechanism that inosituria in the STZ-rat model arises primarily through impaired renal function but is also potentiated by the hyperglycemic state.
Our study design was confined to a single point analysis in the diabetic time course at a stage where kidney function had markedly declined as indicated by a reduction in GFR. The early stages of diabetic nephropathy are characterized by an increase in proximal tubule size resulting in increased reabsorption and glomerular hyperfiltration. 22 This initial process leads in later stages to tubular senescence and hypertrophy, interstitial fibrosis, and ultimately renal impairment. 22 In STZ-induced diabetes in rats, the change from tubular growth to senescence occurs following 10 days of diabetes 23 leading to reductions in GFR by 14 days. 24 Although we did not monitor the progression of renal function decline prior to four weeks of diabetes, the presence of glomerular damage was clearly evident from the observed 3-fold decrease in GFR in four-week diabetic vs. non-diabetic kidneys.
Interestingly, decreased inositol reabsorption occurred despite increased expression of SMIT1 and SMIT2. Of these, SMIT2 displays similar affinities for MI and DCI transport and is primarily responsible for inositol reabsorption in the proximal tubule.17,20 Increased expression of SMIT2 in four-week diabetic kidneys therefore points to other pathophysiological processes underlying impaired transport of MI and DCI in the proximal tubules such as potential post-translational modifications leading to changes in transporter function.
The selective increase in DCI urinary excretion can be potentially explained by preferential MI reabsorption in the short loop of Henle. This compartment possesses a high capacity for MI reabsorption and can transport >90% of the physiological MI glomerular load. 17 Essentially, no MI absorption occurs beyond the short loop of Henle. 12 In contrast to inositol uptake studies in brush border membranes where SMIT2 is primarily expressed, 20 co-infusion of DCI into the late proximal segments of superficial nephrons only partially inhibits MI reabsorption in the loop of Henle. 17 These observations suggest that preferential reabsorption of MI compared to DCI occurs in the short loop thereby leading to the observed increase in DCI vs. MI excretion.
Although SMIT1 expression was increased in four-week diabetic kidneys, its role in the reabsorption of MI is currently uncertain. SMIT1 is expressed predominantly in the medulla and cortical thick ascending limbs of Henle’s loop and macula densa cells 25 and does not transport DCI. 20 However, unlike SMIT2 which is apically targeted, SMIT1 is localized to the basolateral membrane 26 suggesting it mediates cellular uptake of MI from the blood, presumably to protect inner medullary tubule cells against increased extracellular osmolarity due to urine concentration. 27 Whether SMIT1 is the transporter that mediates MI uptake in the loop of Henle or whether other transport mechanisms operate requires further elucidation.
Our findings also showed that high glucose ambience further potentiated DCI but not MI urinary excretion. This effect occurred independently of the diabetic phenotype as demonstrated by perfusion of non-diabetic kidneys with 20 mM glucose. Glucose competes for MI uptake via SMIT2 with an inhibition constant in the low millimolar range (ki = 6.1 mM)18,28 and glucose re-absorption also occurs primarily in this compartment mediated through SGLT proteins. 17 The increased urinary excretion of DCI by high glucose ambience is therefore consistent with competitive inhibition of inositol transport by glucose via SMIT2. The lack of a corresponding effect for MI is also consistent with preferential reabsorption of MI in the loop of Henle.
There are acknowledged limitations to the use of the STZ-induced rats as a model of diabetic nephropathy in humans. 29 Nevertheless, this model reproduces the essential features of inosituria observed in both human type-1 and type-2 diabetes mellitus 5 and strongly implicates impaired renal function as the predominant causative factor. Interestingly, increased DCI urinary clearance has been reported in human patients with polycystic ovary syndrome (PCOS). 30 This abnormal partitioning of DCI excretion persists in the absence of hyperglycemia, which is consistent with our finding that high glucose ambience had only a comparatively minor effect on DCI urinary clearance. Potentially, impaired renal function might also contribute to increased DCI urinary clearance in PCOS. This possibility is consistent with the association between increased DCI urinary clearance in PCOS and severity of the syndrome 31 pointing towards progressive impairment of renal function as a primary factor. Increased DCI urinary excretion could also explain the association between PCOS and depletion of DCI-containing mediators of insulin action. 32
In conclusion, our study shows that the diabetic kidney is directly responsible for inosituria and selective increase in DCI urinary excretion in diabetes mellitus. Currently, there are active efforts to identify metabolic markers other than albuminuria that reflect kidney function. 3 The present finding suggests that the well-established phenomenon of inosituria might find utility as an indicator of renal function in diabetes mellitus. This possibility could be pursued through longitudinal studies to investigate the association between MI and DCI partitioning in the urine and the progression of kidney disease in human diabetic populations.
Footnotes
Author contributions
H.C., A.P., K.L. designed research; A.P. reviewed manuscript; H.C. performed research; B.C. provided technical assistance. H.C. and K.L. wrote the paper.
Acknowledgements
We gratefully acknowledge grant support from the Maurice & Phyllis Paykel Trust and the University of Auckland Faculty Research Development Fund.