
Abstract
The complement system is one of the body’s major innate immune defense mechanisms in vertebrates. Its function is to detect foreign bodies and promote their elimination through opsonisation or lysis. Complement proteins play an important role in the immunopathogenesis of several disorders. However, excessive complement activation does not confer more protection but instead leads to several autoimmune and inflammatory diseases. With inappropriate activation of the complement system, activated complement proteins and glycoproteins may damage both healthy and diseased tissues. Development of complement inhibitors represents an effective approach in controlling dysregulated complement activity and reducing disease severity, yet few studies have investigated the nature and role of novel complement inhibitory proteins of viral origin. Viral complement inhibitors have important implications in understanding the importance of complement inhibition and their role as a promising novel therapeutic approach in diseases caused by dysregulated complement function. In this review, we discuss the role and importance of complement inhibitors derived from several viruses in the scope of human inflammatory and autoimmune diseases.
Introduction
Viral-derived complement regulators constitute a potentially novel therapeutic approach to reduce disease-associated inflammation and host tissue damage. Several autoimmune and inflammatory disorders of which dysregulation of complement activation is the major mechanism of pathogenesis indicate the necessity for tight immunomodulation of complement proteins. In this paper, we review the current knowledge of virus-encoded inhibitors of the complement system. The complement system, its physiological regulation and its role in human disease is briefly discussed. In addition, the potential future use of viral-derived complement modulators in therapy is highlighted.
The complement system
Complement or alexin, as it was originally called, was first described by Bucher et al. as humoral heat-labile factors capable of killing bacteria. 1 Bucher’s work was further confirmed by investigations showing the presence of a heat-labile lytic factor and a heat-stable factor termed “sensitiser” (now known as antibody) during the process of immune lysis. Paul Ehrlich later described the role of complement in the antibody neutralisation process. Ehrlich proposed that a complex formed from an antibody and a complement is capable of recognizing and destroying pathogens. 2 Ferrata and Brand later demonstrated that this complement was not a single entity, but could be separated into two fractions: a midpiece (now termed C1) and an endpiece (now termed C2). They showed that the presence of both molecules was necessary for complement activity. 3 With the advancement of technologies, complement proteins were identified, purified, characterized and named based on the order of their discovery, enabling further studies of their roles.
The complement system is an essential component of the innate immune system. It is a family of soluble and membrane-bound proteins capable of detecting pathological agents.4,5 Activation of the complement system results in an immune reaction capable of destroying pathogens and their products. Multiple mechanisms have been documented to clear foreign bodies including cell lysis, immune complexes, opsonization and phagocytosis, T and B cell priming and leukocyte chemotaxis.6–8
There are three pathways involved in activating the complement system: classical, alternative, and lectin pathways (Figure 1). The classical pathway links the adaptive and innate immune systems. It is initiated at the level of C1, a multiprotein complex made of C1q responsible for pathogen recognition and two copies each of C1r and C1s, both of which act as serine proteases.
9
Each of the six C1q subunits is composed of three protein chains A, B, and C forming an N-terminal, triple-helical, collagen-like region (CLR) and a globular head region (GHR). The latter often binds IgG, IgM, C-reactive protein, serum amyloid protein, lipopolysaccharide (LPS), fibromodulin and deoxyribonucleic acid (DNA).10–12 Antigen-antibody complexes recognized by C1q constitute the major mechanism in activating the classical pathway, yet C1 has also been documented to play a crucial role in recognizing and clearing apoptotic cells, cellular debris, beta-amyloid fibrils and the lethal prion protein.13–15 Upon activation of C1, C4 is cleaved into C4a and C4b by the C1 multiprotein complex. On the cell membrane, C2 binds C4b, which is then cleaved by C1s, forming C4b2a, the classical pathway C3 convertase that cleaves C3 into C3a and C3b. C3b binds to C4b2a, forming the C5 convertase (C4b2a3b), which cleaves C5 into C5a and C5b and catalyzes the formation of the membrane attack complex (MAC) on the cell surface, leading to cell lysis and cytotoxicity.
16
By binding to their receptors (C3aR and C5aR) on inflammatory and non-inflammatory cells, the generated C3a and C5a act as anaphylatoxins by attracting inflammatory cells, such as neutrophils, to the site of damage.
17
C4a is a weak anaphylatoxin that has no known receptor.
18
A high level of anaphylatoxins causes the secretion of pro-inflammatory cytokines and chemokines resulting in chemotaxis.19–21 C5a is considered the principal anaphylatoxin since besides recruiting inflammatory cells, it promotes endothelial cell aggregation and adhesiveness by up-regulating adhesion molecules such as integrin, selectin, intercellular adhesion molecule-1 (ICAM-1) as well as vascular cell adhesion molecule-1 (VCAM-1).22–24 While C3b fuels the formation of more complement convertases ultimately forming the MAC complex, deposition of C3b on activated surfaces results in phagocytosis.
25
Also, C3b degradation fragments, iC3b and C3d, promote opsonisation and adaptive immune signaling via complement receptors (CRs).
