
Abstract
Endothelial dysfunction occurs when there are imbalances between factors that regulate the synthesis and degradation of nitric oxide (NO•), and has been reported in patients with hyperglycemia and insulin resistance. We reported that supplementation with γ-tocopherol (γ-T) in humans limits impairments in endothelial function otherwise induced by postprandial hyperglycemia. Given the rapid metabolism of γ-T into γ-carboxyethyl hydroxychroman (γ-CEHC), we hypothesized that the vasoprotective activities of γ-T could be attributed to its metabolite γ-CEHC. To test this, human aortic endothelial cells (HAECs) treated with 0 (vehicle control) or 3 µM γ-CEHC for 24 h prior to incubation with normal (5 mM) or high (25 mM) glucose for 48 h. High-glucose increased levels of uncoupled endothelial nitric oxide synthase (eNOS) as evidenced by reduced (p < 0.05) eNOS dimer:monomer. High glucose also prevented insulin-stimulated increases in p-AktSer473: total Akt, p-eNOSSer1177: total eNOS, and NO• production. These adverse changes were accompanied by increased (p < 0.05) reactive oxygen species and mRNA expression of inflammatory mediators (VCAM-1, E-selectin, IL-8). However, each deleterious response evoked by high glucose was prevented when HAECs were incubated with γ-CEHC prior to the high glucose challenge. Taken together, our data support the hypothesis that vascular protection provided by γ-T in vivo may be elicited through the bioactivity of its metabolite, γ-CEHC. Furthermore, it is possible that the antioxidant and anti-inflammatory activities of γ-CEHC may mediate this protective activity.
Introduction
Endothelial dysfunction is a systemic pathological state resulting from imbalances between factors that regulate the synthesis and degradation of nitric oxide (NO•). 1 The endothelial isoform of nitric oxide synthase (eNOS) is a critical source of NO• to the vascular endothelium, a cellular protective molecule with anti-inflammatory properties. 2 Endothelial dysfunction is commonly associated with diminished NO• bioavailability, due to impaired NO• production by the endothelium and/or increased scavenging of NO• by reactive oxygen species (ROS). In addition, decreased NO• bioavailability is well described in patients with type 1 diabetes 3 and type 2 diabetes 4 and also in cardiovascular disorders such as hypertension and atherosclerosis associated with insulin resistance. 5
Many previous clinical studies have examined supplemental Vitamin E, in the form of α-tocopherol (α-T), as a tool to improve endothelial function and cardiovascular disease risk. However, evidence exists that γ-tocopherol (γ-T) is also cardioprotective and anti-inflammatory. For example, γ-T supplementation in humans was shown to limit impairments in endothelial function otherwise induced by postprandial hyperglycemia by preventing increases in both lipid peroxidation and the naturally occurring eNOS inhibitor, asymmetric dimethylarginine. 6 In smokers, γ-T supplementation was shown to improve endothelial function. 7 In rodents, γ-T elicits significant anti-inflammatory activities. 8 In human subjects with metabolic syndrome, nitrotyrosine levels in urine decreased following γ-T supplementation, indicating γ-T may be highly potent in quenching reactive nitrogen species, 9 consistent with its known reactive nitrogen species-scavenging activities that result in the formation of nitro-γ-tocopherol.10,11
The lipophilic molecule γ-T exerts direct antioxidant and anti-inflammatory activity, but it is readily metabolized by the liver in a cytochrome P450-dependent manner to its physiological water-soluble metabolite γ-carboxyethyl hydroxychroman (γ-CEHC). 12 Because γ−CEHC also exerts anti-inflammatory activities, 13 it is possible that the observed vasoprotective activities may be mediated through γ-T directly and/or through complementary activities of γ−CEHC. While studies support the beneficial effects of γ-T itself in rodents and humans, the effects of its physiologic metabolite γ-CEHC are not well established. It also remains unknown whether γ-CEHC can preserve eNOS signaling in endothelial cells during hyperglycemia. Therefore, the present study tested the hypothesis that γ-CEHC would prevent impairments of NO• bioavailability in human aortic endothelial cells (HAECs) subjected to hyperglycemia-like conditions.
Materials and methods
Cells and reagents
Reagents were obtained commercially from the following companies. HAECs, endothelial cell basal medium-2 (EBM-2), and endothelial growth supplements (EGM-2) were purchased from Lonza (Walkersville, MD). 2,7,8-trimethyl-2 -(beta-carboxyethyl)-6-hydroxychroman (γ-CEHC) was purchased from Encore Pharmaceuticals (Riverside, CA, USA).
