
Abstract
The inhibitors of apoptosis proteins are implicated in promoting cancer cells survival and resistance toward immune surveillance and chemotherapy. Second mitochondria-derived activator of caspases (SMAC) mimetics are novel compounds developed to mimic the inhibitory effect of the endogenous SMAC/DIABLO on these IAPs. Here, we examined the potential effects of the novel SMAC mimetic BV6 on different human cancer cell lines. Our results indicated that BV6 was able to induce cell death in different human cancer cell lines. Mechanistically, BV6 dose dependently induced degradation of IAPs, including cIAP1 and cIAP2. This was coincided with activating the non-canonical NF-kappa B (NF-κB) pathway, as indicated by stabilizing NF-κB-inducing kinase (NIK) for p100 processing to p52. More interestingly, BV6 was able to sensitize some of the resistant cancer cell lines to apoptosis induced by the death ligands tumor necrosis factor-α (TNF-α) and TNF-related apoptosis-inducing ligand (TRAIL) that are produced by different cells of the immune system. Such cell death enhancement was mediated by inducing an additional cleavage of caspase-9 to augment that of caspase-8 induced by death ligands. This eventually led to more processing of the executioner caspase-3 and poly (ADP-ribose) polymerase (PARP). In conclusion, therapeutic targeting of IAPs by BV6 might be an effective approach to enhance cancer regression induced by immune system. Our data also open up the future possibility of using BV6 in combination with other antitumor therapies to overcome cancer drug resistance.
Introduction
Most tumor cells inactivate apoptosis (the programmed cell death) to achieve a malignant state and overcome natural killer and T-cells-mediated tumor surveillance orchestrated by the host immune defense. 1 Tumor cells suppress apoptosis via increasing the anti-apoptotic proteins and/or decreasing the pro-apoptotic proteins. Such strategies hinder the processing of the extrinsic and intrinsic pathways of apoptosis. 2 The former pathway requires binding of death ligands like tumor necrosis factor-α (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL) and FAS ligand to their corresponding cell surface death receptors, leading to the activation of the initiator pro-caspase-8.2,3 The latter pathway, however, initiates when the mitochondria releases cytochrome c and SMAC (second mitochondria-derived activator of caspases)/DIABLO (direct inhibitor of apoptosis-binding protein with low isoelectric point) into the cytosol. Apoptosis-inducing factor-1 (Apaf-1) and pro-caspase-9 subsequently bind to cytochrome c, resulting in the assembly of apoptosome and activation of the initiator pro-caspase-9.4–6
There is a class of proteins known as inhibitors of apoptosis proteins (IAPs) that inhibit apoptosis via their baculoviral IAP repeat (BIR) domains.7,8 These IAPs include cellular inhibitor of apoptosis protein (cIAP1 and cIAP2), X-linked inhibitors of apoptosis (XIAP) and survivin that exert anti-apoptotic effect because of their ability to ubiquitinate caspase proteins.9,10 cIAP1 and cIAP2 behave as E3 ubiquitin ligase and mediate either hetero- or auto-ubiquitination due to their gene (RING) domain.9,10 Moreover, cIAP1 and cIAP2 regulate TNF-α-induced NF-κ B (NF-κB) signaling that modulates their anti-apoptotic activity.9,11,12 It is recently proved that the level of IAPs is highly elevated in several tumor types and is associated with poor outcome.13,14
SMAC/DIABLO is considered an endogenous inhibitor of IAPs that enhances the activation of caspases and cell execution.6,15 In the last decade, several research efforts were exerted to develop new small molecules mimicking SMAC activity for the use in cancer therapy.16–20 Interestingly, recent studies have shown the ability of SMAC mimetics to induce degradation of cIAPs, leading to activation of the noncanonical NF-κB pathway and TNF-dependant apoptosis.21,22 Moreover, there are several ongoing clinical studies to prove the antitumor efficacy of SMAC mimetics either alone or in combination with chemotherapy in different tumor types.23,24
One of the recently developed SMAC mimetics is BV6, which binds to BIR domains of IAPs leading to their ubiquitination, followed by proteosomal degradation and inhibition of XIAP-mediated inhibition of caspases.25,26 In the present study, we sought to explore the cytotoxic effects of BV6 in numerous human cancer cells. Besides, we studied the sensitizing potential of BV6 on TNF-α and TRAIL-induced apoptosis as a novel strategy for combinational antitumor therapy.