26
Activation pathways of the complement system. The classical pathway is often initiated by Ag–Ig complexes binding to C1, the lectin pathway is activated by microbial surfaces (e.g. mannose) and the alternative pathway is activated either spontaneously or by microbes, parasites, and viruses. The C3 convertase is formed via the C1-mediated formation of C2a and C4b through the classical pathway, MBL/MASPs-mediated formation of C2a and C4b through the lectin pathway and the C3b-Bb interaction through the alternative pathway. The generated C3b participates in the formation of the C5 convertase, which eventually ends in the assembly of MAC causing cell lysis. Ag: antigen; Ig: antibody; MBL: mannose-binding lectin; MAC: membrane attack complex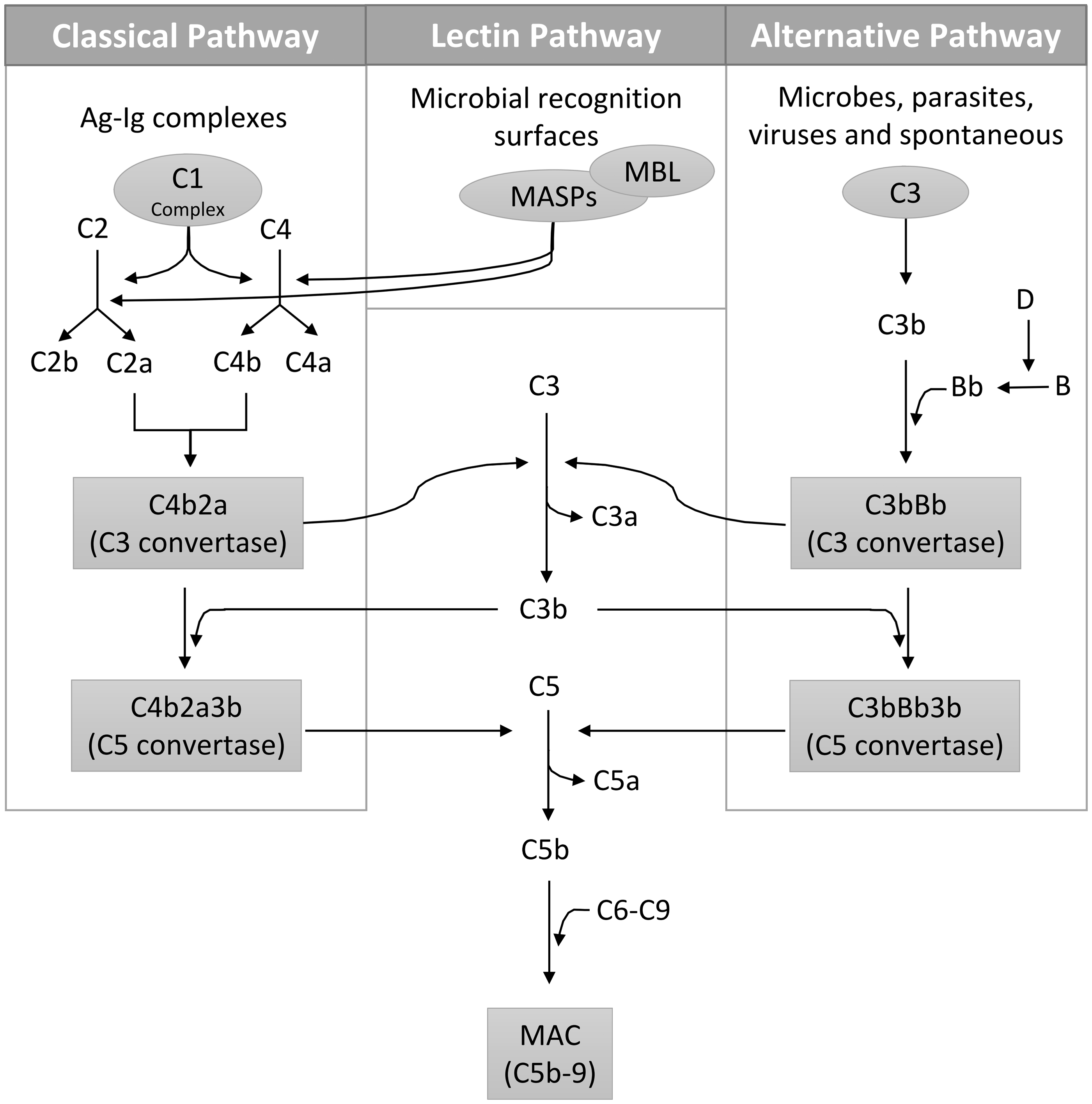
Fifty years after the discovery of the classical pathway, the ‘properdin pathway’ (now termed the alternative pathway) was proposed by Pillemer et al. who demonstrated that the complement system was activated by the direct binding of bacteria or yeast, independent of antibody recruitment. 27 C3 has a highly reactive thioester bond that can undergo spontaneous hydrolysis in the presence of water, forming a C3b-like C3(H2O) molecule. C3(H2O) then binds to factor B, forming the alternative pathway C3 convertase. This pathway is controlled by factor D, a peptidase that cleaves factor B, thus regulating the formation of the alternative C3 convertase. 28 Properdin, the only known positive regulator of the complement system, promotes the assembly of the alternative pathway convertases by stabilizing the binding of factor B to C3b. 29
The lectin pathway was later described by Kawasaki et al. and Kozutsumi et al.5,30 This pathway is initiated by multimeric lectin protein complexes termed “collectins” such as MBL and ficolin, that recognize carbohydrate patterns distinct from those of the host. 30 The interaction between collectins and foreign carbohydrate patterns activates the lectin pathway through the enzymatic activity of MBL-associated serine proteases (MASPs). For instance, MASP-2 cleaves C4 and C2 into C4b and C2a, thereby forming the C3 convertase. 31 Besides cleaving C2, MASP-1 has also been reported to directly cleave C3 into C3b, thereby forming the C5 convertase, although at a very slow rate. 31
Physiological regulators of the complement system
Physiological inhibition of C1 is achieved by four well-documented families including C1-Inhibitor (C1-INH), decorin and biglycan, neutrophil defensins, and C1q receptor proteins. C1-INH, a serine protease inhibitor (serpin), is a glycosylated serum protein that can modulate the coagulation cascade, the kinin system, the lectin pathway and the alternative pathway besides its major role as an inhibitor of activated C1.10,31–34 Recombinant C1-INH has been recently approved by the FDA for the treatment of hereditary angioedema and tracheal obstruction, which may result from either insufficient secretion of C1-INH or production of its inactive form.35,36 Biochemically, C1-INH binds to the activated C1r and C1s, facilitating their dissociation from C1q, which in turn prevents C4 activation. 37 Decorin and biglycan are abundant extracellular matrix proteoglycans, both of which specifically bind to the GHR and CLR regions of C1q, resulting in inhibition of the classical pathway.38,39 Both decorin and biglycan are capable of binding strongly to MBL, but only biglycan can inhibit the lectin pathway. 39 Also, evidence suggests an overlap between heparin binding sites and complement inhibitory sites. 40 Binding of complement inhibitors to heparin and glycosaminoglycans presents a mechanism for these molecules to anchor to their targets, thus modulating the outcome of the complement reactions. Neutrophil defensins are contained within the neutrophils azurophilic granules and often participate in neutrophil-mediated antimicrobial clearance. They have been shown to bind to the CLR C1q region and MBL, thus inhibiting the classical and lectin pathways, respectively.41,42 C1q receptor proteins such as the GHR-binding gC1qR/p33 receptor and the CLR-binding cC1qR/calreticulin receptor are present on cell surfaces and can inhibit C1-mediated complement activity.43,44