HAECs culture and treatment
Cells were cultured in EBM-2 media containing 2% FBS and endothelial growth supplements in a humidified atmosphere (5% CO2/95% O2, 37℃) as previously described. 14 HAECs were passaged when they were 70–80% confluent and transferred into appropriately sized culture plates. Cells were pre-treated with 3 µM γ-CEHC dissolved in dimethyl sulfoxide (DMSO), or DMSO alone as a vehicle control for 24 h. Next, cells were treated with 25 mM glucose for 48 h. We have previously found ∼3 µM of γ-CEHC in human plasma after supplementation with a γ-tocopherol-rich vitamin E supplement. 6 Cells cultured in 5 mM glucose + 20 mM mannitol were used as osmotic controls. In selected experiments, cells were treated for 10 min with 100 nM insulin, or saline (vehicle control) to evoke eNOS phosphorylation and NO• production as previously described. 15
Nitrite and nitrate production
After the appropriate treatment, cells were washed gently with ice-cold PBS twice prior to NO• generation being estimated by measuring the stable metabolites of NO•, nitrate and nitrite, using a kit (Cayman Chemical, Ann Arbor, MI, USA) as previously described. 16
Reactive oxygen species production
Cells were treated appropriately, gently washed with iced PBS three times, and ROS were assessed using 10 μM H2DCFDA-AM fluorescent probe for 1 h at 37℃ as previously described.15,17 Fluorescence intensity was measured at excitation wavelength 485 nm and emission wavelength 530 nm.
Indices of inflammation and adhesion
Expression of inflammatory markers was assessed by quantitative reverse transcriptase polymerase chain reaction (q-RT PCR). After treatments, cells were washed twice with iced PBS and collected in PBS. RNA was extracted using the RNAeasy mini kit (Qiagen Inc., Valencia, CA, USA), then reverse transcribed to cDNA using oligo dt primers and superscript reverse transcriptase III. Primers specific for human ICAM (intracellular adhesion molecule), VCAM-1 (Vascular cell adhesion molecule-1), E-Selectin, MCP-1 (Monocyte chemoattractant protein-1) and IL-8 (Interleukin-8) were commercially obtained (Qiagen Inc., Valencia, CA, USA). The assay was completed in a 25 µL reaction, with 50 ng of template DNA, 0.5 µM of the forward and reverse primers, power SYBR green master mix and PCR grade water. The PCR cycling conditions were 95℃ for 10 min, followed by 40 cycles of 95℃ for 3 min, annealing 61℃ for 30 s, and extension at 72℃ for 30 s. A melt curve analysis was performed to confirm the formation of a single product and the absence of nonspecific amplifications and primer dimers. Data were analyzed using the delta–delta CT method and GAPDH was the normalizing gene among the samples as described.15,17
Western blotting
HAECs were processed for immunoblotting as described. 15 Briefly, samples were harvested, lysed (in mM, 150 NaCl, 20 Tris-HCl, 2 EDTA, 50 NaF, 10 HEPES, 50 Na2H2P2O7, and 1% Triton X-100), and protein concentrations were determined using a Bio Rad protein assay kit (BioRad, Hercules, CA, USA). Proteins were separated on 10–12% SDS-polyacrylamide gel, transferred to FL-PVDF membranes and subjected to immunoblot analyses using antibodies directed against eNOS, p-eNOSSer1177, Akt, p-AktSer473, ERK, p-ERKThr2023/Tyr204, AMPK, p-AMPKThr172, GSK3β, p-GSK3βSer9, and ACC, p-ACCSer79 (Cell Signaling Technologies, Beverley, MA, USA). Membranes were probed for GAPDH as a loading control and bands densities were quantified using Odyssey Imaging Software.
Measurement of eNOS dimers and monomers
Low-temperature SDS-PAGE (LT-PAGE) was performed for detection of eNOS dimers as described.15,17 Briefly, total proteins, mentioned above, were incubated in Laemmli buffer without 2-mercaptoethanol at 37℃ for 5 min. The samples were then subjected to SDS-PAGE with 8% gel. Gels and buffers were equilibrated at 4℃ before electrophoresis, and the buffer tank was placed in an ice bath during electrophoresis to maintain the temperature of the gel < 15℃. After LT-PAGE and transfer to membranes, blots were probed with an antibody directed against eNOS (Cell Signaling Technology, Beverly, MA,USA). A control sample treated with 2-mercaptoethanol was used as an eNOS monomer control.