Materials and methods
Chemicals
Killer TRAIL was purchased from Enzo Life Sciences, Germany. Cycloheximide (CHX) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from SERVA Electrophoresis GmbH, Germany. All cell culture media and supplements were purchased from PAA Laboratories, Germany. BV6 and TNF-α (a recombinant human TNF-α) were kind gifts from Prof. Harald Wajant, Wuerzburg University, Germany. The used in vitro concentration of BV6 (10 µM) was based on our preliminary screening experiments with incremental concentrations and previous reports.26,27
Cell death assay
The cell lines used in the current study are human cells from different origins. KMS12.BM, L363, MMI.s, OPM2, RPMI, Amo-1 and KMS.11cells are multiple myeloma cell lines. HT29 is a human colorectal adenocarcinoma cell line. HT1080 is a fibrosarcoma cell line. HeLa is a cervical carcinoma cell line. Jurkat is a T cell lymphoma cell line. All these cell lines were grown in RPMI medium with 10% FCS. Adherent cell lines (HT1080, HT29 and HeLa) were counted 20 × 103 cells per well and seeded in 96-well plates and stimulated on the next day. The other suspension or semi-adherent cell lines were counted 60–80 × 103 cells per well, seeded in 96-well plates and stimulated on the same day. BV6 or CHX was added to the cells 30 min before stimulating the cells with TNF-α or killer TRAIL. PBS:methanol (1:1) was used as a control for BV6 vehicle. After 24 h or 48 h, cell viability was assessed using MTT assay.
Gel electrophoresis and Western blotting
The different cell lines were harvested in ice-cold PBS and then washed twice in PBS. Afterwards, cells were centrifuged at 2300 r/min (4℃) for 4 min to prepare cell pellets. Cell pellets were lysed in 1 × Laemmli buffer (pH 8.0) containing 8% sodium dodecyl sulfate (SDS), 0.2 M Tris, 10% β-mercaptoethanol, 40% glycerol, phosphatase inhibitor mixture II (Sigma) and protease inhibitor (Roche Diagnostics). Then, the lysates were sonicated for 20 s to obtain total cell lysates. Total cell lysate proteins were fractionated using vertical SDS-polyacrylamide gel electrophoresis according to their molecular weight. The fractionated proteins were subsequently transferred to a nitrocellulose membrane by semi-dry blotting. The blotted membrane was then incubated overnight (4℃) with primary antibodies specific for p100 (05-361, Merck Millipore, 1: 2000), NIK (4994, Cell Signaling Technology, 1:2000), cIAP1 (4952, Cell Signaling Technology, 1:2000), cIAP2 (3130, Cell Signaling Technology, 1:2000), caspase-3 (9662, Cell Signaling Technology, 1:2000), caspase-9 (9502, Cell Signaling Technology, 1:2000), caspase-8 (clone C15, Enzo Life Sciences, 1:2000), PARP (clone 7D3-6, BD Biosciences, 1:2000) and tubulin (Dunn Labortechnik, 1:10,000). Anti-mouse-HRP (Dako-Cytomation, 1: 10,000) and anti-rabbit-HRP (Cell Signaling Technology, 1:5000) were used to detect antigen-primary antibody complexes that were further visualized using ECL Western Blotting detection system (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis in the current study was performed by the GraphPad Prism 5.0 program (GraphPad Software, Inc., CA, USA). Student’s t-test was used to determine the extent of significance (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001).
Results
BV6 induces IAPs degradation and NIK accumulation
Different human cell lines were challenged with the indicated concentrations of BV6. Interestingly, BV6 induced significant degradation of cIAP1 and/or cIAP2 that may be attributed to proteosomal degradation (Figure 1). Next, p100 processing to p52 and NIK accumulation were assessed to test whether BV6 can activate the non-canonical NF-κB pathway. Notably, BV6 was able to induce significant NIK accumulation in all indicated cell lines and moderately stimulated p100 processing in KMS12.BM, HT29 and HT1080 cells only (Figure 1).