C4b binding protein (C4BP) is a large soluble glycoprotein that regulates complement activity by binding to C4b and accelerating the decay of the classical and lectin C3 convertases whereas factor H binds to C3b and accelerates the disintegration of the alternative C3 convertase.45–47 Factor I, supported by factor H, CD35, CD46 and C4BP as cofactors, cleaves cell-bound or soluble C3b and C4b generating iC3b, C3c, C3dg, C4c and C4d, preventing the formation of C3 and C5 convertases. 48 Carboxypeptidase N regulates the complement system as an inhibitor of anaphylatoxins by cleaving arginines and lysines located at the carboxy-terminus. 49
Among the membrane-bound complement regulators is CD55 (decay accelerating factor [DAF]) whose main function is to facilitate the decay of C3 convertases generated from the classical and alternative pathways. 50 By interacting with C4b, CD55 interferes with the cleavage of C2 into C2a and prevents the formation of the C3 convertase of the classical and lectin pathways. Additionally, by recognizing C3b of the alternative pathway, CD55 prevents the conversion of factor B to Bb by factor D, thereby preventing the formation of the alternative C3 convertase. 50 The transmembrane glycoprotein CD35 (CR1) is another membrane-bound complement regulator that facilitates the decay of the classical and alternative C3/C5 convertases and promotes factor I-mediated degradation of C3b and C4b. 51 CD35 is also essential for the clearance of complement-generated and opsonized immune complexes. 52 Similar to CD35, CD46 (membrane cofactor protein [MCP]) acts as a cofactor for factor I in the cleavage of C3b and C4b. 53 CD59 (protectin) is universally expressed on all body cells and regulates complement activation by inhibiting the insertion of C9 into the lipid bilayer, thereby preventing MAC assembly.54,55
Human regulators of the complement system

Regulator acts as a cofactor for factor I.
Role of complement in inflammatory diseases
The interplay between the inflammatory manifestation and complement system has been well documented in several diseases such as vascular disease as in atherosclerosis; renal disease as in glomerulonephritis; pulmonary disease as in lung injury; neurological diseases as in age-related macular degeneration (AMD), myasthenia gravis (MG), Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), Pick’s disease, Guillain-Barré syndrome (GBS), Miller Fisher syndrome (MFS), prion disease and neurotrauma (traumatic brain injury [TBI] and spinal cord injury [SCI]); bone disease as in rheumatoid arthritis and other diseases as in diabetic retinopathy, atypical hemolytic uremic syndrome (aHUS), systemic lupus erythematosus (SLE), ischemia/reperfusion injury, transplant rejection and many other pathologies (Figure 2).56–65 Such inflammatory diseases involve dysregulated control of the complement pathways which results in inappropriate secretion of reactive oxygen species, eicosanoids, cytokines and chemokines, inducing the influx of inflammatory cells. A few examples are discussed below.
Spectrum of inflammatory diseases due to uncontrolled complement activation. (A color version of this figure is available in the online journal).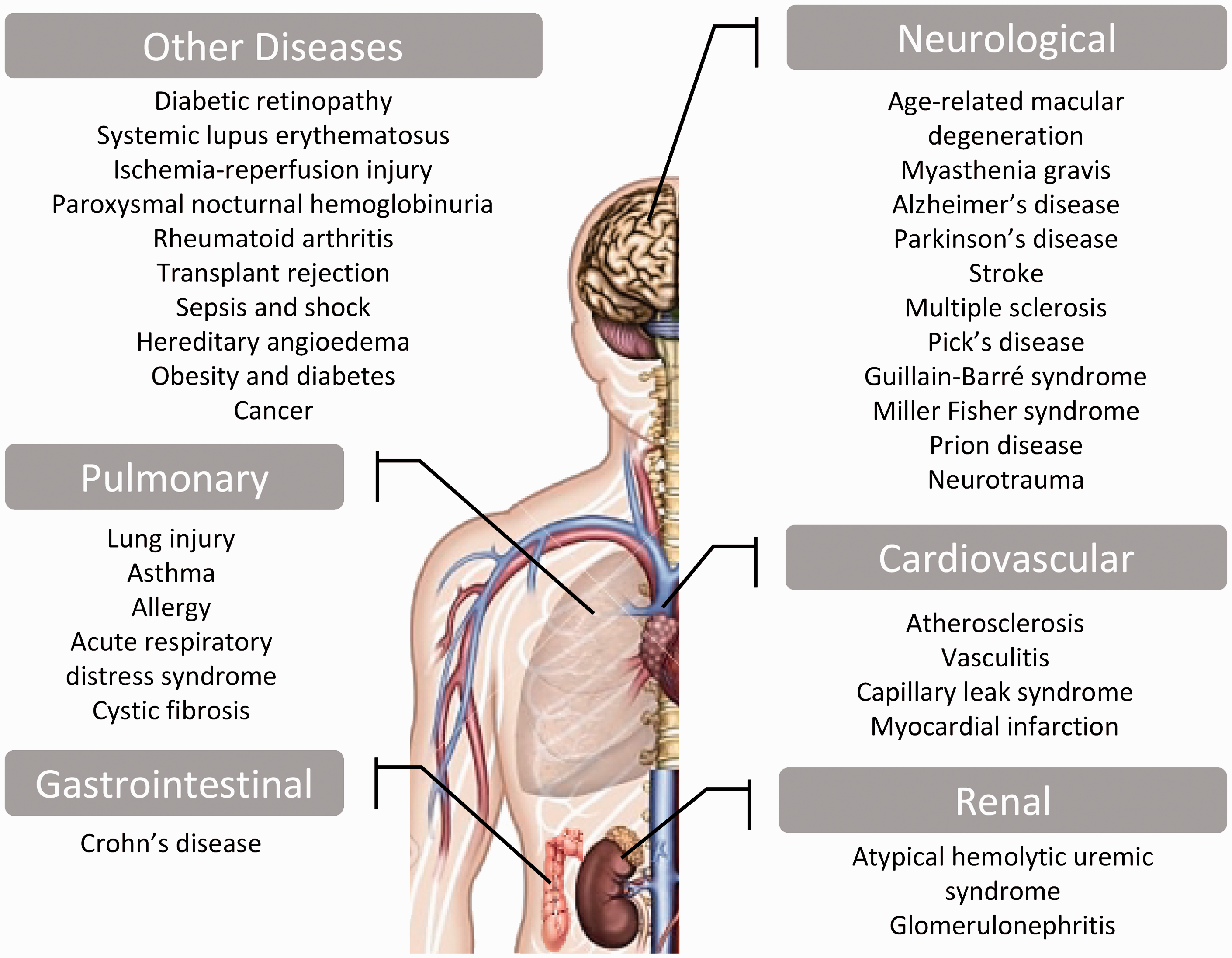
Atherosclerosis, a chronic inflammatory disease of fat deposition on the tunica intima of arterial walls, is characterized by several inflammatory mechanisms that result in endothelial cell dysfunction. A study by Wu et al. demonstrated that the loss of CD59, which inhibits MAC assembly, in apolipoprotein E (ApoE)-deficient mice facilitated atherogenesis, occlusive atherosclerosis, and earlier death. Consistently, the phenotype was reverted by the over-expression of CD59. 66 The study indicates the atherogenic role of MAC, suggesting that its inhibition via either over-expression of CD59 or inhibition of C5 may be a potential therapy in the management of atherosclerosis.