Cell viability
Cell viability was assessed in HAECs and compared to 0.25% trypsin as a positive control as previously described.17,18 The number of attached (live) cells were counted in a fixed area before and after treatment.
Statistical analyses
Data are presented as mean ± standard error of the mean. A one-way ANOVA was used to analyze all data (SPSS v.20). Tukey’s post hoc test was performed for to detect differences between groups. Significance was accepted at p < 0.05 for all tests.
Results
γ-CEHC prevents impairments in nitric oxide production otherwise induced by high glucose
Under control conditions (5 mM glucose), insulin-stimulated NO• production increased, whereas high glucose conditions reduced NO• production by ∼50%. γ-CEHC treatment restored insulin stimulated NO• production in cells challenged with high glucose conditions to levels similar to that of control conditions (Figure 1(a)). Neither condition evoked cell death (data not shown). Similarly, high glucose also impaired insulin-stimulated eNOS phosphorylation at Ser1177 (Figure 1(b)) and Akt phosphorylation at Ser473 that were otherwise increased under control conditions (Figure 3). Similar to γ-CEHC improving NO• production, it also restored insulin-stimulated eNOS phosphorylation despite high glucose conditions.
Insulin-stimulated nitric oxide (NO•) production and endothelial nitric oxide synthase (eNOS) phosphorylation in human aortic endothelial cells incubated in 5 mM glucose, 25 mM glucose, and 25 mM glucose + 3 µM γ-CEHC. (a) NO• production after insulin stimulation. (b, top): Total and insulin-stimulated phosphorylation of eNOS at serine 1177 (p-eNOS Ser1177). (b, bottom): Ratio of eNOS phosphorylation at Ser473 to total eNOS expression. All values are presented as means ± SE; n = 4–6 independent experiments. *P < 0.05 vs. 5 mM glucose (a to e) Insulin-stimulated signal transduction in human aortic endothelial cells incubated in 5 mM glucose, 25 mM glucose and 25 mM glucose + 3 µM γ-CEHC. (a, top): total and insulin-stimulated phosphorylation of Akt at Serine 473 (p-Akt Ser473). (a, bottom): Ratio of phosphor-Akt at to total Akt expression. (b, top): Total and insulin-stimulated phosphorylation of ERK at threonine Threonine 202/Tyrosine 204 (p-ERKThr202/Tyr204). (b, bottom): ratio of phosphor-ERK to total ERK expression. (c, top): total and insulin-stimulated phosphorylation of AMPK at Threonine 172 (p-AMPKThr172). (c, bottom): ratio of phosphor-AMPK to total AMPK expression. (d, top): total and insulin-stimulated phosphorylation of GSK3β at Serine 9 (p-GSK3βSer9). (d, bottom): ratio of phosphor-GSK3β to total GSK3β expression. (e, top): total and insulin-stimulated phosphorylation of ACC at Serine 79 (p-ACCSer79). (e, bottom): ratio of phosphor-ACC to total ACC expression. All values are presented as means ± SE; n = 4–6 independent experiments. *P < 0.05 vs. 5 mM glucose Reactive oxygen species (ROS) in human aortic endothelial cells incubated in 5 mM glucose, 25 mM glucose, and 25 mM glucose + 3 µM γ-CEHC. All values are presented as means ± SE; n = 4–6 independent experiments. *P < 0.05 vs. 5 mM glucose; #P < 0.05 vs. 25 mM glucose
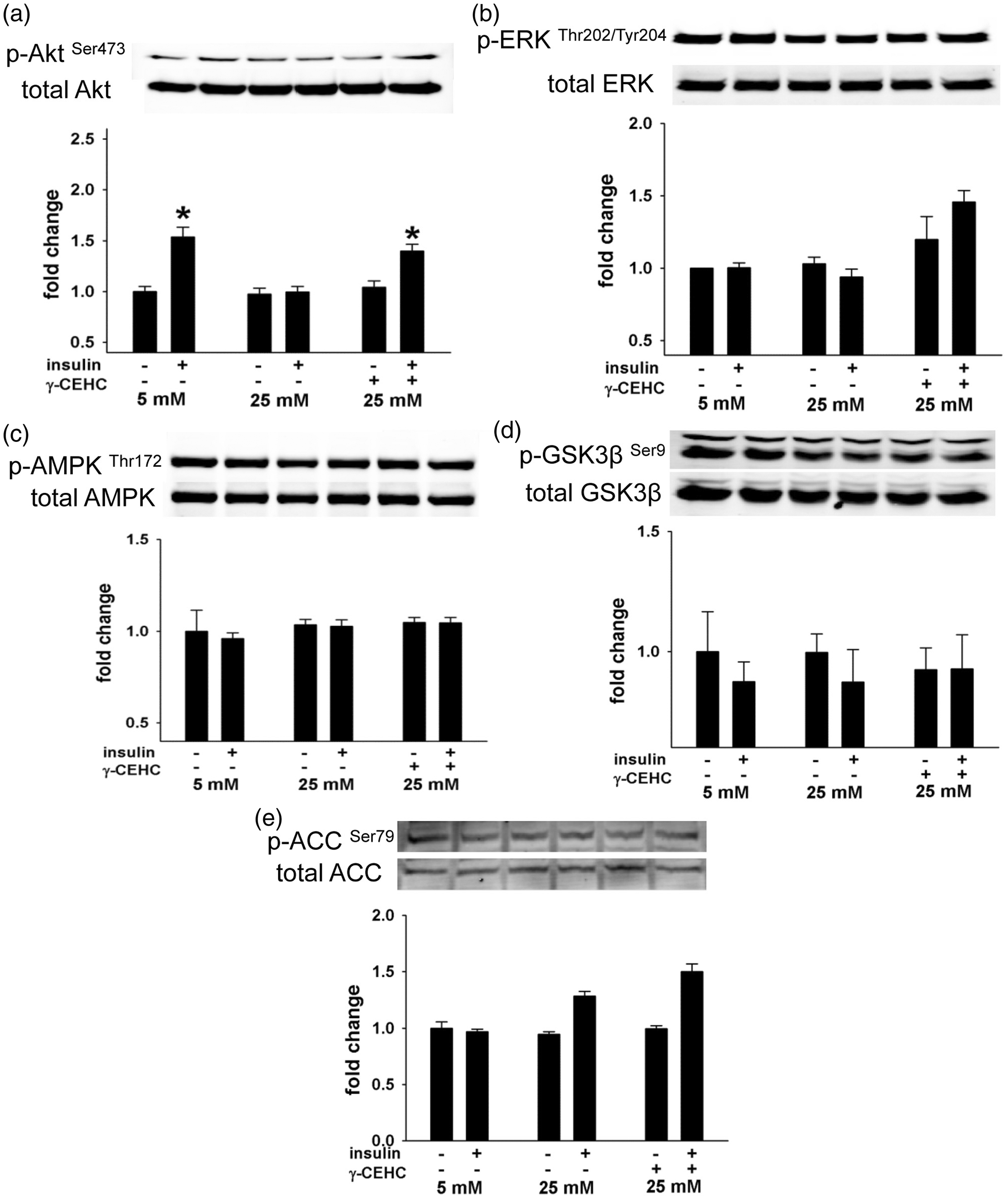

High glucose-induced reductions in eNOS phosphorylation are accompanied by changes in Akt signaling, but not ERK1/2, AMPK, ACC, and GSK3β
We evaluated if Akt, ERK1/2, AMPK, ACC, and GSK3β, kinases that regulate eNOS activity via phosphorylation of its regulatory sites18,19 are operative in mediating the protective effects of γ-CEHC on eNOS during high glucose conditions. Changes in any one or all of these kinases could affect eNOS phosphorylation and subsequent NO• production. Insulin-stimulated phosphorylation of Akt was markedly lower when cells were incubated in 25 mM glucose conditions compared with 5 mM glucose. However, pretreatment with γ-CEHC preserved insulin-stimulated Akt activation (Figure 2) consistent with its effects on NO• production and eNOS phosphorylation. In contrast, ERK1/2, AMPK, ACC, and GSK3β were unaffected by both high glucose and γ-CEHC pretreatment during both low and high glucose conditions (Figure 2).