BV6 induces IAPs degradation and NIK accumulation. The indicated cell lines were challenged with the indicated concentrations of BV6. After 8 h, total cell lysates were analysed by Western blotting for the processing of the indicated proteins
BV6 induces cell death in different human cell lines
Different human cell lines were then challenged with BV6 (10 µM) for 24 h and 48 h. After 24 h, BV6 was able to induce significant cell death in L363, MMI.s, HT1080 and Jurkat cells (Figure 2). In addition, BV6 induced significant cell death in all the indicated cell lines after 48 h, except for Amo-1, KMS.11, and HeLa cells (Figure 2).
BV6 induces cell death in different human cell lines. The indicated cell lines were treated in triplicates with either BV6 (10 µM) or BV6 vehicle (PBS:methanol, 1:1). After 24 h or 48 h, cell viability was determined using MTT assay
BV6 sensitizes different cell lines to TNF-α- and TRAIL-induced cell death
TNF superfamily death ligands can trigger apoptosis in susceptible cancer cells by activating their corresponding death receptors. TNF-α can induce apoptosis in susceptible cancer cells via stimulating the death receptor TNFR-1.
28
TRAIL is also a death ligand of TNF superfamily that is produced by different cells of the immune system and can induce apoptosis via binding to its corresponding death receptors, TRAILR1 and TRAILR2.
29
Different cell lines were then challenged with TNF-α (100 ng/mL) or killer TRAIL (20 ng/mL), a commercially available oligomerized highly active form of TRAIL, in the presence of BV6 (10 µM). Interestingly, BV6 enhanced TNF-α-induced cell death in MMI.s, HT1080, HeLa and Jurkat cells (Figure 3). Likewise, TRAIL-induced cell death was enhanced by BV6 in L363, MMI.s, OPM2, RPMI, HT29, HT1080, HeLa, and Jurkat cells (Figure 3). On the other hand, BV6 was unable to enhance TNF-α- or TRAIL-induced cell death in KMS12.BM, Amo-1 or KMS.11 cells (Figure 3).