Immunohistochemical investigations revealed extensive deposits of the C5b-9 and C3d complement complexes in the eyes of deceased patients with diabetic retinopathy.67,68 Detection of terminal complement proteins like C5b-9 indicated that the complement pathway took place to completion, resulting in the pathologic sequelae of diabetic retinopathy.
The role of the complement cascade has also been implicated in AMD. AMD was shown to be the result of multiple dysregulated complement proteins, including polymorphisms in the factor H gene and increased MAC formation, resulting in macular degeneration and detrimental loss of choriocapillaries. 69
At the level of the renal system, membranoproliferative glomerulonephritis (MPGN) is a kidney disease caused by paramesangial deposits and thickening of the glomerular basement membrane. MPGN type I and type II are associated with uncontrolled activation of the classical complement pathway and the alternative pathway, respectively.70,71 Similar to glomerulonephritis, aHUS has been associated with dysregulation of complement activation particularly through factor H. 72 Different mutations in factor H protein have been linked to both glomerulonephritis and aHUS. 73
Sun et al. demonstrated that acute lung injury (ALI) induced by infection of mice with highly pathogenic influenza A/H5N1 virus is caused by excessive activation of the complement system. In their study, inhibition of complement activation by an anti-C5a antibody yielded less neutrophil infiltration, less lung injury and an increased survival rate in H5N1-induced ALI. 74
MG is a neuromuscular disease caused by the presence of autoantibodies against the acetylcholine receptor (AChR). The severity of MG is determined by the complement fixation capacity at the neuromuscular junction.75,76 A study by Soltys et al. reported a novel C5 complement inhibitor, rEV576, derived from tick saliva that reduced disease severity and increased survival rate, indicating inappropriate complement activity in MG. 77
In AD, both the classical and alternative pathways are activated by the A beta peptide of the amyloid precursor protein (APP). 78 APP is the hallmark of AD with an A beta peptide that binds C1q and C3, mediating MAC assembly. As expected, injection of C-100, a recombinant polypeptide containing A beta, into C5-sufficient mice recruited complement proteins C1q and C3, causing an increase in chemotactic factors and neutrophil infiltration. 79 In a similar fashion, tau protein which is the main component of neurofibrillary tangles was shown to be an activator of the classical pathway by directly binding to C1q, thus contributing to brain inflammation observed in AD. This result indicates that targeting the interaction between both of the A beta and tau proteins and the complement proteins in therapy provides a potential approach in reducing AD-associated neurodegeneration. In addition, the early appearance of A beta and tau proteins makes them promising early targets for complement inhibition and early diagnostic markers of AD. 80 Levels of activated microglia, C-reactive protein and all components of the MAC are also elevated in PD, thus accounting for the chronic neuroinflammation and neurodegeneration seen in PD.81,82
Elevated levels of the terminal complement complex protein were noted in the cerebrospinal fluid of patients with MS, an autoimmune demyelinating central nervous system (CNS) disease. 83 Similar findings were also reported in Pick’s disease, a neurodegenerative disease of frontotemporal dementia. 84 In GBS, a demyelinating peripheral nervous system disease, complement-mediated inflammatory infiltrates were noted in patient autopsies. 85 Decreased deposition of MAC at neuromuscular junctions was observed in MFS, a GBS variant, in mouse models treated with rEV576 compared to controls. 86 In general, inflammation that occurs due to activated CNS astrocytes and microglial cells secreting cytokines, eicosanoids and radicals during the pathogenesis of CNS disorders ultimately accounts for the observed neurodegenerative and neuroinflammatory processes and neurologic disability. 87
This overview on the essential role of complement activation in mediating the pathogenesis of an array of diseases highlights the therapeutic potential of complement inhibitors to reduce complement-induced inflammation and host damage.