γ-CEHC prevents high glucose-mediated increases in ROS and eNOS uncoupling
ROS increased in HAECs that were incubated in high vs. low glucose (Figure 3). Protein accumulation of eNOS dimer, the catalytically active form, was reduced after high glucose treatment, resulting in a decreased ratio of eNOS dimer to monomer, indicative of eNOS uncoupling (Figure 4). Pre-treatment with γ-CEHC for 24 h prevented uncoupling of eNOS by preserving a normal dimer to monomer ratio (Figure 4) and prevented the increase in ROS normally produced under high glucose conditions (Figure 3).
Endothelial nitric oxide synthase (eNOS) uncoupling in human aortic endothelial cells incubated in 5 mM glucose, 25 mM glucose, and 25 mM glucose + 3 µM γ-CEHC. Top: Western blots of eNOS dimer, monomer, and GAPDH. Bottom: Ratio of dimer/monomer to GAPDH expression. All values are presented as means ± SE; n = 5 independent experiments. *P < 0.05 vs. 5 mM glucose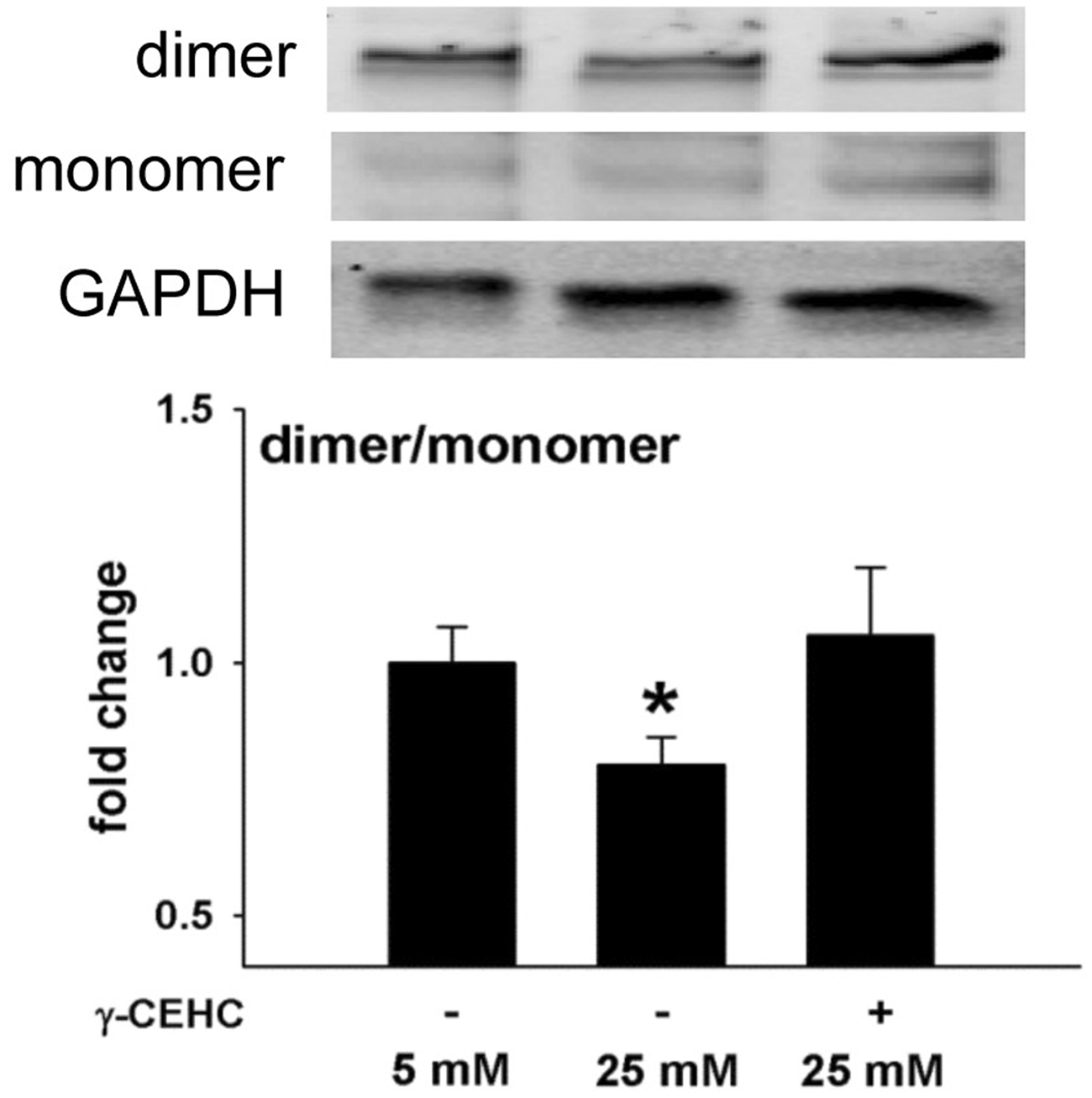
Inflammation resulting from high glucose conditions is prevented by γ-CEHC pre-treatment
We determined the effect of γ-CEHC on high glucose-induced endothelial inflammation by measuring mRNA expression of NFκB-dependent proinflammatory targets (ICAM, VCAM-1, E-Selectin, MCP-1 and IL-8) in HAECs. High glucose increased expression of VCAM-1, E-Selectin, and IL-8, whereas treatment with γ-CEHC prior to high glucose challenge attenuated expression of these molecules (Figure 5(a) to (e)).