BV6 enhances TNF- and TRAIL-induced cell death. The indicated cell lines were stimulated with BV6 (10 µM). After 30 min, the cells were stimulated with TNF (100 ng/mL) or killer TRAIL (20 ng/mL). After 24 h, cell viability was determined using MTT assay
BV6 enhances TNF-α- and TRAIL-induced apoptosis
As demonstrated by our results, BV6 was able to enhance both TNF-α- and TRAIL-induced cell death (Figure 3). Hereby, we next investigated the effect of BV6 on TRAIL- and TNF-α-induced apoptosis and the processing of caspases and PARP in HT1080 and HT29 cells. As assessed by Western blotting analysis, BV6 was able to sensitize HT1080 cells toward TNF-α and TRAIL-induced apoptosis (Figure 4(a)), while this effect was limited in HT29 cells to TRAIL-induced apoptosis only (Figure 4(b)). Thus, BV6 when combined with TNF-α and/or TRAIL was able to induce a significant processing of pro-caspases and the caspase-3 substrate PARP.
BV6 enhances TNF- and TRAIL-induced apoptosis. HT1080 and HT29 cells were treated with BV6 (10 µM). After 30 min, HT29 cells were treated with killer TRAIL (100 ng/mL) and HT1080 cells were treated with killer TRAIL (100 ng/mL) and TNF (100 ng/mL). After 6 h, total cell lysates were analyzed by Western blotting for the processing of the indicated proteins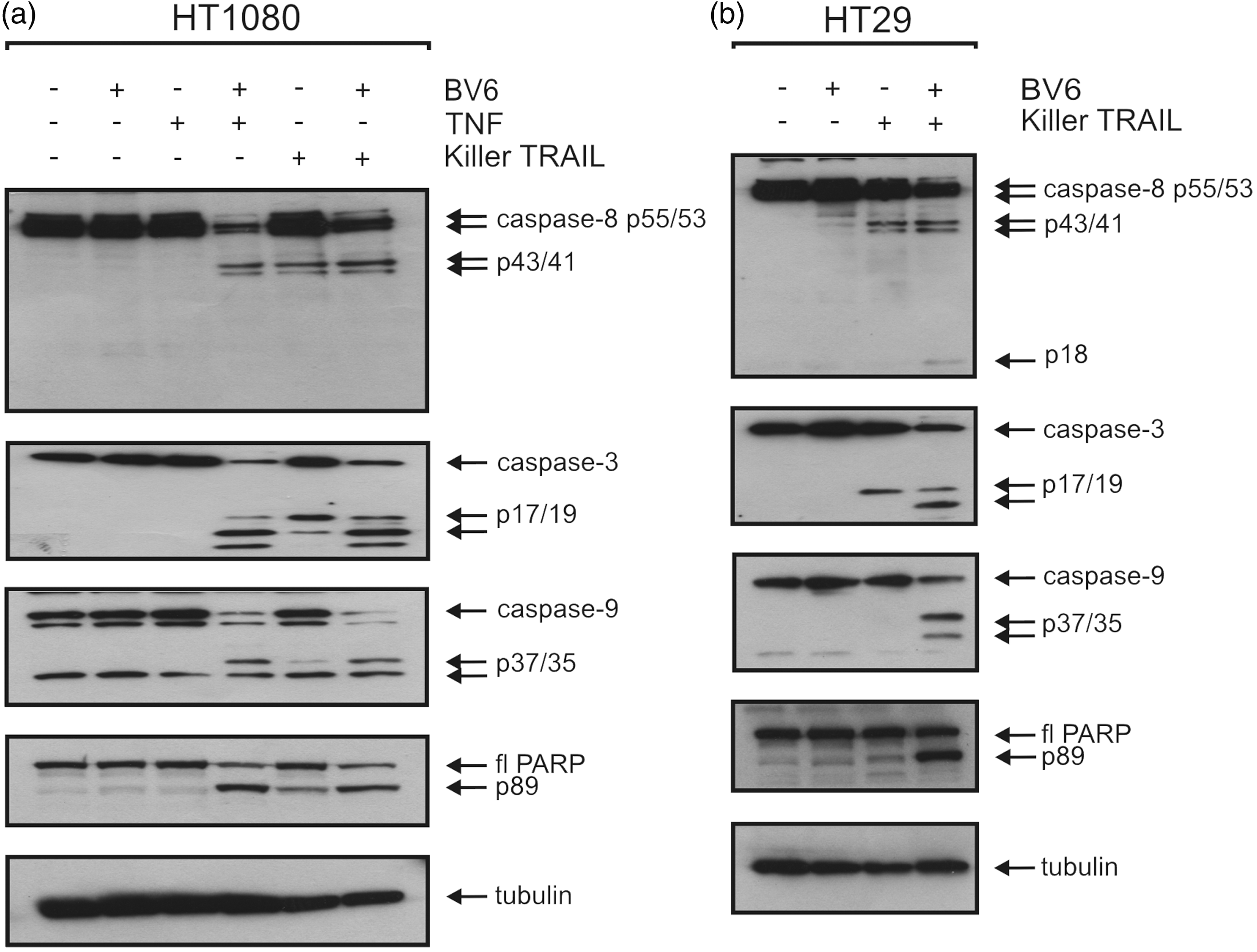
Comparison between BV6 and CHX sensitizing effects on TRAIL-induced cell death
Although TRAIL induces cell death in different types of cell lines, it may require a sensitizing agent like the protein synthesis inhibitor CHX to overcome TRAIL resistance exhibited by certain types of tumors.30–33 Therefore, we compared the sensitizing effect of BV6 on TRAIL-induced cell death to that of CHX in different human cell lines showing little or no cell death-induced by TRAIL alone (Figure 5). While HT1080 cell line was sensitized by BV6 and CHX to TRAIL-induced cell death almost to the same extent, HT29 cell line was sensitized by BV6 to TRAIL-induced cell death slightly less than CHX (Figure 5).