Viral-derived complement inhibitors
The link between viral infection and complement reactions is well studied. Upon infection, viral evasion of complement-mediated clearance is not uncommon. Recruiting viral complement control proteins, viruses evade the complement system by one of three mechanisms: (1) interference with the binding of complement to antibody-antigen complexes (e.g. herpesviruses and coronaviruses), (2) viral mimicry of host CRPs (e.g. poxviruses and herpesviruses), and (3) incorporation of complement inhibitors into the viral envelope and/or infected cell surface (e.g. herpesviruses). 88
Mechanisms and outcomes of viral-derived complement modulators
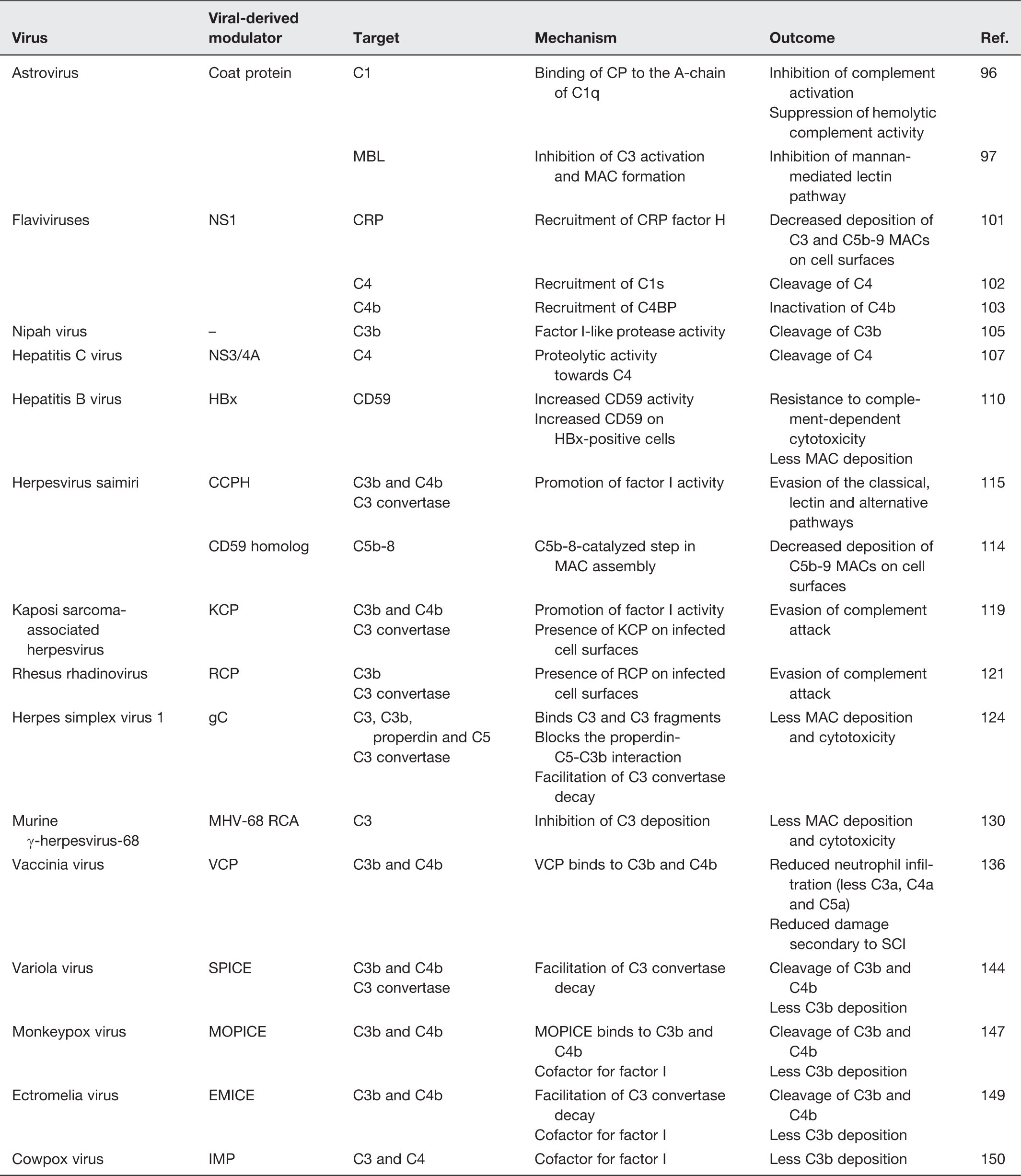
Astrovirus
Human astroviruses (AstVs) are nonenveloped, icosahedral viruses with a single-stranded positive-sense 7 kb-long ribonucleic acid (RNA) genome made up of three open reading frames (ORFs): ORF1a and 1b encode non-structural (NS) proteins and ORF2 encodes the coat protein (CP).89–92 Human AstVs are among the leading causative agents of viral gastroenteritis in young children. 93
Although AstV particles have been found in intestinal epithelial cells of children with diarrhea, indicating shedding through feces, the pathology and immunity to AstV infection remains unclear. 94 The fact that AstV, unlike rotavirus, does not significantly cause cell death or inflammation, has triggered researchers to scrutinize the action of AstVs on the complement system. 95
In 2008, Bonaparte et al. described a novel AstV-derived inhibitor of C1, the first non-enveloped icosahedral viral agent inhibiting complement activation and the first viral C1 inhibitor. 96 In their study, virions of AstV serotypes 1, 2, 3 and 4 and purified CP suppressed complement activity using a standard hemolytic complement assay. Moreover, the AstV CP was found to bind the A-chain of C1q and inhibited the classical complement pathway as well as all subsequent complement-mediated downstream immune reactions such as C4b and terminal C5b-9 formation. Another study showed that AstV CP can bind to purified MBL and was able to inhibit the lectin pathway in addition to inhibiting C3 activation and MAC formation. 97
In ABO incompatible blood transfusion reactions or hemolytic diseases, E23A, a derivative of the AstV CP, was found to inhibit antibody-mediated complement attack and hemolysis.98,99 A peptide formed of 15 amino acid residues and derived from the AstV CP resulted in a superior inhibition, compared to E23A, of the classical complement pathway and ABO incompatibility besides a promising protective role from kidney injury.99,100
Products of complement activation play a crucial role in triggering inflammation and infiltration of inflammatory cells. Thus, by inhibiting the cleavage of C3 and C5 by AstV CP into the chemoattractants C3a and C5a, respectively, it is likely that the influx of inflammatory cells into the site of infection will also be inhibited. Thus, AstV CP-mediated complement inhibition may justify the subtle inflammation observed upon AstV infection.