Expression of inflammation markers in human aortic endothelial cells incubated in 5 mM glucose, 25 mM glucose, and 25 mM glucose + 3 µM γ-CEHC. (a) VCAM-1 (vascular cell adhesion molecule-1); (b) E-Selectin; (c) ICAM-1 (intracellular adhesion molecule-1); (d) IL-8 (interleukin-8); (e) MCP-1 (monocyte chemoattractant molecule). All values are presented as means ± SE; n = 3 independent experiments. *P < 0.05 vs. 5 mM glucose; #P < 0.05 vs. 25 mM glucose
Discussion
Based on clinical studies demonstrating that γ-T protects against vascular dysfunction resulting from postprandial hyperglycemia, 6 the present study examined the hypothesis that the vasoprotective activities of γ-T during hyperglycemic conditions may be mediated through its physiological metabolite γ-CEHC. To evaluate the protective effects of γ-CEHC during hyperglycemic conditions, we tested both basal- and insulin-stimulated NO• production in HAECs cultured for 48 h in 5 and 25 mM glucose. While it could be argued that 25 mM glucose concentrations are greater than those experienced during post-prandial hyperglycemia, our objective was to use a model system that has been validated to produce endothelial cell dysfunction and impaired NO• production. 20 Here we show first direct evidence that vasoprotective activities of γ-T during hyperglycemia are at least partly attributable to γ-CEHC preserving NO• bioavailability. We propose that γ-CEHC elicited anti-inflammatory and antioxidant effects that prevented eNOS uncoupling. To our knowledge, this is the first study to demonstrate that a metabolite of γ-T can rescue the suppression of insulin-stimulated NO• production evoked by hyperglycemia. There are clinical implications for our studies because the γ-CEHC concentrations that produced these benefits in vitro were based on blood levels reported in our previous study that demonstrated supplemental γ-T protects against endothelial dysfunction induced by postprandial hyperglycemia in healthy men. 6
Phosphorylation of eNOS is involved in regulation of eNOS activity, and consequently NO• production. 21 For example, increased eNOSSer1177 phosphorylation results in greater NO• production in endothelial cells. Therefore, we hypothesized that alterations in insulin-stimulated NO• production were a consequence of altered eNOS phosphorylation at these regulatory sites. We determined that under low glucose conditions (5 mM), insulin stimulation led to an increase in eNOSSer1177 phosphorylation; however, high glucose conditions (25 mM) abolished this response. Akt, a kinase downstream of the insulin receptor that phosphorylates eNOS at Ser1177 2 , also had augmented phosphorylation at Ser473 after insulin stimulation under low glucose conditions, but no change in phosphorylation during high glucose conditions. Abrogation of insulin-mediated Akt phosphorylation under hyperglycemic conditions as we observed in our study is also consistent with an insulin-resistant state.22,23 Importantly, pre-treatment with γ-CEHC preserved insulin-stimulated Akt and eNOS phosphorylation in HAECs that were challenged with high glucose conditions. Other kinases such as ERK, AMPK, ACC, and GSK3β have also been reported to regulate eNOS activity via phosphorylation of regulatory sites. However, we found that the phosphorylation state of these kinases remained similar regardless of glucose conditions or insulin stimulation. Taken together, our data indicate that γ-CEHC may preserve insulin-stimulated signaling during high glucose conditions, as evidenced by normalized insulin-stimulated Akt phosphorylation that ultimately allows for eNOS phosphorylation at Ser1177 and NO• production.