Comparison between BV6 and CHX sensitizing effects on TRAIL-induced cell death. HT1080 and HT29 cells were challenged in triplicates with the indicated concentrations of killer TRAIL in the presence and absence of BV6 (10 µM) or CHX (2.5 µg/mL). After 24 h, cell viability was determined using MTT assay and normalized according to cells treated with a a cocktail of cytotoxic substances and untreated cells or cells treated only with BV6 or CHX in case of pretreatment with BV6 or CHX, respectively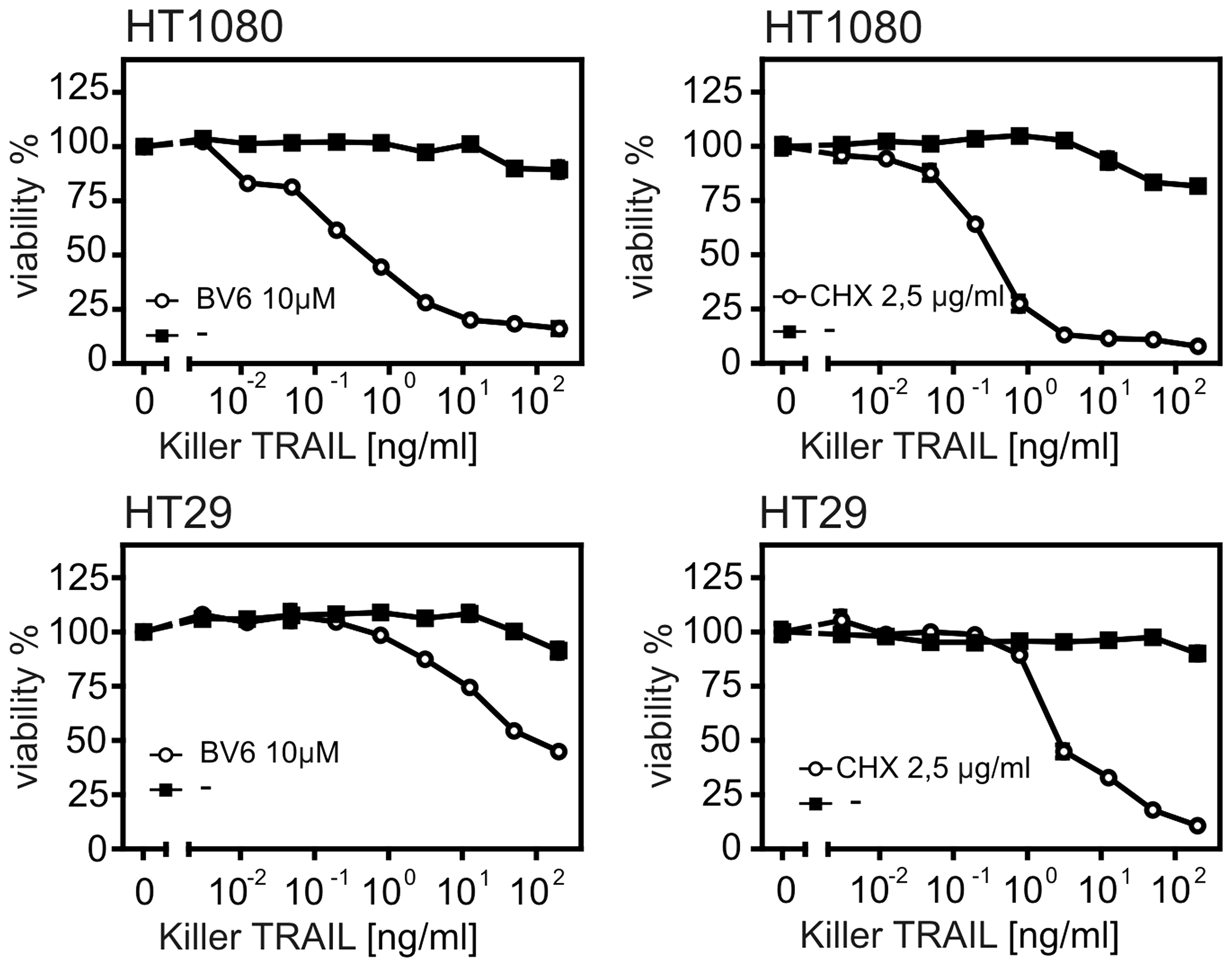
Discussion
Tumor cell resistance to apoptosis induced by different chemotherapeutic agents is a major hindrance for cancer cure. Overexpression of IAPs is one of the possible causes implicated in this feature. Increased IAPs and/or decreased IAPs antagonists expressions are highly associated with cancer progression, poor prognosis, and resistance for therapy.34–38
BV6 is a novel bivalent SMAC mimetic that has the capability antagonize IAPs and induce processing of caspases.26,39 In the current work, the ability of BV6 to induce cIAP1 and cIAP2 degradation was explored not only in multiple myeloma cells, but also in other cell lines of different cancer entities. Our results showed that BV6 was able to induce significant degradation of cIAP1 and cIAP2 ensued by activation of the non-canonical NF-κB pathway as indicated by NIK accumulation.