Flaviviridae
West Nile virus (WNV), a member of flaviviruses whose infections are often subclinical, is an 11-kb long single-stranded RNA enveloped virus encoding three structural proteins and seven NS proteins including NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. 101 Interestingly, both soluble and membrane-bound NS1 proteins were reported to employ CRP factor H, resulting in decreased deposition of C3 and C5b-9 MACs on cell surfaces. Recent studies also revealed that NS1 of flaviviruses such as Dengue, West Nile and yellow fever can antagonize complement and lectin pathways by recruiting and activating C1s and C4BP in an attempt to promote the cleavage of C4 and inactivate C4b, respectively.102,103
Nipah virus
Nipah virus (NiV) is an emergent paramyxovirus that is deadly to humans and animals, with symptoms ranging from asymptomatic to fatal encephalitis and acute respiratory syndrome. It possesses an 18.2 kb-long negative-sense single-stranded RNA in a pleomorphic viral structure. 104
Johnson et al. recently demonstrated that treatment of purified NiV with normal human serum resulted in cleavage of C3b into iC3b. 105 Despite the fact that NiV was capable of activating the complement pathways, virus particles had very little deposition of C3 on their surface. Using electron microscopy with anti-factor I antibodies, the study proved that NiV virions had a factor I-like protease activity capable of cleaving C3 in a time- and dose-dependent manner. This was the first study to report this novel mechanism of a viral-derived factor I-like protease activity to escape complement attack.
Interfering with the anti-complement protease activity of NiV may form the basis of future effective therapies and vaccines targeting such a highly pathogenic virus. Furthermore, the NiV-derived complement inhibition may be utilized as a potential treatment for diseases of dysregulated complement activation.
Hepatitis C virus
Hepatitis C virus (HCV), which causes viral hepatitis and some lymphomas, is a small, enveloped positive-sense single-stranded RNA virus. 106 The NS 3/4A protease protein of HCV was shown to cleave C4 and consequently inhibit the classical pathway. 107 This finding was further confirmed by the specific inhibition of the NS 3/4A protease, which reduced C4 cleavage. Further studies found that iC3b expression was inhibited and that of the classical C3 convertase was suppressed in the sera of HCV patients. 108 Although not virally derived, CD59 has also been reported to be incorporated into HCV particles and to inhibit complement activation as a mechanism of viral escape from the host immunity. 109
Hepatitis B virus
Other hepatitis viruses were also described to modulate the activation of the complement system. For instance, hepatitis B virus (HBV), an enveloped DNA virus, encodes the HBV X protein (HBx), which was shown to upregulate CD59, impairing MAC assembly on cell membranes of HBx-positive cells. 110 HBx was found to activate C4BPα, a potent inhibitor of complement system, thereby enhancing the protection of hepatoma cells. 111
Herpesviruses
Herpesviridae, a family of icosahedral double-stranded DNA viruses, includes eight herpesvirus types capable of infecting humans.
Herpesvirus saimiri (HVS) is a T-lymphotropic gammaherpesvirus type 2, which is associated with leukemias of T cell origin, lymphosarcomas, and lymphomas in New World monkeys. 112 HVS encodes two distinct functional homologs of CRPs: a regulator of complement activation (RCA) homolog named complement control protein homolog (CCPH) encoded by ORF4 and a CD59 homolog encoded by ORF15.113,114 The CD59 homolog inhibits MAC assembly, whereas CCPH was shown to promote factor I-mediated cleavage of C3b and C4b besides facilitating the decay of the C3 convertases of the classical, lectin and alternative pathways.115–117
Kaposi’s sarcoma-associated herpesvirus (KSHV) and its closest known homologue, Rhesus rhadinovirus (RRV), encode two complement inhibitors: KSHV complement control protein (KCP) and RRV-complement control protein (RCP). 118 KCP and RCP modulate the activation of the complement system by promoting the cleavage of C3 and facilitating the decay of C3 convertase.119–121 It is also worth mentioning that KCP and RCP are incorporated into infected cell surfaces, leading to viral evasion of complement attack.
Herpes simplex virus 1 (HSV-1) and HSV-2 encode glycoprotein C (gC) protect the virus from neutralization. Using two distinct domains, gC binds to C3 and C3b inhibiting the activation of the complement cascade and blocks the interaction between properdin, C5 and C3b, thus interfering with the activation of the classical and alternative pathways.122–125 gC was also found to accelerate the decay of the alternative pathway C3 convertase. 126 Furthermore, C3 reconstitution experiments showed that C3 resulted in a reduction in the virulence of the gC-mutant virus compared to the wild-type virus suggesting a C3-mediated mechanism of HSV-1 pathogenesis against the host complement system. 127 Although both HSV-1 and HSV-2 encode gC, HSV-2-infected cells displayed no gC C3b receptor activity.123,128 Multiple mechanisms accounting for this difference between the HSV-1 and HSV-2 gC proteins have been suggested: (1) gC-2 (encoded by HSV-2) receptor activity may be blocked by components of the cell membrane; (2) gC-2 binding to C3b may be inhibited by membrane glycoproteins expressed during HSV-2 infection; or (3) that gC-2 has lower affinity for C3b than gC-1, a possibility that was refuted by Rux et al. 129
Murine γ-herpesvirus-68 (MHV-68) belongs to the γ-herpesvirinae subfamily and is known to cause B-cell lymphoma and Kaposi’s sarcoma. 130 Sequence analysis revealed an ORF 4-coded RCA that shares high homology to RCAs of HVS, KSHV, and VV as well as the human DAF and CD46. 130 Three isoforms of the MHV-68 RCA protein (60-65 kDa, 50-55 kDa, and 40-45 kDa) were detected in infected fibroblasts in addition to a soluble 40–45 kDa isoform potentially generated from the proteolysis of the membrane form. MHV-68 RCA protein was found to regulate complement activity via the inhibition of C3 deposition of both the classical and alternative pathways. Kapadia et al. showed that the deletion of the MHV-68 RCA protein caused a decrease in virulence during acute CNS infection. 131
Poxviruses
Poxviridae, a family of enveloped double-stranded DNA viruses, is one of the well-studied family of viruses encoding complement inhibitors of viral origin.