ROS has been implicated as a mediator of endothelial dysfunction during hyperglycemia. For example, monocytes from patients with diabetes showed an increase in production of superoxide relative to normal controls, 24 and increased lipid peroxidation has been reported in patients with type 1 diabetes. 25 In our study, high glucose increased the production of ROS in HAECs, a result consistent with Nishikawa et al. who also reported increased ROS in endothelial cells cultured in 25 mM glucose. 26 Importantly, our data indicate that pre-treatment of HAECs with γ-CEHC prevents ROS accumulation in response to 25 mM glucose. Elevated ROS might result from uncoupled eNOS and subsequent production of superoxide. Elevation of ROS has been reported in diabetic patients, and reduction of ROS after treatment of such patients with an eNOS inhibitor (L-NAME) implicates uncoupled eNOS as a generator of superoxide. 24 Uncoupled eNOS, characterized by a lower dimer (active eNOS) to monomer (inactive) ratio, has also been found to increase ROS in endothelial cells cultured in high glucose (30 mM) conditions. 27 In agreement with these studies, we also found that high glucose challenge elevated ROS and decreased ratio of the eNOS dimer to monomer. It was notable that this uncoupling of eNOS was prevented by γ-CEHC pretreatment, and it occurred in parallel with reduced ROS.
The parent compound γ-T exerts anti-inflammatory actions in vitro and in vivo28,29 and evidence exists that the physiological metabolite γ-CEHC has anti-inflammatory effects in vitro. In the present study, concurrent incubation of HAECs with 25 mM glucose and γ-CEHC prevented increased expression of VCAM, E-Selectin, and IL-8 that otherwise occurs when HAECs incubated with 25 mM glucose per se.30,31 VCAM is expressed on the surface of endothelial cells and mediates binding of leukocytes to the endothelial cells. 32 Similarly, E-Selectin is a C-type lectin that regulates tethering and rolling of lymphocytes along the intimal layer. 33 As such, our data confirm that γ-CEHC has anti-inflammatory properties in HAECs in the context of hyperglycemia. Furthermore, our findings provide strong proof of concept that the anti-inflammatory effects previously reported in clinical studies using supplemental γ-T9,34 may be mediated, at least in part, by the physiological metabolite γ-CEHC.
Given the rapid metabolism of γ-T to γ-CEHC, data presented here provide important mechanistic insight to our previous observation that γ-T is protective concerning endothelial dysfunction evoked by postprandial hyperglycemia. These data concerning γ-CEHC contribute importantly to the evolving body of evidence that γ-CEHC might possess antioxidant, anti-inflammatory, and vasculoprotective effects. This finding has myriad implications regarding treatment and prevention of vascular complications associated with healthy aging, and endothelial dysfunction associated with pathological conditions such as obesity, insulin resistance, and type 2 diabetes. In conclusion, though past supplement studies on Vitamin E have generally focused on the parent compound γ-T, our data suggest strongly that the physiological metabolite of γ-T, i.e. γ-CEHC, has vasculoprotective properties, at least in HAECs challenged with high glucose.
Footnotes
Authors’ contributions
YL conducted experiments, analyzed data, and wrote the manuscript; LPBL conducted experiments and analyzed data; YQ and TR conducted experiments; PVAB provided scientific direction; RSB provided scientific direction and edited the manuscript; JDS provided scientific direction and edited the manuscript; and TJ provided scientific direction, analyzed data, and wrote the manuscript.
Acknowledgements
This work was completed in partial fulfillment for the PhD degree of Youyou Li. All work was completed at the University of Utah. Abstracts of data contained in this manuscript were presented at Experimental Biology 2013 (Boston), 2014 (San Diego), and 2015 (Boston). This work was funded in part by the American Diabetes Association (ADA: 1-12-BS-208, ADA 7-08-RA-164) to JDS, the National Institutes of Health (NIH: 2R15HL091493) to JDS, and Seed Grants from the UU Office of the Vice-President for Research and the UU College of Health to JDS and TJ. Student support was provided by the American Physiological Society (APS) Undergraduate Research Program, APS Undergraduate Research Excellence Fellowship Program, and APS STEP-UP Program; the American Heart Association, Western States Affiliate, Undergraduate Student Summer Research Program; the ADA Minority Undergraduate Internship Program; The Science without Borders Program of the Government of Brazil, the Native American Research Internship Program, and the UU Undergraduate Research Opportunities Program.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.