cIAPs and XIAP are able to bind caspases−3, −7 and −9 via BIR domains and can trigger their ubiquitination via RING domain.40–42 On the other hand, the IAPs antagonist SMAC/DIABLO works by preventing XIAP from binding to caspases and inducing auto-ubiquitination and proteasomal degradation of cIAPs via binding to their BIR domains.37,38 Furthermore, IAPs are considered key regulators not only for the canonical NF-κB pathway, but also for the non-canonical pathway. 43 The role of IAPs in the non-canonical NF-κB pathway is to promote NIK degradation via cIAP/TRAF complex.44,45 Hence, BV6-induced degradation of cIAPs led to NIK accumulation and processing of p100 to p52.
There is growing evidence that drug resistance in multiple myeloma is largely attributed to disruption of SMAC release. 46 Thus, it is not surprising to see considerable research efforts to characterize SMAC mimetics in multiple myeloma.47,48 Although the anti-apoptotic role of XIAP in multiple myeloma is well verified, the roles of cIAP1 and cIAP2 have not been clarified.49–51 As far as the ability of BV6 to induce significant cIAP1 and cIAP2 degradation was confirmed, we were motivated in this study to screen the ability of BV6 to induce cell death not only in multiple myeloma cell lines (KMS12.BM, L363, MMI.s, OPM2, RPMI, Amo-1 and KMS.11), but also in other different cells, such as colorectal adenocarcinoma (HT29), fibrosarcoma (HT1080), cervical carcinoma (HeLa) and T cell lymphoma (Jurkat). Interestingly, BV6 induced significant cell death in most of the cell lines screened in this study.
Induction of apoptosis by stimulating death receptors is considered one of the most attractive approaches for cancer therapy. In particular, the death ligand TRAIL has received more research interest than other members of TNF superfamily due to its ability to induce apoptosis in cancer cells with little or no cytotoxic effect on normal cells. 29 Nevertheless, certain types of cancer can develop resistance against TRAIL released by the host immune cells because of the accumulation of IAPs. 52 Multiple lines of evidence have recently reported the success of SMAC mimetics in cancer combination therapy.53–58 Accordingly, it was worth in this study to investigate the sensitizing effect of BV6 on TNF (recombinant human TNF-α) and killer TRAIL (a highly active form of TRAIL) in different cell lines.
Our results explicitly indicated that BV6 was able to sensitize different cell lines to TNF-α and/or TRAIL-induced cell death. Moreover, the ability of BV6 to enhance TNF-α and TRAIL-induced caspase activation and apoptosis was proved in this study. Additionally, the sensitizing effect of BV6 on TRAIL-induced cell death was comparable to that of the potent protein synthesis inhibitor CHX. Thus, incorporating BV6 in cancer combination therapy may be clinically applicable to improve the cure rate of cancer.
In conclusion, the use of the small-molecule SMAC mimetic BV6 represents a novel therapeutic approach for enhancing the anti-tumor immunity mediated by the endogenously released TNF-α and TRAIL upon immune system activation. Besides, BV6 may provide an alternative therapeutic intervention to overcome TRAIL-apoptosis resistance in different cancers. Overall, this study highlights the cytotoxicity and sensitizing effects of BV6 on diverse cancer cell lines that indeed will help clinically in deciding which type of cancer is optimal for BV6 combination therapy. Further studies are also required to elucidate the different responses of cancer cell lines to BV6 cytotoxicity and sensitizing effects.
Footnotes
Authors’ contributions
ME designed the experiments; ME, MS and AE performed the experiments, analyzed the data and wrote the manuscript.
Acknowledgements
This work was supported in part by financial support to Dr Mohamed El-Mesery (Application number 41590) via the Returning Experts Programme that is implemented by the Centre for International Migration and Development (CIM). The authors acknowledge Prof. Dr Harald Wajant, Division of Molecular Internal Medicine, University Hospital Wuerzburg, Germany for the generous gifts of materials.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.