Vaccinia virus (VV) is a large enveloped, 190 kbp-long virus. 132 VV is often used in gene therapy and genetic engineering. Four infectious forms of VV result from its replication cycle: the intracellular mature virion (IMV) responsible for spreading between hosts and is the most abundant, the intracellular enveloped virion (IEV), the cell-associated enveloped virion (CEV) responsible for cell-to-cell spreading, and the extracellular enveloped virion (EEV) responsible for long range dissemination. 133 VV complement control protein (VCP), a 35-kDa 244 amino acid-long nonglycosylated soluble protein was first identified in 1988. 134 VCP helps the virus escape the host immune response by binding to C3b and C4b, blocking both the classical and alternative pathways of complement. Subsequent downregulation of proinflammatory chemoattractants (C3a, C4a and C5a) leads to a reduced influx of inflammatory cells. In addition, VCP can bind to heparin, blocking macrophage inflammatory protein (MIP)-1α and can inhibit monocyte infiltration. 135 In an experimental model of SCI, rats receiving VCP had less neutrophil infiltration to the site of the lesion as compared to rats receiving saline. In the same study, VCP binding to heparin and heparan sulfate resulted in blockage of monocyte infiltration. 136 In rats subjected to severe head trauma followed by injection of VCP at the site of injury, significant improvement in sensorimotor function was observed compared to saline-treated controls. 137 At the level of vascular disease, VCP treatment of C57BL/6 mice with fatty diet-induced atherosclerosis resulted in a reduction in the development of fatty streaks. 138
Al-Mohanna et al. demonstrated that treating cultured pig aortic endothelial cells (PAECs) with VCP results in a dose-dependent blockage of complement-mediated PAECs cytotoxicity. Interestingly, VCP was also found to prevent VCP-treated PAECs’ interaction with neutrophils and NK cells due to its heparan sulfate binding ability. 139 In an animal model of xenograft transplantation, VCP was able to block hyperacute rejection of the transplanted cervical cardiac xenograft, thereby prolonging graft survival. Upon histological examination, cardiac tissue damage was significantly reduced as compared to controls, as well as less C3, IgG and IgM deposition, thereby preventing complement fixation and activation. 140 In an attempt to uncover a therapeutic role of VCP following ischemia/reperfusion injury, rats were treated with recombinant VCP (rVCP), natural VCP, humanized recombinant VCP (hrVCP) or a vehicle. Biochemical findings indicated an increase in the urea and creatinine levels in the vehicle-treated group compared to the groups receiving VCP. Also, histopathologic examination revealed focal necrosis in the VCP-treated groups as compared to diffuse necrosis observed in the vehicle-treated group. Deposition of C3 in the renal tubule of vehicle-treated rats suggests a potential role of VCP in reducing complement-mediated ischemia/reperfusion injury. 141
The VCP protein may be beneficial in the prevention, treatment, and follow-up of disorders with inappropriate complement activation. Indeed, the striking diversity of functions that VCP exhibits may be of promising therapeutic potential in reducing secondary damage following traumatic SCI, ischemia/reperfusion injury, and other inflammatory conditions. This increases the chance of earlier recovery and regenerative repair. VCP has been shown to be neuroprotective from inflammatory damage in animal models of AD, TBI, SCI, and arthritis. However, VCP cannot cross the blood–brain barrier due to size-limitations, thus constituting a limitation in CNS therapeutics and calls for alternative methods for CNS delivery in cases of exaggerated CNS complement activation. 142
Variola virus, the causative agent of smallpox, is a 186-kb long virus. 143 The smallpox inhibitor of complement enzymes (SPICE), which utilizes heparan sulfate and chondroitin sulfate as attachment sites to regulate complement on the cell surface, was purified and expressed as transmembrane and soluble forms. Both forms were found to regulate human complement by inhibiting complement activation at the level of the C3 convertase. 144 SPICE is a more potent inhibitor of the complement system compared to VCP due to its significantly greater binding capacity to C3b residing within four of the 11 residues differentiating the two proteins. 145
Monkeypox virus (MPV), carried by both humans and animals, is a poxvirus that causes a milder form of the smallpox disease and has a lower death rate. 146 Similar to SPICE, MPV inhibitor of complement enzymes (MOPICE) is capable of binding C3b and C4b and acting as a cofactor for factor I in facilitating C3b cleavage. Compared to SPICE and VCP, MOPICE has a more efficient C3b binding capacity than VCP but less efficient compared to SPICE. MOPICE has a similar cofactor activity as VCP but both were 100-fold less efficient than SPICE. Unlike SPICE and VCP, MOPICE does not possess decay-accelerating activity towards the classical pathway C3 and C5 convertases. 147
Ectromelia virus (ECTV) causes mousepox, a fatal disease in mice. 148 Ectromelia virus inhibitor of complement enzymes (EMICE) was characterized as a complement inhibitory protein that protects intracellular virions from neutralization. 149 This is achieved by the binding of EMICE to C3b and C4b, acting as a cofactor for factor I in the cleavage of C3b and by mediating the dissociation of the classical C3 convertase catalytic domain.
Cowpox virus (CPV) is a poxvirus with a zoonotic potential that causes a mild form of the smallpox disease. CPV encodes a highly conserved homolog of VCP termed the inflammation modulatory protein (IMP). 150 In a study where a recombinant CPV lacking the IMP protein was injected into the footpads of mice, macroscopic examination showed greater damage, hemorrhage and induration compared to controls. 150 Further studies examining CPV infection in C3 −/− mice revealed a C3-dependent immunopathogenesis of the IMP protein. 151 IMP has significant homology to RCAs of other poxviruses and was found to block the classical and alternative pathways by binding to C3 and C4 and acting as a cofactor for factor I. 152
Other poxviruses such as camelpox virus, leporipoxvirus, suipoxvirus, and yatapoxvirus also encode proteins resembling RCAs that have not been fully characterized.
Strategic approaches of complement inhibition
Numerous strategies of complement inhibition have been devised to prevent inflammation and tissue damage. The critical role that serine proteases (e.g. C1r, C1s, MASPs and factor I) play in the complement cascade made them a primary target for inhibitors used in drug therapy. 35 Nafamostat is a synthetic broad spectrum protease inhibitor not specific for complement and is currently in use for disseminated intravascular coagulation and pancreatitis; however, nafamostat is neurotoxic which limits its use as a therapeutic agent.153,154 C1-INH, previously discussed, is the only complement protease inhibitor that is currently in use for hereditary angioedema. 155 A randomized, double-blind, placebo-controlled study in 125 patients suffering from type I or II hereditary angioedema demonstrated that administration of C1 esterase inhibitor concentrate (Berinert) was highly tolerated and resulted in rapid relief of acute attacks. 156
One of the most successful therapeutic strategies is achieved by targeting complement components with antibody-based agents. Targeting C5 with anti-C5 antibodies and the subsequent reduction in the anaphylatoxin C5a-mediated cellular infiltration was effective in the treatment of paroxysmal nocturnal hemoglobinuria (PNH), myocardial infarction (MI) and arthritis.157–160 Eculizumab, an anti-C5 monoclonal antibody, is FDA approved for the treatment of PNH and aHUS. 161 The development of antibodies targeting factors of the complement pathways also offer additional tools to regulate complement activation and reduce inflammation.162–165 Other pre-clinical experimental studies and clinical trials such as the use of eculizumab in diabetes, GBS, and MPGN are underway to find the best delivery approach and uncover potential benefits of using complement inhibitors in numerous human diseases (ClinicalTrials.gov Identifiers: NCT02727608, NCT02029378, and NCT02093533, accessed on 1 August 2016).
Soluble forms of membrane-bound complement regulators possess a promising therapeutic effect. Weisman et al. developed a soluble form of CR1 (sCR1) which prolonged graft survival when injected into diabetic mice in the absence of immunosuppression therapy.166,167 Also in SCI, sCR1 decreased inflammatory reactions and improved motor function post-acute neurotrauma. 168 In a clinical trial of 59 lung transplant patients, the administration of TP-10, a soluble complement receptor 1 inhibitor, resulted in a decrease in the duration of mechanical ventilation compared to patients receiving a placebo. 169 Another clinical trial of 564 high-risk patients undergoing cardiac surgery showed that TP-10 reduced the incidence of mortality and MI particularly in male patients. 170
The complement system can be also inhibited by interfering with the interaction between activated complement proteins. Compstatin, for instance, discovered through phage-display libraries, binds to C3 effectively hindering the formation of C3 convertase and C3 cleavage into C3a and C3b fragments.171,172 Compstatin was recently found to inhibit complement dysregulation in renal disease. 173
Viruses have been engineered as vectors to deliver human complement inhibitors. In an experimental model of rheumatoid arthritis, mice treated with an adenovirus engineered to express human MBL-associated protein of 44 kDA (AdhMAp44), an alternatively spliced RNA of the MASP-1/3 gene, showed reduced histopathologic damage due to inflammation in the cartilage, synovium and bone. 174 Leaderer et al. cloned genes encoding the CD46, CD55 and CD59 (SACT) or CD55 and CD59 (DTAC) complement regulatory domains into an adeno-associated virus (AAV) vector. 175 Since several viruses pack host cell proteins to evade complement attack, delivering this AAV vector to mice resulted in a significant reduction in human complement-mediated damage. 176 Also, subretinal delivery of human proline/arginine-rich end leucine-rich repeat protein (PRELP), a complement C9 polymerization inhibitor, using an AAV2/8 vector inhibited choroidal neovascularization (CNV) and reduced the deposition of MAC in an experiment model of AMD. 177 Similar findings were also obtained in an AMD animal model using an adenovirus expressing soluble CD59 (sCD59). 178 Adhi et al. proposed the use of AAV2/8-sCD59 as a potential therapy that combats MAC in diabetic retinopathy. 179 In their study, injection of AAV2/8-sCD59 into diabetic mice showed a reduction in retinal blood vessel leakage and retinal ganglion cell apoptosis, in addition to decreased MAC deposition.
Concluding remarks
Previous studies demonstrating the therapeutic utility of complement inhibitors suggest that viral-derived complement inhibitors could also provide novel therapeutic approaches. The use of complement inhibitors of viral origin may be advantageous over current inhibitors due to their higher affinity to components of the host complement system as compared to host regulatory proteins. 129 We now know that SPICE, MOPICE, and VCP are potent inhibitors of the complement system; however, their inhibitory potential and affinity compared to current inhibitors are still to be uncovered. 147 Identifying and characterizing complement inhibitors of viral origin allow the use of combination therapy between human inhibitors and new viral proteins which could be more protective than using a single approach. However, the effectiveness of combining known inhibitors as compared to their co-administration with those of viral origin remains to be understood.
A critical aspect of using viral-derived complement inhibitors is the host’s immune response. This is because administering viral proteins can elicit an immune response that might clear the administered protein before achieving its potential therapeutic effect, particularly with a previous exposure to the same viral antigen. The immunogenicity of viral proteins, bioavailability, dosage (single versus multiple), safety, and purification as well as devising ways to overcome the host’s immunity by means of nanotechnology and targeted therapy represent critical areas for further research.
Viruses have evolved to overcome the host immune response. Further research is warranted to uncover yet undiscovered classes of complement regulatory proteins including those of viral origin. The application of viral-derived complement inhibitors in a clinical setting is still at its infancy but provides promise for novel therapeutic approaches in immunomodulation.
Although physiological inflammation has a protective role, in severe autoimmune and inflammatory diseases, much of the damage results from uncontrolled inflammatory processes. Complement inhibitors may be utilized to reduce inflammation-induced damage to host tissue since inappropriate regulation of the complement pathways has been reported in numerous metabolic, neurological and other pathological conditions.
Virally-encoded complement inhibitors may be themselves a target in viral therapy as an attempt to control viral infectivity and pathogenesis. Equally important, viral-derived complement regulators have the potential to immunomodulate diseases where dysregulated complement activation is a major cause of tissue damage.
Footnotes
Authors’ contributions
HAEH wrote the manuscript, collected data and reviewed all articles cited in the paper. HZ supervised and critically revised the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.